Introduction
Carcinogenesis is a multistep and multifactorial
process in which inflammation is important. Clinical observations
have indicated that the use of non-steroidal anti-inflammatory
drugs (NSAIDs) may reduce tumor growth and development, and may
improve patient survival (1,2). The main effect of NSAIDs is the
inhibition of cyclooxygenase (COX) enzymes, which exist in two
isoforms, COX-1 and COX-2, and catalyze the conversion of
arachidonic acid to prostanoids. Prostanoid-promoted tumor growth
and subsequent host survival are driven by the signaling of
prostaglandin E2 (PGE2), which acts via
E-prostanoid 1–4 (EP1-4) subtype receptors (3).
It is well-recognized that COX-2/PGE2
signaling involves several different processes of tumor
progression, including the inhibition of apoptosis, increased
angiogenesis and metastatic formation (4). Thus, the actions of PGE2
depend on the pattern or mix of EP receptor expression in normal
and cancer cells, since each receptor promotes distinctive
downstream signaling. In a previous study on human colorectal
cancer, we reported high levels of EP1 and EP2 receptor protein in
human tumor tissue, with considerably less content in the stroma.
In the same study, EP3 receptor protein occurred mostly in the
submucosa of such tumors and occasionally in the tumor cells, while
EP4 receptor protein was usually not visible at all in the human
colorectal tumor cells (5). It was
also found that disease-specific mortality in colorectal cancer
showed a negative correlation with EP2 receptor expression
(5,6).
Moreover, several studies have displayed that EP2-knockout mice
have reduced tumor growth compared with tumors in wild-type mice
(7–11). Therefore, we speculate that the
differences in gene expression among tumors growing on EP2-knockout
mice versus tumors growing on wild-type mice may indicate important
signaling between the tumor and stroma tissue, which subsequently
determines tumor growth and progression. Thus, the aim of the
present study was to screen for potentially important transcript
alterations in tumor tissue associated with host EP2 receptor
signaling.
Materials and methods
Animals
The animal experimental protocol was approved by the
Committee for Ethics at the University of Gothenburg (Gothenburg,
Sweden). Adult, male and female (6-7-month-old, 20±1 g) age-matched
EP2−/− (n=7) and EP2+/+ (n=8) mice (C57BL/6
genetic backgrounds) were in-house bred (12) and housed in plastic cages in a
temperature controlled room with a 12 h dark/light cycle, and
received laboratory rodent chow (B&K Universal AB, Stockholm,
Sweden). Tumors were implanted subcutaneously in the bilateral
flanks with a 3-5-mm3 syringe containing a
transplantable MCG-101 methylcholanthrene-induced sarcoma while the
mice were under general anesthesia (isoflurane, inhaled
concentration of 2.7%). This tumor model has been used for several
years at the Department of Surgery, University of Gothenburg
(Gothenburg, Sweden) and is known to produce increased levels of
PGE2 in the systemic circulation (10). The model originates from a chemically
induced tumor on C57BL mice and appears now to be an
epithelial-like poorly-differentiated tumor. All mice were
sacrificed on day 10 post-tumor implantation, when blood samples
were obtained during general anesthesia by cardiac puncture for
plasma PGE2 determination and additional analyses.
Plasma samples were collected in ethylenediaminetetraacetic acid
tubes and stored at −80°C until use. Tumor biopsies were collected
in RNA-later according to the manufacturer's protocols
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany). Wet tumor
weight was determined by direct weighing immediately upon
resection.
Plasma analysis
Multiplex enzyme-linked immunosorbent assays
(Luminex®; Merck KGaA, Darmstadt, Germany) were
performed on plasma samples from the EP2-knockout mice and
wild-type mice using two different panels: i) Mouse acute phase
magnetic bead panel 2 with antibodies towards serum amyloid
protein/pentraxin-2 (SAP), adipsin (complement factor D),
α-2-macroglobulin (A2M) and haptoglobin; and ii) a mouse cytokine
magnetic bead panel with antibodies towards IL-1α, IL-1β, IL-6,
IL-10, tumor necrosis factor α (TNFα), vascular endothelial growth
factor and interferon γ (IFNγ) (Milliplex® Map; Merck
KGaA). Analyses were performed according to the manufacturer's
protocols for plasma samples and all samples were run in duplicate;
plasma samples used for the acute phase panel were diluted 1:1,000
in assay buffer and those for the cytokine panel were diluted 1:2
in assay buffer.
Tumor analysis: RNA extraction
Total RNA was extracted from tumor tissue using an
RNeasy Fibrous Tissue kit according to the manufacturer's protocols
(Qiagen GmbH, Hilden, Germany). The quality and quantity of RNA
were checked in an Agilent 2100 Bioanalyzer using an RNA 6000 Nano
assay kit with an RNA Integrity number of 7 as the limit for
further analyses (Agilent Technologies, Inc., Santa Clara, CA,
USA). Concentrations of RNA were measured in a NanoDrop ND-1000A
spectrophotometer (NanoDrop Technologies, Inc.; Thermo Fisher
Scientific, Inc., Wilmington, DE, USA). RNA from the wild-type mice
(n=8) was pooled and used as reference RNA in the gene expression
analyses.
Tumor analysis: Gene expression
microarray
Microarray analyses were performed on 7 biological
replicates of tumor mRNA from EP2−/− mice, with pooled
wild-type RNA as reference RNA. mRNA (500 ng) from the tumors was
labeled with Cy-3-dCTP in a cDNA synthesis reaction with Agilent
Flourescent Direct Label (Agilent Technologies, Inc.). Pooled
wild-type tumor RNA samples (500 ng) were labeled with Cy-5-dCTP.
