Introduction
DNA methylation in mammalian cells is a conserved
epigenetic silencing mechanism, which is associated with numerous
significant biological processes (1–3). Aberrant
patterns of DNA methylation affect the expression of oncogenes or
tumor suppressor genes (TSGs), encoding proteins involved in
genomic instability, malignant cell growth, cell differentiation
and metastasis (4–6). Therefore, DNA methylation plays a
crucial role in tumorigenesis. Similar to other members of the DNA
methyl transferase (DNMT) family, abnormal levels of DNMT3A have
been identified in numerous types of malignancies (7,8). High
expression of DNMT3A has been observed in non-small cell lung
carcinomas (NSCLCs) (9). However, the
mechanism underlying DNMT3A's role in lung cancer requires further
investigation.
MicroRNAs (miRNAs) are a class of
non-protein-coding, endogenous, small RNAs that cause mRNA
degradation or inhibition of mRNA translation by interacting with
the 3′-untranslated region (3′-UTR) of the target gene mRNA
(10). miRNAs of 22 nucleotides (nt)
play significant regulatory roles in various fundamental biological
processes, including development, differentiation and apoptosis,
sharing common pathways with cancer (11–14). Loss
and gain of miRNA functions contribute to the development of cancer
through the upregulation of oncogenes and silencing of TSGs.
miR-101 acts as a tumor suppressor, which regulates growth,
apoptosis, migration and invasion of various tumor cells (15–17).
However, miR-101 has not been clearly identified as a selective
regulator of DNMT3A in NSCLC.
In the present study, we analyzed targets of miR-101
using bioinformatics, and observed that miR-101 targeted DNMT3A.
Then, using dual-luciferase reporter assays, we further
demonstrated that DNMT3A is a novel target of miR-101. We also
identified that methylation of the phosphatase and tensin homolog
(PTEN) promoter was reduced in A549 cells transfected with miR-101.
Furthermore, in vitro experiments proved that re-expression
of miR-101 inhibited NSCLC proliferation significantly, arrested
the NSCLC cell cycle at the S/G2 phase, and induced cell apoptosis.
Silencing of DNMT3A by miR-101 or siDNMT3A not only downregulated
the cell cycle-related proteins phospho-AKT (p-AKT), cyclin A2
(CCNA2), cyclin-dependent kinase 2 (CDK2) and the cell
apoptosis-related protein B-cell lymphoma 2 (Bcl-2), but also
significantly upregulated bax. The same results were observed in
vivo. These findings demonstrate that miR-101 suppresses the
progression of A549 cells by targeting DNMT3A via the PTEN/AKT
pathway.
Materials and methods
Cell line culture
A549 cells were cultured in RPMI-1640 medium (PAA
Laboratories GmbH, Pasching, Austria) supplemented with 10% fetal
bovine serum (FBS; PAA Laboratories GmbH). Cells were maintained at
37°C in a humidified chamber with 95% air and 5%
CO2.
Plasmid constructions
The vector pcDNA™ 6.2-GW/EmGFP-miR (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to
generate vectors of re-expression of miR-101. First, EcoRI
and HindIII sites were inserted into the multiple cloning
site of the vector. Then, the gene coding for miR-101 was
chemically synthesized and cloned into pcDNA™6.2-GW/EmGFP-miR,
between the EcoRI and HindIII sites
(Pri-miR-101-S5′-TGCCCTGGCTCAGTTATCACAGTGCTGATGCTGTCTATTCTAAAGGTACAGTACTGTGATAACTGAAGGATGGCA-3′,
Pri-miR-101-A5′-TGCCATCCTTCAGTTAGTTATCACAGTACTGTACCTTTAGAATAGACAGCATCAGCACTGTGATAACTGAGCCAGGGCA-3′).
The software programs RegRNA (regulatory RNA motifs and elements
finder; http://regrna.mbc.nctu.edu.tw/), TargetScan
(http://www.targetscan.org/) and DIANA
(http://diana.cslab.ece.ntua.gr/microT/) were used to
predict gene-related specified microRNA. Using bioinformatics
analyses, we identified the fragments of DNMT3A by miR-101
targeting. Specific fragments of DNMT3A were chemically synthesized
(DNMT3A 3′UTR-S 5′-CTATATATATAAAAGGTACTGTTC-3′, DNM T3A 3′UTR-A
5′-TCGAGAACAGTACCTTTTATATATATAGAGCT-3′, DNM T3A 3′UTR-MS
5′-CTATATATATAAAAGACACTGTTC-3′, DNM T3A 3′UTR-MA
5′-TCGAGAACAGTGTCTTTTATATATATAGAGCT-3′). The luciferase-UTR
reporter constructions were generated by introducing the wild-type
(wt)/mutant (mut) EGFR 3′-UTR, carrying a putative miR-101 binding
site, into the pmirGLO Dual-Luciferase miRNA Target Expression
vector (Promega Corporation, Madison, WI, USA), between the
XhoI and SacI sites.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol solution
(Invitrogen Life Technologies) according to the manufacturer's
instructions, and RNAse-free DNase was used to remove DNA
contamination. Total RNA concentration and quantity were assessed
using a DNA/protein analyzer (GeneQuant pro RNA/DNA; GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). cDNA was synthesized from RNA
using a PrimeScript™ RT reagent kit (Takara Biotechnology Co.,
Ltd., Dalian, China). Specific primers were used to synthesize
miR-101 cDNA (miR-101 RT
5′-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTTCAGTT-3′).
cDNA was amplified using SYBR Premix Ex Taq™ II (Takara
Biotechnology Co., Ltd.). The PCR primers used in this study were
miR-101-F 5′-ATCCAGTGCGTGTCGTG-3′ and miR-101-R
5′-TGCTTACAGTACTGTGAT-3′). PCR amplification was performed with an
IQ5 Optical System real-time PCR machine. β-actin and U6 were used
to normalize mRNA and miRNA, respectively (β-actin-F
5′-CGGGAAGCTTGTCATCAATGG-3′, β-actin-R 5′-GGCAGTGATGGCATGGACTG-3′;
U6 RT 5′-GTCGTATCCAGTGCAGGGTCCGAGGTGCACTGGATACGACAAAATATGG-3′, U6-F
5′-TGCGGGTGCTCGCTTCGGCAGC-3′, U6-R 5′-CCAGTGCAGGGTCCGAGGT-3′).
Relative quantification of mRNA expression levels was determined
using the relative standard curve method according to the
manufacturer's instructions (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
MTT assay was performed as described previously
(18). Following transfection with
miR-101, miR-101-inhibitor, si-DNMT3A or their respective controls,
the cells were further cultivated for an additional 1–3 days. Cell
viability was assessed using an MTT assay and a FLUOstar OPTIMA
microplate reader (BMG Labtech, Aylesbury, UK). Each experiment
contained three replicates and was repeated at least twice. The
data were summarized as the means ± standard deviation.