The samples were then hybridized using a SurePrint G3 Mouse GE
8×60K Microarray kit, design ID 028005 [detects 4,623 long
non-coding RNAs (lncRNAs); Agilent Technologies, Inc.] according to
the manufacturer's instructions. Microarrays were quantified on an
Agilent G2505C microarray scanner and fluorescence intensities were
extracted using the Feature Extraction software program (v10.7.1.1;
Agilent Technologies, Inc.). Dye-normalized, outlier- and
background-subtracted values were analyzed in the GeneSpring GX10
software program (Agilent Technologies, Inc.) according to standard
procedures. Quality control was performed with QC metrics. A
fold-change (FC) of >1.5 of log2-transformed ratios was
considered a statistically significant change in gene expression.
The analyses used were ‘filter on flags’ (present or marginal) and
the t-test (P<0.05). The results are presented as the mean FC
value of 7 replicates.
Statistical analysis
Statistical analyses were performed with StatView
for Windows version 5.0.1 (SAS institute Inc., Cary, NC, USA). The
results are presented as the mean ± standard error of the mean.
Cytokine and acute phase results were analyzed with an analysis of
variance followed by Fisher's protected least significant
difference test. P≤0.05 was considered to indicate a statistically
significant difference in two-tailed tests.
Results
Tumor growth and systemic
inflammation
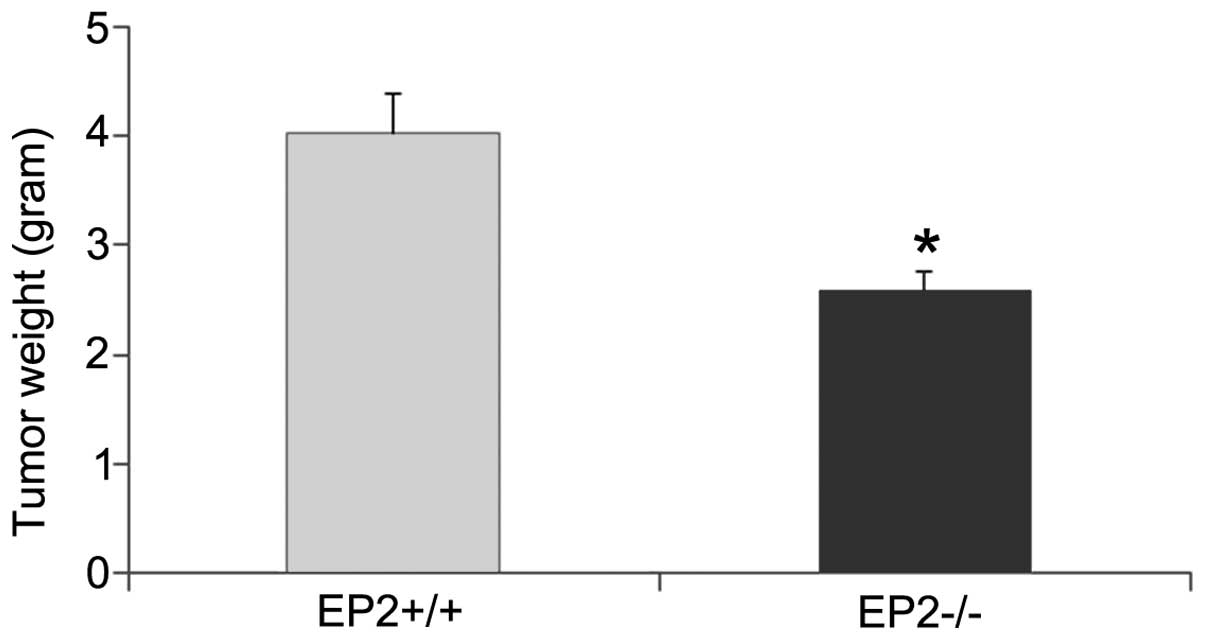
Tumor growth in the EP2−/− mice was
significantly reduced compared with tumor growth in the wild-type
mice at 10 days post-tumor implantation (Fig. 1). No significant difference in plasma
PGE2 was detected between the EP2-knockout and wild-type
mice, as previously reported (10).
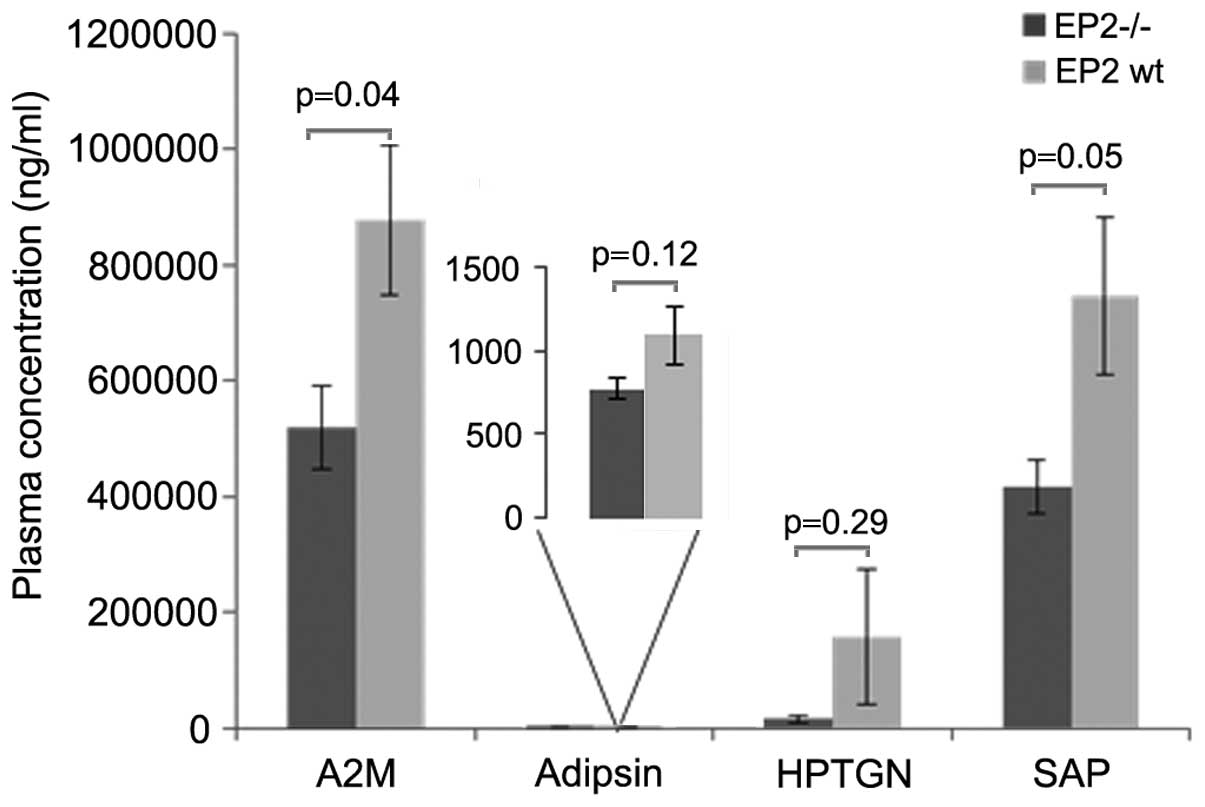
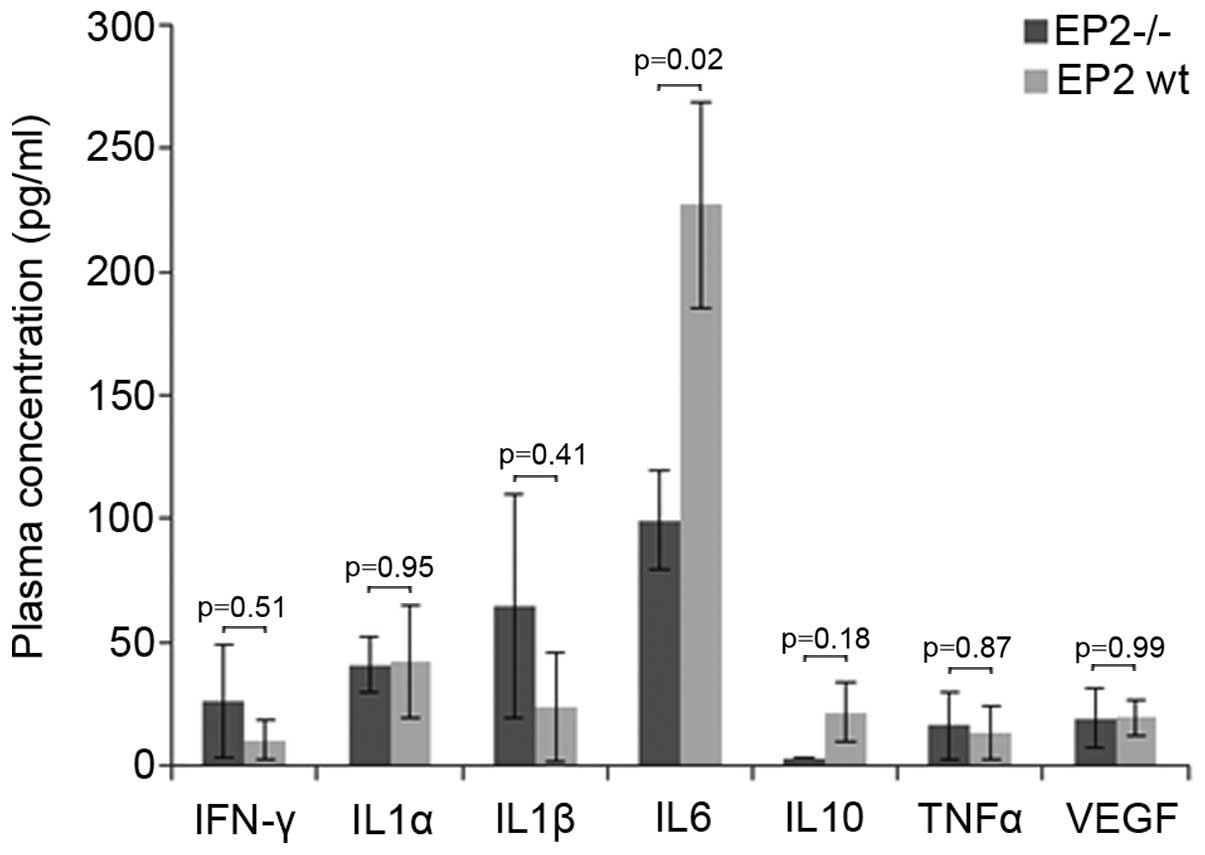
Systemic inflammation was decreased in the EP2−/− mice,
as displayed by significantly reduced levels of the acute phase
proteins A2M and SAP in the blood (Fig.
2). The cytokine assay displayed non-detectable levels (<3.2
ng/ml) of IL-1β (in 6 out of 15 mice), IFNγ (in 12 out of 15 mice),
IL-10 (in 13 out of 15 mice) and TNFα (in 11 out of 15 mice), while
IL-6 was detected in all samples and was significantly reduced in
plasma from EP2−/− mice (Fig.
3). Non-tumor-bearing animals showed no signs of acute phase
reactions (13,14).
Tumor gene expression
A total of 55,821 gene expression microarray
entities passed the ‘filter on flags’ analysis, while 16,127
entities exhibited significant alterations upon t-test (P<0.05).