Colony formation assay
Post-transfected A549 cells were re-suspended and
seeded onto 12-well plates at a density of 2,000 cells/well,
incubated for two weeks, and then stained with 0.5% crystal violet
for 30 min. Excess dye was rinsed off twice with phosphate-buffered
saline (PBS). Images were obtained using the computer software
Quantity One® from Bio-Rad Laboratories, Inc.
Cell cycle analysis
Cell cycle analysis was performed as described
previously (18). A549 cells were
transfected with miR-101 re-expression vector, miR-101 inhibitor,
si-DNMT3A or their controls. Cells were harvested by
trypsinization, and 1×106 cells were used for analysis
after 24, 48 and 72 h. The cells were washed with PBS and fixed in
ice-cold ethanol overnight at 4°C. The cells were then washed with
PBS and incubated in 1 ml staining solution (20 µg/ml propidium
iodide and 10 U/ml RNaseA) for 30 min at room temperature. Cell
cycle distributions were assayed by fluorescence-activated cell
sorting using a flow cytometer (FACSort; BD Biosciences, Franklin
Lakes, NJ, USA).
Cell apoptosis analysis
Cell apoptosis analysis was performed with an
Annexin-V/fluorescein isothiocyanate apoptosis detection kit
(Invitrogen Life Technologies), according to the manufacturer's
instructions. Cells were seeded onto 12-well plates at a density of
1×106 cells per well in triplicate, transfected with DNA
vectors or siRNAs for 48 h, and then analyzed using the flow
cytometer (BD Biosciences). Apoptotic populations were determined
using ModFit software (Verity Software House, Inc., Topsham, ME,
USA).
Western blot analysis
Western blot analysis was performed as described
previously (18). A549 cells were
lysed using RIPA buffer supplemented with protease inhibitor
(Invitrogen Life Technologies). Protein concentration was estimated
using a quantitative analyzer (GeneQuant pro RNA/DNA). Proteins
were then separated with 8 to 10% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis (Invitrogen Life
Technologies), transferred to a nitrocellulose membrane, and
incubated with DNMT3A, PTEN, p-AKT, AKT, CCNA2, CDK2, Bcl-2, Bax
and β-actin antibodies (diluted 1/500; Bioworld Technology, Inc.,
St. Louis Park, MN, USA). The membrane was washed three times with
Tris-buffered saline and Tween-20 and incubated with a goat
anti-rabbit antibody (Bioworld, diluted 1/5000). Relative protein
expression was then normalized to β-actin levels in each
sample.
Dual-luciferase assay
Dual-luciferase assay was performed as described
previously. Reporter gene assays were performed 24 h
post-transfection using the Dual Luciferase® reporter
assay system (Promega Corporation) according to the manufacturer's
instructions. Firefly luciferase activity was normalized to
Renilla luciferase activity. All experiments were performed
at least three times.
DNA extraction and
methylation-specific PCR
DNA was extracted using a Qiagen DNeasy tissue kit
(Qiagen Inc., Valencia, CA, USA). DNA (1 µg) was placed in 100 µl
water and denatured by adding 7 µl 3 M NaOH for 10 min at 37°C. To
each denatured DNA solution was added 550 µl freshly prepared
sodium bisulfite mixture (Qiagen, Inc.). The resulting mixtures
were then incubated at 50°C for 16 h. During bisulfite
modification, unmethylated cytosines are deaminated and converted
to uracils, whereas 5-methyl-cytosines remain unaltered. DNA
samples were then purified by ethanol precipitation and
re-suspended in 25–50 µl TE buffer (10 mM Tris/0.1 mM EDTA, pH
7.5). The bisulfite-treated DNA was amplified with
methylation-specific primers (using an annealing temperature of
60°C for 40 cycles) or unmethylated-specific primers (using an
annealing temperature of 58°C for 40 cycles). The primer sequences
were PTENM-F 5′-TTTTTTTTCGGTTTTTCGAGGC-3′, PTE NM-R
5′-CAATCGCGTCCCAACGCCG-3′; PTE NUM-F
5′-TTTTGAGGTGTTTGGGTTTTTGGT-3′, PTENUM-R
5′-ACACAATCACATCCCAACACCA-3′).
Tumorigenicity assay in nude mice
Five-week-old female nude mice were used to analyze
tumorigenicity. A549 cells were transfected with lentiviral vector
(LV)-miR-101 and control (LV-CN) and re-suspended in PBS, then
1×106 cells were injected subcutaneously into both
posterior flanks of nude mice. Tumor size was measured using a
vernier caliper every 3 days for 30 days and monitored by
bioluminescent imaging using IVIS Spectrum (Xenogen Corp., Alameda,
CA, USA). The mice were anesthetized by intra-peritoneal injection
with 1% pentobarbital sodium (50 mg/kg). The tumor was removed
following induction of deep anesthesia and the incision was closed
with surgical staples. Mice were euthanized 3 weeks after the
injection. Tumor volumes (V) were calculated by measuring the
length (L) and width (W) of tumors, using the formula:
V=(LxW2)/2. All animal experiments were approved by the
Institutional Animal Care and Use Committee of Xi'an Jiaotong
University, China.
Statistical analysis
Each experiment was repeated at least three times.
Numerical data are presented as the means ± standard deviation.
Unless indicated, the differences between the two groups were
analyzed using Student's t-test (two-tailed). All statistical
analyses were performed using SPSS 13.0 software (SPSS Inc.,
Chicago, IL, USA).