Results displayed the changed expression of several genes as well
as lncRNA expression (Table I). At
2-fold change [FC 2], several entities were changed; 1,685 genes
and 234 lncRNA were upregulated while 800 genes and 465 lncRNA were
downregulated. Increasing FC, representing greater differences,
indicated a trend towards downregulation in the knockout mice. At
FC 5, 153 entities were downregulated, and at FC 10, 7 genes and 7
lncRNAs were downregulated compared with the tumors in the
wild-type mice. Genes with highly changed expression were
associated with a range of different cellular functions (Table II).
 | Table I.Significantly changed number of
lncRNAs per chromosome in tumors from E-prostanoid 2−/−
mice compared with tumors from wild-type mice. |
Table I.
Significantly changed number of
lncRNAs per chromosome in tumors from E-prostanoid 2−/−
mice compared with tumors from wild-type mice.
| Chromosome | FC 2.0 | FC 5.0 | FC 10 |
|---|
| 1 | 21↓, 24↑ | 2↓ | 1↓ |
| 2 | 26↓, 14↑ | 3↓ | – |
| 3 | 39↓, 12↑ | – | – |
| 4 | 35↓, 12↑ | – | – |
| 5 | 34↓, 11↑ | 3↓ | 2↓ |
| 6 | 25↓, 12↑ | – | – |
| 7 | 20↓, 8↑ | 1↓ | – |
| 8 | 25↓, 17↑ | 2↓ | – |
| 9 | 26↓, 6↑ | 2↓ | – |
| 10 | 21↓, 15↑ | 1↓ | – |
| 11 | 21↓, 16↑ | – | – |
| 12 | 19↓, 8↑ | – | – |
| 13 | 24↓, 7↑ | 2↓ | 1↓ |
| 14 | 21↓, 12↑ | – | – |
| 15 | 15↓, 14↑ | – | – |
| 16 | 20↓, 9↑ | 1↓ | – |
| 17 | 13↓, 12↑ | – | – |
| 18 | 17↓, 7↑ | – | – |
| 19 | 11↓, 7↑ | 1↓ | – |
| 20a | – | – | – |
| 21a | – | – | – |
| 22a | – | – | – |
| X | 32↓, 10↑ | 1↓ | – |
| Y | – | – | – |
| Table II.Genes with the most changed
expression in tumor tissue from EP2−/− mice versus
tumors in wild-type mice. |
Table II.
Genes with the most changed
expression in tumor tissue from EP2−/− mice versus
tumors in wild-type mice.
| Gene | Regulation | FC | Name/known
function |
|---|
| Npl | ↑ |
9.4 | N-acetylneuraminate
pyruvate lyase |
| Ptplad2 | ↑ |
8.6 | Protein tyrosine
phosphatase-like A domain-containing 2 |
| Ptger2 | ↑ |
7.9 | PGE2
receptor EP2 |
| Ear11 | ↑ |
7.6 | Ribonuclease,
RNase2A |
| Gm5150 | ↑ |
7.0 | Predicted gene
5150, protein coding |
| Slc40a1 | ↑ |
6.8 | Solute carrier
family 40 (iron-regulated transporter) |
| Cybb | ↑ |
6.6 | Cytochrome b-245,
important in the innate immune response, increased activity leads
to the generation of reactive oxygen species that result in
oxidative stress and can cause tissue damage. |
| Dock10 | ↑ |
6.5 | Dedicator of
cytokinesis 10 |
| Gstm5 | ↑ |
6.5 | Glutathione
S-transferase |
| Cd79b | ↑ |
6.4 | CD79B antigen also
known as B29, Igb, Igβ or Ig-β |
| Cpne5 | ↓ | 16.0 | May regulate
molecular events at the interface of the cell membrane and
cytoplasm |
| Krt18 | ↓ | 14.9 | Protects epithelial
tissue from injury |
|
Tnfrsf13b | ↓ | 12.7 | TNF receptor
superfamily, regulation of B cell function |
| Fndc7 | ↓ | 12.3 | Fibronectin type
III |
| Lrit1 | ↓ | 12.0 | Leucine-rich
repeat, immunoglobulin-like transmembrane domains 1 |
| Spesp1 | ↓ | 10.6 | Required for fully
fertile sperm in mice |
| Sntg1 | ↓ | 10.0 | Syntrophins are
cytoplasmic peripheral membrane proteins |
| Vmn2r69 | ↓ |
9.9 | Vomeronasal 2,
receptor 69, Vomeronasal organ |
| Rn18s | ↓ |
8.7 | 18S ribosomal
RNA |
| Slc7a15 | ↓ |
8.7 | Solute carrier
family 7 (cationic amino acid transporter) |
PGE2-related transcripts were
significantly changed in the tumor tissues from the
EP2−/− mice (Table III);
Cox-1 and 15-hydroxyprostaglandin dehydrogenase
(15-Pgdh) expression was upregulated, while Cox-2
expression was downregulated. EP subtype receptors were
upregulated, except EP3, in tumors on EP2−/− mice,
particularly EP2 receptor (Ptger2; FC 7.9). Another
prostanoid receptor with highly changed expression was Ppara
(PPARα), which was 4-fold downregulated.
| Table III.Effect of host EP2−/− on
prostanoid-related transcripts in tumor tissue.a |
Table III.