Results
miR-101 targets DNMT3A
We searched for miR-101 target genes using three
computer-aided miRNA target prediction programs: RegRNA, DIANA and
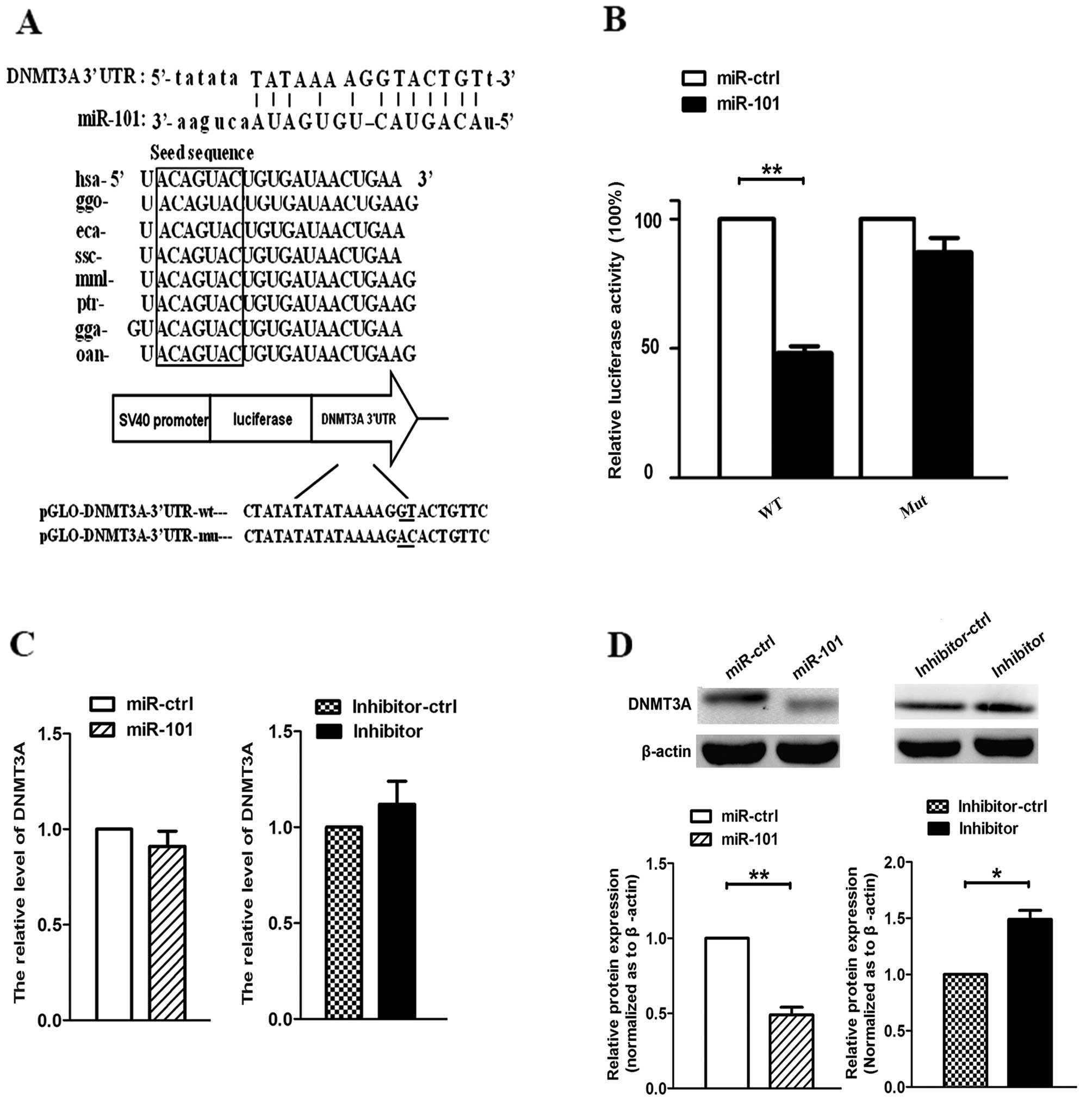
TargetScan. As shown in Fig. 1A, we
identified an miR-101 binding site at 3891–3912 nt of the DNMT3A
3′-UTR. By comparing the human sequence with those of other
species, we observed that the sequence of miR-101 was highly
conserved among different species. To determine whether DNMT3A was
a target gene of miR-101, we constructed pmirGLO-DNMT3A-3′-UTR-wt
and pmirGLO-DNMT3A-3′-UTR-mut. Furthermore, we co-transfected A549
cells with miR-101 or miR-ctrl, and pmirGLO-DNMT3A-3′-UTR-wt or
pmirGLO-DNMT3A-3′-UTR-mut. The results revealed that miR-101
suppressed the firefly luciferase activity of
pmirGLO-DNMT3A-3′-UTR-wt after 24 h, whereas miR-ctrl did not
(Fig. 1B). Next, we demonstrated that
re-expression of miR-101 or expression of miR-101 inhibitor did not
affect the mRNA expression of DNMT3A (Fig. 1C). However, when cells were
transfected with miR-101 and miR-101 inhibitor, the protein levels
of DNMT3A were decreased and increased, respectively (Fig. 1D). These data suggest that miR-101
inhibits DNMT3A expression at the translational but not the
transcriptional level in A549 cells.
DNMT3A affects the expression of a
downstream gene
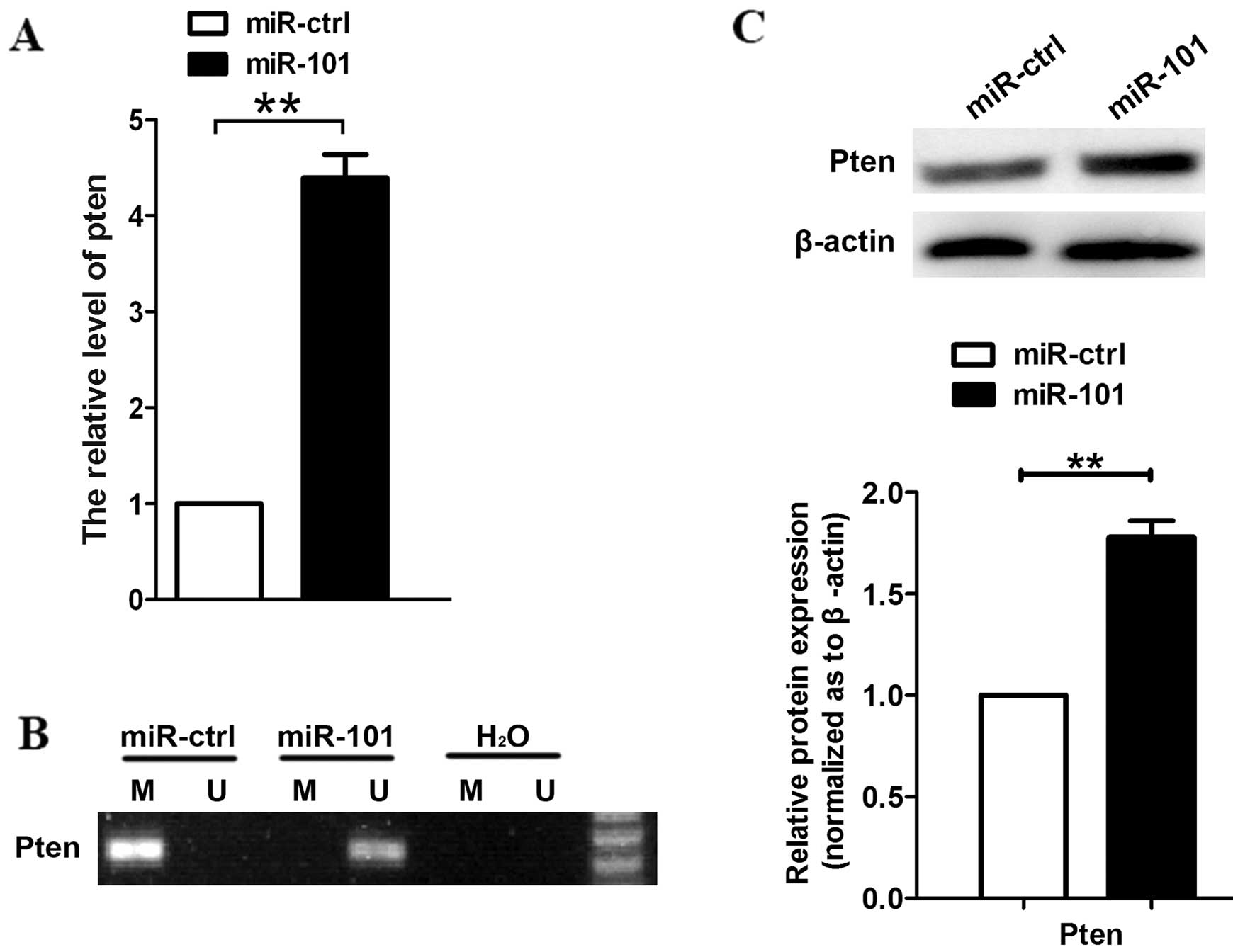
Using RT-qPCR, we measured the transcript levels of
PTEN following transfection with miR-101, and noted that PTEN
expression was increased (Fig. 2A).
Since DNMT3A affects the expression of oncogenes or TSG-encoding
proteins, we examined DNA methylation at the upstream region of the
PTEN coding sequence using methylation-specific PCR. The results
revealed that the CpG sites of PTEN were highly unmethylated in
A549 cells transfected with miR-101, but not in
miR-ctrl-transfected cells (Fig. 2B).
We also observed that protein levels of PTEN were increased upon
transfection with miR-101 (Fig.
2C).
miR-101 inhibits the growth of A549
cells by suppressing the PTEN/AKT signaling pathway
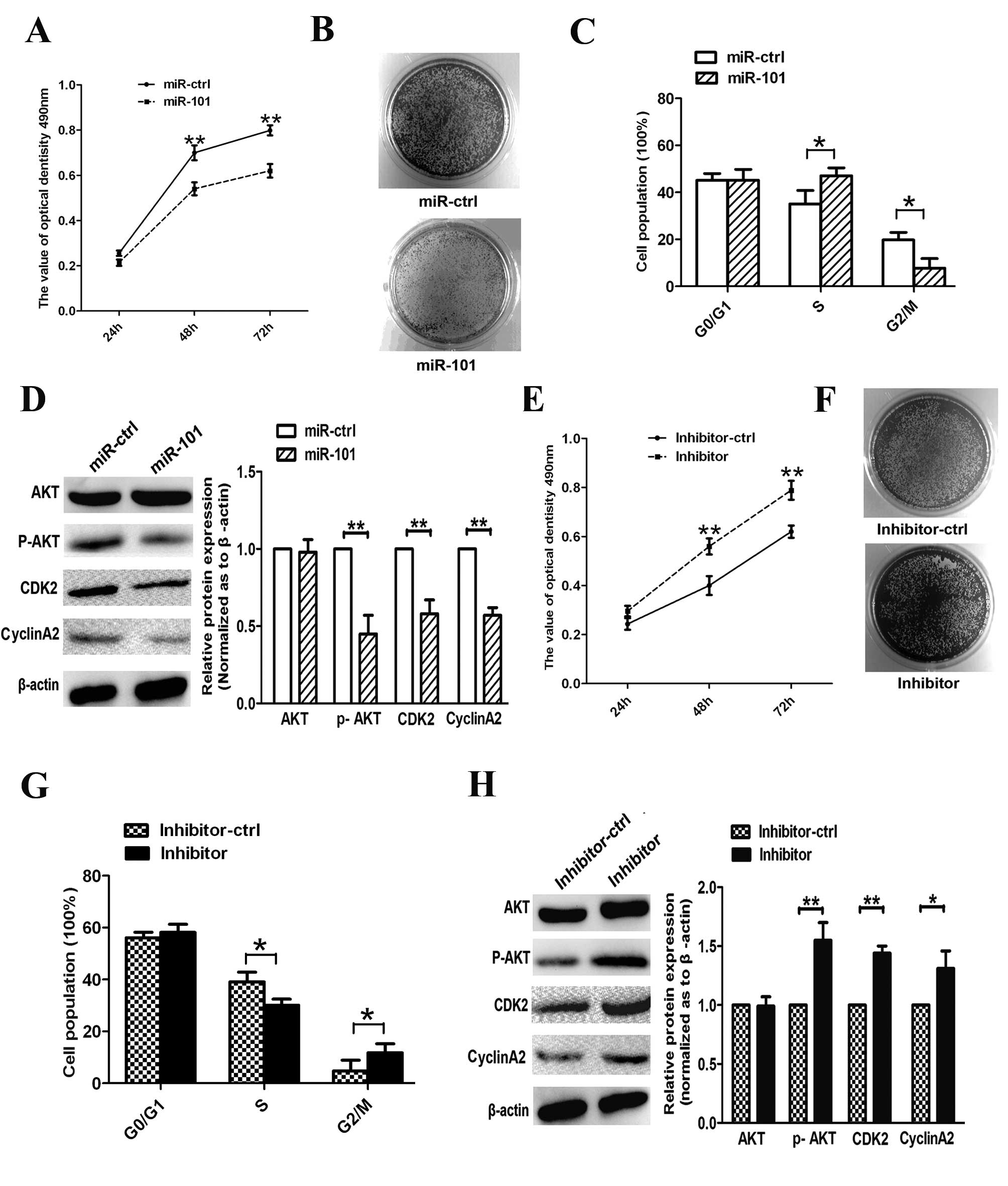
To study the role of miR-101 in A549 cell
proliferation, we performed MTT, clone formation and cell cycle
assays. The results demonstrated that overexpression of miR-101
inhibits the proliferation of A549 after 48 and 72 h (Fig. 3A); miR-101-transfected cells exhibited
fewer and smaller colonies compared with miR-ctrl-transfected cells
(Fig. 3B). Moreover, re-expression of
miR-101 resulted in a marked repression from the S phase to the G2
phase in A549 cells (Fig. 3C). In
addition, we measured the expression of cell cycle regulators
associated with the PTEN/AKT pathway. We observed that the
expression of p-AKT, CCNA2 and CDK2 was suppressed upon
transfection with miR-101 (Fig. 3D),
suggesting that miR-101 arrested the cell cycle at the S/G2
transition phase and suppressed cell proliferation in
vitro.