Effect of host EP2−/− on
prostanoid-related transcripts in tumor tissue.a
| Gene |
Enzyme/receptor | FC | Regulation |
|---|
| PG metabolism |
|
|
|
|
Ptgs1 | COX-1 | 2.6 | ↑ |
|
Ptgs2 | COX-2 | 1.5 | ↓ |
|
Hpgd | 15-PGDH | 3.1 | ↑ |
|
Pgt | PGT | 2.1 | ↓ |
|
Mrp4 | MRP4 | – | – |
|
PGE2 |
|
|
|
|
Ptges | mPGES-1 | 1.5 | ↓ |
|
Ptges2 | mPGES-2 | – | – |
|
Ptges3 | cPGES | – | – |
|
Ptger1 | EP1 receptor | 1.7 | ↑ |
|
Ptger2 | EP2 receptor | 7.9 | ↑ |
|
Ptger3 | EP3 receptor | 3.7 | ↓ |
|
Ptger4 | EP4 receptor | 3.2 | ↑ |
|
PGD2 |
|
|
|
|
Ptgds | PGD2 synthase | – | – |
|
Ptgdr | DP1 receptor | 1.5 | ↓ |
|
Gpr44 | DP2 receptor | – | – |
|
PGF2α |
|
|
|
|
2810405K02Rik | PGF synthase | 2.1 | ↑ |
|
Ptgfr | FP receptor | 2.0 | ↑ |
|
Ptgfrn | Neg. regulator
PGF2 | 1.5 | ↑ |
|
PGI2 |
|
|
|
|
Ptgis | PGI synthase | – | – |
|
Ptgir | IP receptor | 1.8 | ↑ |
|
TXA2 |
|
|
|
|
Tbxas1 | TXA synthase | 3.1 | ↑ |
|
Tbxa2r | TP receptor | 1.7 | ↑ |
| Nuclear
receptors |
|
|
|
|
Ppara | PPARα receptor | 4.3 | ↓ |
|
Ppard | PPARδ receptor | – | – |
|
Pparg | PPARγ receptor | – | – |
Genes associated with tumor progression, including
stemness [cluster of differentiation 133/prominin-1
(Cd133/Prom1) and Cd44], displayed decreased
expression in the tumor tissue of the EP2−/− mice, while
the majority of cytokines (Il1b, Il6, Il10 and Ifng)
and metalloproteinase 13 (Mmp13) showed increased expression
(Table IV). Only genes with
significantly changed expression are reported in Table IV. Other genes that may be important
for tumor progression did not show significantly altered
expression, including TNFα (Tfab2a), Nfκb,
Nfat and Ras-family transcripts.
| Table IV.Transcripts of genes associated with
tumor progression, transcription and cancer stemness that were
significantly changed in tumor tissue from EP2−/−
mice.a |
Table IV.
Transcripts of genes associated with
tumor progression, transcription and cancer stemness that were
significantly changed in tumor tissue from EP2−/−
mice.a
| Gene | FC | Regulation |
|---|
| Cytokines |
|
|
|
IL1B | 2.1 | ↑ |
|
IL6 | 2.8 | ↑ |
|
IL6R | 1.6 | ↓ |
|
IL10 | 1.9 | ↑ |
|
IL10RA | 2.2 | ↑ |
|
IFNg | 2.3 | ↑ |
| Stemness |
|
|
|
CD133/Prom1 | 1.5 | ↓ |
|
CD44 | 1.9 | ↓ |
| Tumor
progression |
|
|
|
Mmp13 | 6.1 | ↑ |
|
Mmp7 | 1.5 | ↓ |
|
Hif1a | 1.8 | ↑ |
|
Dnmt3a | 1.6 | ↓ |
|
Apc | 1.5 | ↓ |
|
Gsk3b | 1.6 | ↓ |
|
Egfr | 2.0 | ↑ |
|
Prkcb | 2.9 | ↑ |
| Transcription |
|
|
|
Batf | 3.5 | ↑ |
|
Cebpa | 3.4 | ↑ |
|
Creb5 | 1.9 | ↓ |
|
Fos | 2.7 | ↑ |
|
Tcf4 | 1.8 | ↓ |
Discussion
The elevated production of PGE2 has been
linked to tumor progression in numerous types of cancer, and
reduced PGE2 activities with COX-inhibiting NSAIDs have
been reported to reduce tumor growth in animal models, as well as
in clinical studies (4–6,15,16). However, the non-specific drug
inhibition of COX can also induce adverse side effects (4–6,15,16).
Therefore, it is of interest to find more specific targets to
inhibit the actions of prostaglandins in cancer. PGE2
activates subtype receptors, EP1-4, which induce downstream
signaling upon activation. EP2 receptor signaling is involved in
several different physiological and disease-related events, which
are of interest in targeted therapy (17). In tumors, EP2 and EP4 have gained the
most attention, as tumor-host knockout models have been shown to
display reduced tumor growth (7,18).
Previous studies have focused on polyp formation, proliferation,
apoptosis and angiogenesis, while the present study evaluated
overall transcription in tumor cells (7–9,19–22).
Indeed, tumor growth was significantly reduced in the present
EP2-knockout tumor-bearing mice where the host lacked EP2
expression, while tumor cells displayed EP2 receptor expression.
Thus, a response circuit between host and tumor cells appears to be
interrupted by a lack of EP2 signaling from the host stroma, as
illustrated in Fig. 4. The systemic
effects in tumor-bearing EP2-knockout mice were reduced
inflammation and a reduced plasma IL-6 level, as expected.
Accordingly, inflammation and inflammatory factors, such as IL-6,
have been recognized to drive tumor progression (23), which may be one of several factors
behind reduced tumor growth in EP2 receptor-knockout mice. IL-6 and
PGE2 have been shown to affect the
epithelial-to-mesenchymal transition (EMT) in tumor cells via the
EP2 and EP4 receptors (23). Also,
EMT was shown to affect the expression of EP subtype receptors in
human bronchial epithelial cells with reduced EP2, EP4 and one of
the EP3 subtype receptors (24). In
the same study, an altered response to PGE2 regarding
cell migration was detected after EMT from stimulation to
inhibition, mediated by EP2 and EP4 receptors (24).