To examine the anti-proliferative role of miR-101 in
human lung cancer cells, we eliminated endogenous miR-101 in A549
cells using a miR-101 inhibitor. The inhibitor reduced endogenous
expression of miR-101 in A549 cells and increased cell viability
and colony forming numbers (Fig. 3E and
F). We further investigated the effects of miR-101 inhibitor on
cell cycle progression in A549 cells and revealed that transfection
with this inhibitor decreased the amount of cells in the S phase
and increased the percentage of cells in the G2 phase (Fig. 3G). Furthermore, expression levels of
p-AKT, CCNA2 and CDK2 were increased in inhibitor-transfected cells
(Fig. 3H). These results suggest that
endogenous miR-101 plays an essential anti-carcinogenic role in
A549 cells during lung cancer progression.
miR-101 induces apoptosis in A549
cells
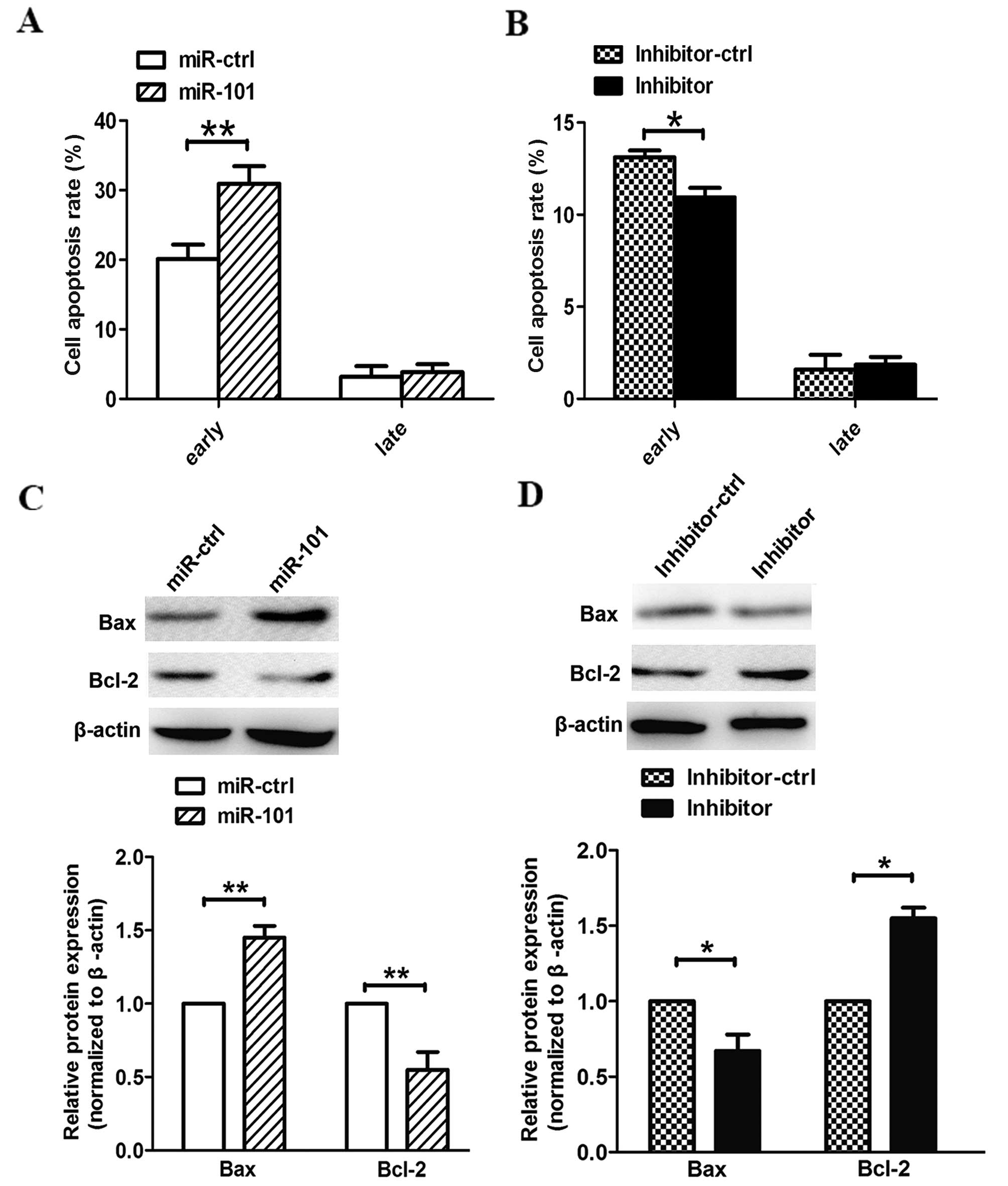
In our in vitro experiments, the
overexpression of miR-101 also induced cell apoptosis. Compared
with cells transfected with a control vector,
pre-miR-101-transfected cells exhibited higher apoptotic rates at
an early phase. In addition, cells transfected with the miR-101
inhibitor exhibited lower apoptotic rates than cells transfected
with a control vector (Fig. 4A and
B). Our data demonstrate that miR-101 induces apoptosis in
human lung cancer cells in vitro. Furthermore, we observed
that miR-101 modified the expression of apoptosis-associated genes
in the PTEN/AKT signaling pathway. As a result, suppression of
p-AKT promoted apoptosis by accelerating Bax and inactivating Bcl-2
(Fig. 4C). Notably, we observed the
opposite phenomenon when A549 cells were transfected with the
miR-101 inhibitor (Fig. 4D).
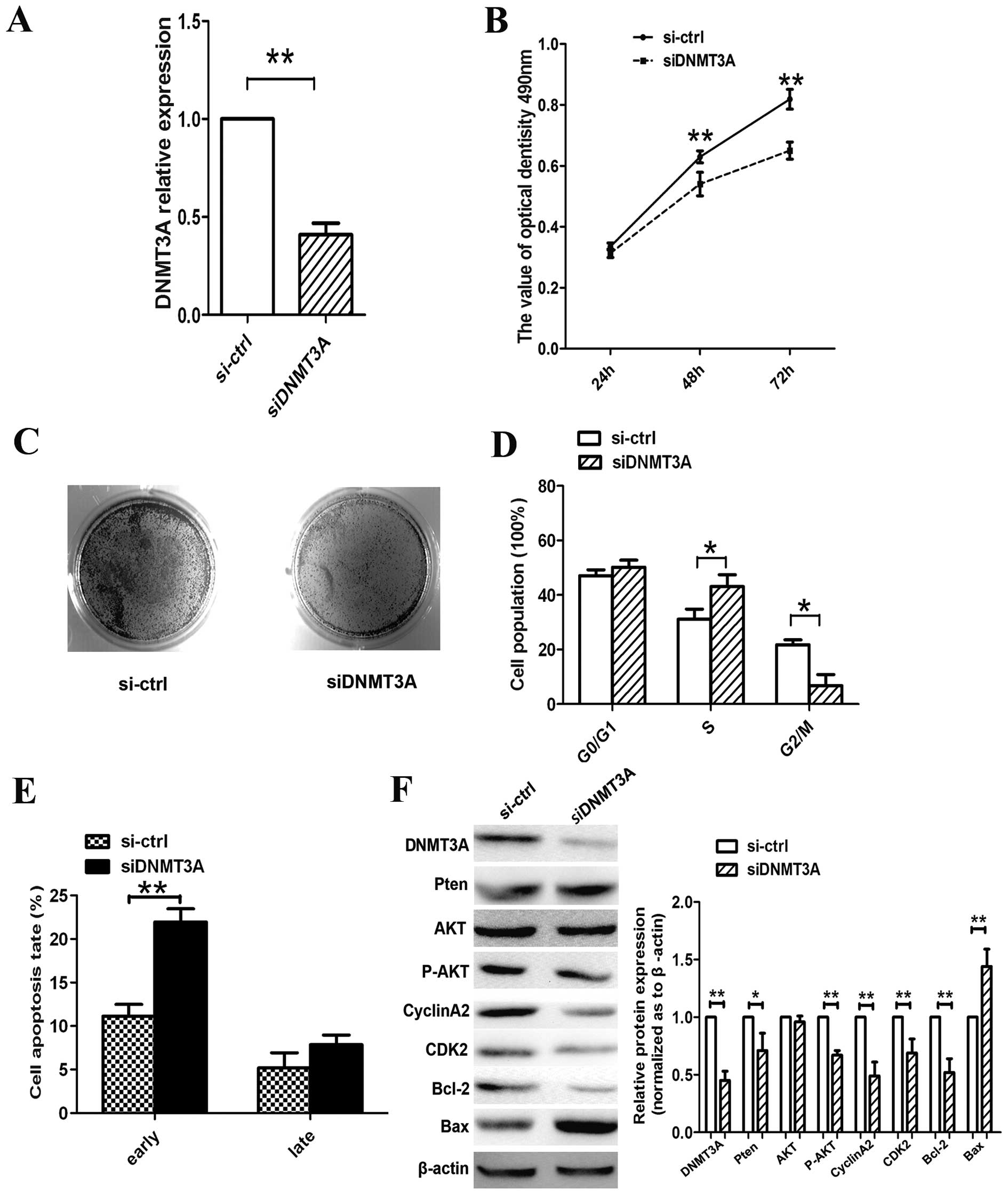
DNMT3A silencing suppresses lung
cancer cell growth and induces G1/S-phase arrest and cell
apoptosis
As demonstrated previously, overexpression of
miR-101 affects cell growth, cell proliferation, cell cycle and