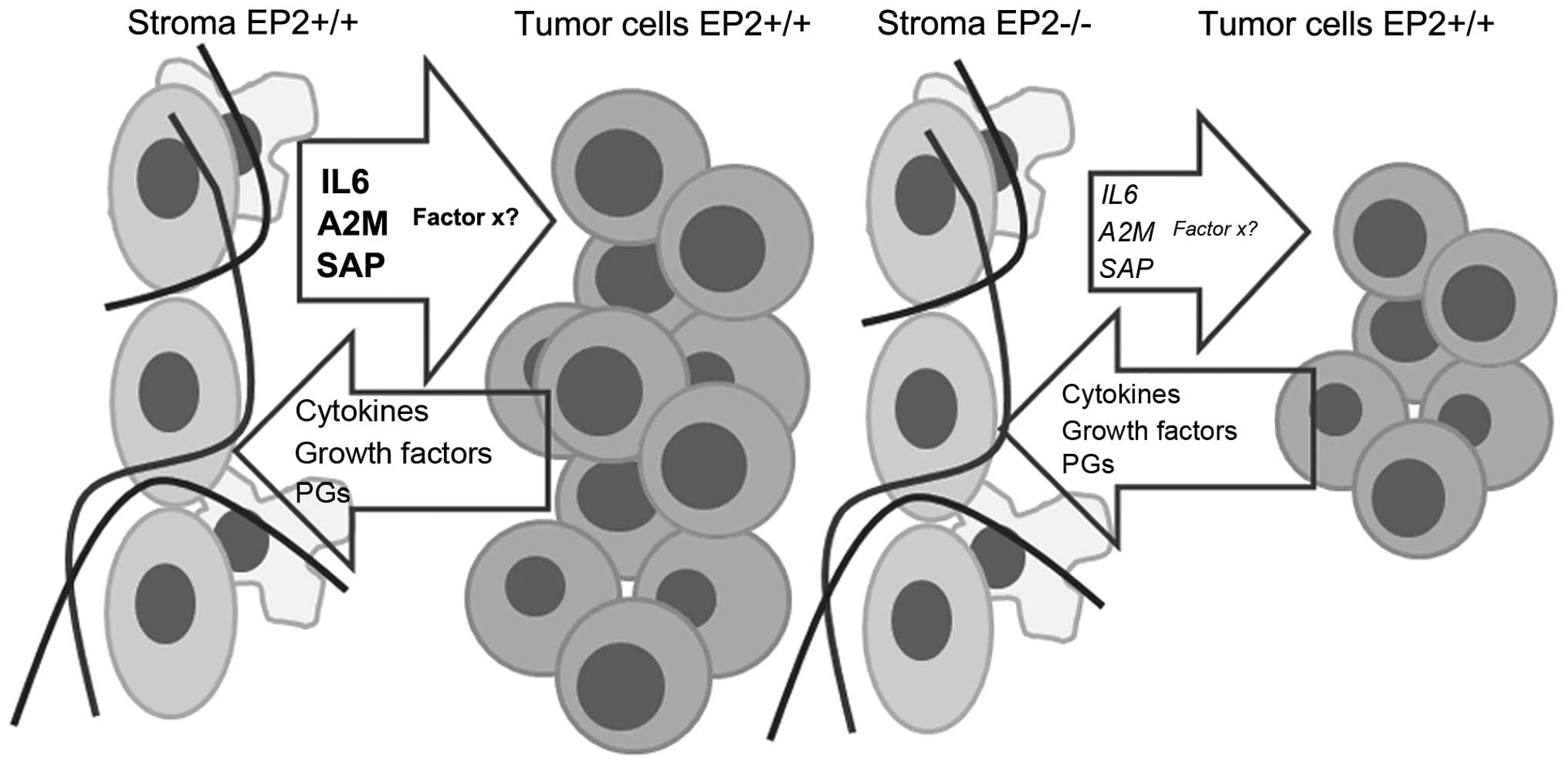 | Figure 4.Summary of a speculative
interpretation of the findings in the present study, as well as
knowledge from the literature. Host EP2-knockout reduced systemic
inflammation and plasma cytokine levels, as well as tumor gene
expression, resulting in significantly reduced tumor growth in the
EP2-knockout mice. These alterations indicated that stroma-tumor
interactions based on IL-6 and inflammatory signaling were
important for tumor growth, perhaps associated with an undiscovered
‘factor x’, as suggested by the present illustration. EP2,
E-prostanoid 2; A2M, α-2-macroglobulin; SAP, serum amyloid
protein/pentraxin-2; HPTGN, haptoglobin; IL, interleukin. |
In the present study, several hundreds of genes
showed changed expression in the tumor tissues from the
EP2-knockout mice compared with tumor tissues from the wild-type
mice. A reason for this could be that EP2 signaling in host tissues
is involved in several different pathways that are important events
in tumor cells associated with tumor progression, including
angiogenesis, the upregulation of COX-2, the loss of E-cadherin
expression and anti-apoptosis in stem cells (16,22,25–28).
Prom-1, also known as CD133, and CD44 are well-known markers of
cancer stem cells, indicating reduced gene expression in the tumor
tissues from the EP2-knockout mice in the present study. This
suggests that PGE2 and EP2 receptor signaling may be
involved in tumor progression associated with cancer stem cells. In
agreement with this, our earlier study reported that pre-operative
COX inhibition with NSAIDs reduced the expression levels of
stemness factors (CD133) in human colon cancer tissue (16).
Ptger2 transcripts were increased almost
8-fold in the tumors of the EP2-knockout mice, probably due to a
changed feedback-loop between COX-2, PGE2 and EP2
(7). G-proteins start signaling via
protein kinase A and CREB mainly upon activation of EP2 by
PGE2. This phenomenon has been reported to be a
PGE2-dependent pathway for cell proliferation and COX-2
induction (21). Creb and
Cox-2 gene expression were downregulated in the tumors from
the EP2-knockout mice in the present study. By contrast,
Cox-1 and 15-Pgdh were upregulated. COX-1 has similar
effects as COX-2 in tumor models (29,30), while
15-PGDH degrades prostaglandins. The transporter of prostaglandins
across cell membranes was also downregulated in the tumors from the
EP2-knockout mice in the present study. This could result in
reduced levels of PGE2 in tumors from EP2-knockout mice,
which may explain the increase in EP receptor transcripts, as well
as the reduced tumor growth.