cell apoptosis in A549 lung cancer cells. We also confirmed that
DNMT3A was a direct target of miR-101. Therefore, we silenced
DNMT3A expression using RNA interference to confirm that DNMT3A is
involved in the antitumor effects of miR-101. On the mRNA
expression level, siDNMT3A knocked down DNMT3A (Fig. 5A). Moreover, DNMT3A silencing resulted
in cell growth suppression, arrest of the S/G2 phase and promotion
of cell apoptosis (Fig. 5B-E). These
results follow the same trend as those obtained with
miR-101-transfected A549 cells.
Furthermore, we examined the expression of genes
associated with the PTEN/AKT pathway. As shown in Fig. 5F, the expression levels of DNMT3A and
p-AKT and of the cell cycle regulators CCNA2 and CDK2 were reduced,
whereas PTEN expression was increased. Moreover, siRNA promoted
apoptosis by activating the pro-apoptotic protein Bax and
inactivating the anti-apoptotic protein Bcl-2. Therefore, we
concluded that miR-101 regulates lung cancer cell progression by
directly targeting DNMT3A through the PTEN/AKT signaling
pathway.
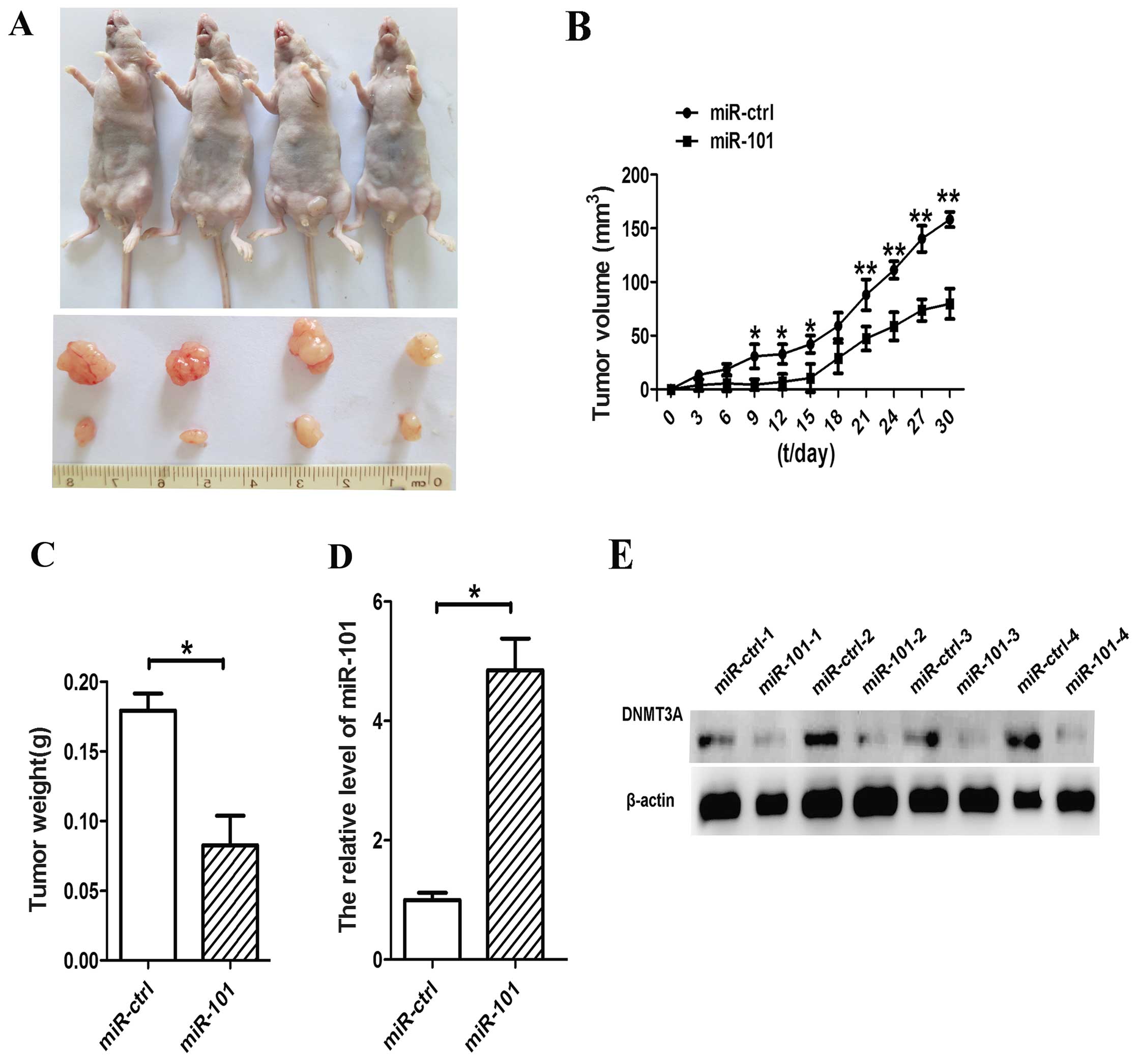
miR-101 induces growth inhibition of
A549 cells in vivo
To further confirm the growth inhibitory function of
miR-101 in lung cancer, we used lentiviral vectors to stably
restore the expression of miR-101 in A549 cells. The
LV-miR-101-infected and LV-CN-infected cells were injected
subcutaneously into the left and right posterior flank of the same
nude mice, respectively. We measured xenograft tumor growth for
four weeks. As shown in Fig. 6A,
tumor growth was significantly suppressed by LV-miR-101 compared
with the control during the experiment. On day 30, the average
volume of miR-101-treated tumors was much smaller than that of
control tumors (Fig. 6B). The average
tumor weights for the control and miR-101 groups on day 30 were
0.18 and 0.07 g, respectively (Fig
6C). Furthermore, expression levels of miR-101 and DNMT3A in
tumor tissues were examined by RT-qPCR and western blot analysis.
Consistent with the in vitro data, the in vivo data
revealed that the expression of miR-101 was increased and the
expression of the DNMT3A protein was decreased in miR-101-treated
tumors (Fig. 6D and E).
Discussion
Lung cancer is one of the most lethal malignant
diseases in the world. NSCLC, constituting ~80% of lung cancer
cases, is a primary type of lung cancer. Current treatments
including chemotherapy have limited efficacy, leading to poor
prognosis and metastasis of lung cancer. Therefore, it is essential
to investigate the underlying molecular mechanisms to improve the
situation. Previous studies have revealed that hypermethylation of
promoters, mediated by DNMTs, is the main reason for epigenetic
inactivation of TSGs. Hypermethylation is responsible for the
silencing of TSGs involved in tumorigenesis (19,20).
DNMT3A, like other DNMT family members, is involved in
tumorigenesis, differentiation and metastasis (21,22), but
the correlation between DNMT3A and NSCLCs remains largely unknown.
In the last ten years, miRNAs have been noted to play a significant
role in the initiation and progression of NSCLCs (23,24).
miR-101 is a miRNA that regulates a variety of tumor-related
biological processes by modulating the expression of several target
genes at the transcript or protein level (25,26).
Normally, miR-101 inhibits the expression of lung cancer promoting
genes (17,27). In this study, we observed a miR-101
binding site at 3891–3912 nt of the DNMT3A 3′-UTR. In addition,
dual-luciferase reporter assays demonstrated that miR-101 targeted
directly DNMT3A by recognizing the 3′-UTR of the DNMT3A miRNA, and
inhibited DNMT3A translation (Fig.
1).
Furthermore, P14, P16, RASSF1A and PTEN are all
TSGs; their functions have been investigated in a number of
malignant tumors (28–30). These genes are frequently inactivated
in numerous human malignancies including lung, breast and
esophageal cancers (31–33). DNMT3A regulated the expression of TSGs
by methylating their upstream region. Fig. 2 demonstrates that the expression of
PTEN was increased and the CpG sites of PTEN were less methylated
in A549 cells transfected with miR-101 than in the control cells.
The same results were observed in hepatocellular carcinoma cells
(7).
PTEN is a phosphatase, and mutations of PTEN are
observed in a number of cancers (34). Normally, degradation and inactivation
of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) is due to PTEN
dephosphorylation (35). The
phosphoinositide 3-kinase (PI3K) pathway is one of the most potent
pro-survival pathways in the development of cancer (36). Inactivation of PTEN not only leads to
accumulation of PIP3, but also increases activity of the kinase
AKT, which contributes to oncogenesis in numerous cancers,
including glioblastoma, prostate and liver cancers (37,38). AKT,
a key downstream effector of the P13K signaling pathway, modulates
the function of numerous substrates associated with cell cycle
progression and cell apoptosis, either by direct phosphorylation of
the target proteins themselves or, indirectly, by regulating
protein expression levels (39). In
our study, overexpression of miR-101 or siDNMT3A inhibited DNMT3A
expression, which resulted in PTEN activation and a decline in AKT
phosphorylation. Next, we verified the effects of AKT on the
downstream target genes CCNA2 and CDK2, which are key
transcriptional factors in the S/G2 phase. From these results, we
noted a reduced expression of CDK2 and CCNA2 in A549 cells
transfected with miR-101. Furthermore, in order to investigate the
role of miR-101 and DNMT3A in the apoptosis of A549 cells, we also
measured the expression levels of Bcl-2 and Bax, and demonstrated
that the miR-101-induced PI3K-AKT pathway plays a significant role
in the regulation of cell apoptosis.
Further animal studies indicated that miR-101
suppressed the growth of lung cancer cells in vivo and
decreased DNMT3A expression in treated tumors (Fig. 6). The in vivo studies support
our in vitro observations that miR-101 targets DNMT3A and
suppresses lung cancer cell growth.
In summary, we investigated the roles of miR-101 and
its targeted gene, DNMT3A, in the cell cycle and apoptosis. Our
findings suggest that miR-101 may be a novel tumor suppressor that
blocks the growth of NSCLC cells through the PTEN/AKT signaling
pathway by targeting DNMT3A. Our findings highlight the functional
association of miR-101 and its host genes, provide new insight into
the regulatory network of the cell cycle and apoptosis, and open
possibilities for future therapeutic interventions.
Acknowledgements
This study was funded by the National Natural
Science Foundation of China (81402008, 31100921 and 5143827), the
Fundamental Research Funds for the Central Universities (08142006),
and the Program for Changjiang Scholars and Innovative Research
Team in University (PCSIRT: 1171).
References
|
1
|
Ohgane J, Aikawa J, Ogura A, Hattori N,
Ogawa T and Shiota K: Analysis of CpG islands of trophoblast giant
cells by restriction landmark genomic scanning. Dev Genet.
22:132–140. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song F, Smith JF, Kimura MT, Morrow AD,
Matsuyama T, Nagase H and Held WA: Association of tissue-specific
differentially methylated regions (TDMs) with differential gene
expression. Proc Natl Acad Sci USA. 102:3336–3341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klose RJ and Bird AP: Genomic DNA
methylation: the mark and its mediators. Trends Biochem Sci.
31:89–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: a landscape takes shape. Cell. 128:635–638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turek-Plewa J and Jagodziński PP: The role
of mammalian DNA methyltransferases in the regulation of gene
expression. Cell Mol Biol Lett. 10:631–647. 2005.PubMed/NCBI
|
|
6
|
Li Y and Tollefsbol TO: Impact on DNA
methylation in cancer prevention and therapy by bioactive dietary
components. Curr Med Chem. 17:2141–2151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Z, Wu Q, Cheng J, Qiu X, Zhang J and
Fan H: Depletion of DNMT3A suppressed cell proliferation and
restored PTEN in hepatocellular carcinoma cell. J Biomed
Biotechnol. 2010:7375352010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Starlard-Davenport A, Kutanzi K, Tryndyak
V, Word B and Lyn-Cook B: Restoration of the methylation status of
hypermethylated gene promoters by microRNA-29b in human breast
cancer: a novel epigenetic therapeutic approach. J Carcinog.