Other genes involved in tumor progression that
displayed changed expression in the tumors of the EP2-knockout mice
were hypoxia inducible factor 1α subunit (Hif1a) and
metalloproteinases (Mmps). Hif1a was 1.83-fold
upregulated in the knockout mice, even when the tumors were small.
A previous study reported that HIF-1α is upregulated by
PGE2, contributing to metastasis and chemoresistance, as
well as the promotion of prostate cancer cell migration, invasion
and angiogenesis (31). Mmps degrade
extracellular matrix and facilitate the migration of tumor cells.
In the present study, Mmp-13 gene expression was 6-fold
upregulated in the tumors from the EP2-knockout mice, with reduced
tumor growth. EP2 signaling has been shown to inhibit the
production of MMP-13 in human osteoarthritic chondrocytes (32) and EP2 may be involved in the
regulation of MMPs.
Several lncRNAs showed different expression in the
tumor-bearing EP2 receptor-knockout mice in the present study. The
function of numerous lncRNAs is not known, but in general, lncRNAs
are involved in the regulation of downstream gene expression
(33). This is a novel area of
research and the results require further investigation. It is not
known to what extent changed lncRNAs are conserved between mice and
humans. In general, lncRNAs lack strong conservation, although
several transcripts have conserved elements (34,35). It is
well recognized that mouse models do not completely reflect human
conditions, particularly with regard to inflammation (36). However, human and mouse EP2 receptors
have 88% identity and differ at the NH2 terminus, where
the mouse receptor has 25 extra amino acids. Additionally, the
pharmacological properties of human EP2 are similar to those of
mouse EP2 receptors (37).
Overall, in the present study, EP2 receptor-knockout
in tumor-bearing host tissues reduced tumor growth, systemic
inflammation and IL-6 expression, which affected the expression of
several hundred genes and lncRNAs in tumor tissue. Thus, altered
signaling from the host affected tumor growth with attenuation of
PGE2-related factors, since tumor cells from the
EP2-knockout and wild-type mice possessed the same intrinsic gene
equipment; Cox-1 and 15-Pgdh expression was
upregulated, Cox-2 expression was downregulated and EP2
receptor gene expression was increased in the tumors from the
EP2-knockout mice.
Acknowledgements
This study was supported by grants from the Swedish
Cancer Society (no. CAN 2010/255), the Swedish State under the
LUA/ALF agreement, the Assar Gabrielsson foundation and the Magnus
Bergvall foundation. The authors would like to acknowledge the
expert technical skill of Dr Marianne Andersson, Dr Ludmila
Mackerlova and the BEA Core Facility at the Karolinska Institute
(Stockholm, Sweden).
Glossary
Abbreviations
Abbreviations:
|
COX-1/2
|
cyclooxygenase-1/2
|
|
EP
|
E-prostanoid
|
|
FC
|
fold-change
|
|
lncRNA
|
long non-coding RNA
|
|
PGE2
|
prostaglandin E2
|
References
|
1
|
Rothwell PM, Fowkes FG, Belch JF, Ogawa H,
Warlow CP and Meade TW: Effect of daily aspirin on long-term risk
of death due to cancer: Analysis of individual patient data from
randomised trials. Lancet. 377:31–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rothwell PM, Wilson M, Elwin CE, Norrving
B, Algra A, Warlow CP and Meade TW: Long-term effect of aspirin on
colorectal cancer incidence and mortality: 20-year follow-up of
five randomised trials. Lancet. 376:1741–1750. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenhough A, Smartt HJ, Moore AE, Roberts
HR, Williams AC, Paraskeva C and Kaidi A: The COX-2/PGE2 pathway:
Key roles in the hallmarks of cancer and adaptation to the tumour
microenvironment. Carcinogenesis. 30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang D and Dubois RN: Eicosanoids and
cancer. Nat Rev Cancer. 10:181–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gustafsson A, Hansson E, Kressner U,
Nordgren S, Andersson M, Wang W, Lönnroth C and Lundholm K: EP1-4
subtype, COX and PPAR gamma receptor expression in colorectal
cancer in prediction of disease-specific mortality. Int J Cancer.
121:232–240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gustafsson A, Hansson E, Kressner U,
Nordgren S, Andersson M, Lönnroth C and Lundholm K: Prostanoid
receptor expression in colorectal cancer related to tumor stage,
differentiation and progression. Acta Oncol. 46:1107–1112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sonoshita M, Takaku K, Sasaki N, Sugimoto
Y, Ushikubi F, Narumiya S, Oshima M and Taketo MM: Acceleration of
intestinal polyposis through prostaglandin receptor EP2 in
Apc(Delta 716) knockout mice. Nat Med. 7:1048–1051. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seno H, Oshima M, Ishikawa TO, Oshima H,
Takaku K, Chiba T, Narumiya S and Taketo MM: Cyclooxygenase 2- and
prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc
(Delta716) mouse intestinal polyps. Cancer Res. 62:506–511.
2002.PubMed/NCBI
|
|
9
|
Keith RL, Geraci MW, Nana-Sinkam SP,
Breyer RM, Hudish TM, Meyer AM, Malkinson AM and Dwyer-Nield LD:
Prostaglandin E2 receptor subtype 2 (EP2) null mice are protected
against murine lung tumorigenesis. Anticancer Res. 26:2857–2861.