12:152013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, et al: MicroRNA-29 family reverts aberrant
methylation in lung cancer by targeting DNA methyltransferases 3A
and 3B. Proc Natl Acad Sci USA. 104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiemer EA: The role of microRNAs in
cancer: no small matter. Eur J Cancer. 43:1529–1544. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wienholds E and Plasterk RH: MicroRNA
function in animal development. FEBS Lett. 579:5911–5922. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu W, Mao SY and Zhu WY: Impact of tiny
miRNAs on cancers. World J Gastroenterol. 13:497–502. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oakley EJ and Van Zant G: Unraveling the
complex regulation of stem cells: implications for aging and
cancer. Leukemia. 21:612–621. 2007.PubMed/NCBI
|
|
15
|
Shen Q, Bae HJ, Eun JW, Kim HS, Park SJ,
Shin WC, Lee EK, Park S, Park WS, Lee JY, et al: MiR-101 functions
as a tumor suppressor by directly targeting nemo-like kinase in
liver cancer. Cancer Lett. 344:204–211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiaoping L, Zhibin Y, Wenjuan L, Zeyou W,
Gang X, Zhaohui L, Ying Z, Minghua W and Guiyuan L: CPEB1, a
histone-modified hypomethylated gene, is regulated by miR-101 and
involved in cell senescence in glioma. Cell Death Dis. 4:e6752013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin J, Wang M, Jin C and Qi Q: miR-101
sensitizes A549 NSCLC cell line to CDDP by activating caspase
3-dependent apoptosis. Oncol Lett. 7:461–465. 2014.PubMed/NCBI
|
|
18
|
Wang L, Yao J, Zhang X, Guo B, Le X,
Cubberly M, Li Z, Nan K, Song T and Huang C: miRNA-302b suppresses
human hepatocellular carcinoma by targeting AKT2. Mol Cancer Res.
12:190–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu YY, Chen C, Kong FF and Zhang W:
Clinicopathological significance and potential drug target of RUNX3
in breast cancer. Drug Des Devel Ther. 8:2423–2430. 2014.PubMed/NCBI
|
|
20
|
Wang D, Cui W, Wu X, Qu Y, Wang N, Shi B
and Hou P: RUNX3 site-specific hypermethylation predicts papillary
thyroid cancer recurrence. Am J Cancer Res. 4:725–737.
2014.PubMed/NCBI
|
|
21
|
Ma QL, Wang JH, Wang YG, Hu C, Mu QT, Yu
MX, Wang L, Wang DM, Yang M, Yin XF, et al: High IDH1 expression is
associated with a poor prognosis in cytogenetically normal acute
myeloid leukemia. Int J Cancer. 137:1058–1065. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao XY, Ma HX, Shang YH, Jin MS, Kong F,
Jia ZF, Cao DH, Wang YP, Suo J and Jiang J: DNA methyltransferase3a
expression is an independent poor prognostic indicator in gastric
cancer. World J Gastroenterol. 20:8201–8208. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang P, Ye B, Yang Y, Shi J and Zhao H:
MicroRNA-181 functions as a tumor suppressor in non-small cell lung
cancer (NSCLC) by targeting Bcl-2. Tumour Biol. 36:3381–3387. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mataki H, Seki N, Chiyomaru T, Enokida H,
Goto Y, Kumamoto T, Machida K, Mizuno K, Nakagawa M and Inoue H:
Tumor-suppressive microRNA-206 as a dual inhibitor of MET and EGFR
oncogenic signaling in lung squamous cell carcinoma. Int J Oncol.
46:1039–1050. 2015.PubMed/NCBI
|
|
25
|
Lin C, Huang F, Li QZ and Zhang YJ:
miR-101 suppresses tumor proliferation and migration and induces
apoptosis by targeting EZH2 in esophageal cancer cells. Int J Clin
Exp Pathol. 7:6543–6550. 2014.PubMed/NCBI
|
|
26
|
Liu L, Guo J, Yu L, Cai J, Gui T, Tang H,
Song L, Wang J, Han F, Yang C, et al: miR-101 regulates expression
of EZH2 and contributes to progression of and cisplatin resistance
in epithelial ovarian cancer. Tumour Biol. 35:12619–12626. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lei YM, Zu YF, Wang J, et al:
Interleukin-1β-mediated suppression of microRNA-101 and
upregulation of enhancer of zeste homolog 2 is involved in
particle-induced lung cancer. Med Oncol. 32:3872015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chaar I, Amara S, Elamine OE, Khiari M,
Ounissi D, Khalfallah T, Ben Hmida A, Mzabi S and Bouraoui S:
Biological significance of promoter hypermethylation of p14/ARF
gene: relationships to p53 mutational status in Tunisian population
with colorectal carcinoma. Tumour Biol. 35:1439–1449. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Camacho CV, Mukherjee B, McEllin B, Ding
LH, Hu B, Habib AA, Xie XJ, Nirodi CS, Saha D, Story MD, et al:
Loss of p15/Ink4b accompanies tumorigenesis triggered by complex
DNA double-strand breaks. Carcinogenesis. 31:1889–1896. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Korah R, Healy JM, Kunstman JW, Fonseca
AL, Ameri AH, Prasad ML and Carling T: Epigenetic silencing of
RASSF1A deregulates cytoskeleton and promotes malignant behavior of
adrenocortical carcinoma. Mol Cancer. 12:872013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamada K, Kohno T, Takahashi M, Yamazaki
M, Yamazaki M, Tashiro H, Sugawara C, Ohwada S, Sekido Y, Minna JD,
et al: Two regions of homozygous deletion clusters at chromosome
band 9p21 in human lung cancer. Genes Chromosomes Cancer.
27:308–318. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayashi N, Sugimoto Y, Tsuchiya E, Ogawa M
and Nakamura Y: Somatic mutations of the MTS (multiple tumor
suppressor) 1/CDK4l (cyclin-dependent kinase-4 inhibitor) gene in
human primary non-small cell lung carcinomas. Biochem Biophys Res
Commun. 202:1426–1430. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Burbee DG, Forgacs E, Zöchbauer-Muller S,
Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S,
et al: Epigenetic inactivation of RASSF1A in lung and breast
cancers and malignant phenotype suppression. J Natl Cancer Inst.
93:691–699. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH
and Hung MC: Cytoplasmic localization of p21Cip1/WAF1 by
Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat
Cell Biol. 3:245–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sano T, Lin H, Chen X, Langford LA, Koul
D, Bondy ML, Hess KR, Myers JN, Hong YK, Yung WK and Steck PA:
Differential expression of MMAC/PTEN in glioblastoma multiforme:
relationship to localization and prognosis. Cancer Res.
59:1820–1824. 1999.PubMed/NCBI
|
|
38
|
Buontempo F, Ersahin T, Missiroli S,
Senturk S, Etro D, Ozturk M, Capitani S, Cetin-Atalay R and Neri
ML: Inhibition of Akt signaling in hepatoma cells induces apoptotic
cell death independent of Akt activation status. Invest New Drugs.
29:1303–1313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu N, Lao Y, Zhang Y and Gillespie DA:
Akt: A double-edged sword in cell proliferation and genome
stability. J Oncol. 2012:9517242012. View Article : Google Scholar : PubMed/NCBI
|