2006.PubMed/NCBI
|
|
10
|
Iresjö BM, Wang W, Nilsberth C, Andersson
M, Lönnroth C and Smedh U: Food intake, tumor growth, and weight
loss in EP2 receptor subtype knockout mice bearing PGE2-producing
tumors. Physiol Rep. 3:e124412015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma X, Aoki T, Tsuruyama T and Narumiya S:
Definition of prostaglandin E2-EP2 signals in the colon tumor
microenvironment that amplify inflammation and tumor growth. Cancer
Res. 75:2822–2832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tilley SL, Audoly LP, Hicks EH, Kim HS,
Flannery PJ, Coffman TM and Koller BH: Reproductive failure and
reduced blood pressure in mice lacking the EP2 prostaglandin E2
receptor. J Clin Invest. 103:1539–1545. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andersson C, Gelin J, Iresjö BM and
Lundholm K: Acute-phase proteins in response to tumor growth. J
Surg Res. 55:607–614. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gelin JL, Moldawer LL, Iresjö BM and
Lundholm KG: The role of the adrenals in the acute phase response
to interleukin-1 and tumor necrosis factor-alpha. J Surg Res.
54:70–78. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lundholm K, Gelin J, Hyltander A, Lönnroth
C, Sandström R, Svaninger G, Körner U, Gülich M, Kärrefors I, Norli
B, et al: Anti-inflammatory treatment may prolong survival in
undernourished patients with metastatic solid tumors. Cancer Res.
54:5602–5606. 1994.PubMed/NCBI
|
|
16
|
Lönnroth C, Andersson M, Asting AG,
Nordgren S and Lundholm K: Preoperative low dose NSAID treatment
influences the genes for stemness, growth, invasion and metastasis
in colorectal cancer. Int J Oncol. 45:2208–2220. 2014.PubMed/NCBI
|
|
17
|
Ganesh T: Prostanoid receptor EP2 as a
therapeutic target. J Med Chem. 57:4454–4465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mutoh M, Watanabe K, Kitamura T, Shoji Y,
Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi
K, et al: Involvement of prostaglandin E receptor subtype EP(4) in
colon carcinogenesis. Cancer Res. 62:28–32. 2002.PubMed/NCBI
|
|
19
|
Brouxhon S, Konger RL, VanBuskirk J, Sheu
TJ, Ryan J, Erdle B, Almudevar A, Breyer RM, Scott G and Pentland
AP: Deletion of prostaglandin E2 EP2 receptor protects against
ultraviolet-induced carcinogenesis, but increases tumor
aggressiveness. J Invest Dermatol. 127:439–446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sung YM, He G and Fischer SM: Lack of
expression of the EP2 but not EP3 receptor for prostaglandin E2
results in suppression of skin tumor development. Cancer Res.
65:9304–9311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ansari KM, Sung YM, He G and Fischer SM:
Prostaglandin receptor EP2 is responsible for cyclooxygenase-2
induction by prostaglandin E2 in mouse skin. Carcinogenesis.
28:2063–2068. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kamiyama M, Pozzi A, Yang L, DeBusk LM,
Breyer RM and Lin PC: EP2, a receptor for PGE2, regulates tumor
angiogenesis through direct effects on endothelial cell motility
and survival. Oncogene. 25:7019–7028. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li HJ, Reinhardt F, Herschman HR and
Weinberg RA: Cancer-stimulated mesenchymal stem cells create a
carcinoma stem cell niche via prostaglandin E2 signaling. Cancer
Discov. 2:840–855. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li YJ, Kanaji N, Wang XQ, Sato T,
Nakanishi M, Kim M, Michalski J, Nelson AJ, Farid M, Basma H, et
al: Prostaglandin E switches from a stimulator to an inhibitor of
cell migration after epithelial-to-mesenchymal transition.
Prostaglandins Other Lipid Mediat 116–117. 1–9. 2015. View Article : Google Scholar
|
|
25
|
Pino MS, Nawrocki ST, Cognetti F,
Abruzzese JL, Xiong HQ and McConkey DJ: Prostaglandin E2 drives
cyclooxygenase-2 expression via cyclic AMP response element
activation in human pancreatic cancer cells. Cancer Biol Ther.
4:1263–1269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brouxhon S, Kyrkanides S, O'Banion MK,
Johnson R, Pearce DA, Centola GM, Miller JN, McGrath KH, Erdle B,
Scott G, et al: Sequential down-regulation of E-cadherin with
squamous cell carcinoma progression: Loss of E-cadherin via a
prostaglandin E2-EP2 dependent posttranslational mechanism. Cancer
Res. 67:7654–7664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Díaz-Muñoz MD, Osma-García IC, Fresno M
and Iniguez MA: Involvement of PGE2 and the cAMP signalling pathway
in the up-regulation of COX-2 and mPGES-1 expression in
LPS-activated macrophages. Biochem J. 443:451–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liou JY, Ellent DP, Lee S, Goldsby J, Ko
BS, Matijevic N, Huang JC and Wu KK: Cyclooxygenase-2-derived
prostaglandin e2 protects mouse embryonic stem cells from
apoptosis. Stem Cells. 25:1096–1103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Niho N, Kitamura T, Takahashi M, Mutoh M,
Sato H, Matsuura M, Sugimura T and Wakabayashi K: Suppression of
azoxymethane-induced colon cancer development in rats by a
cyclooxygenase-1 selective inhibitor, mofezolac. Cancer Sci.
97:1011–1014. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kitamura T, Itoh M, Noda T, Matsuura M and
Wakabayashi K: Combined effects of cyclooxygenase-1 and
cyclooxygenase-2 selective inhibitors on intestinal tumorigenesis
in adenomatous polyposis coli gene knockout mice. Int J Cancer.
109:576–580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fernández-Martínez AB and Lucio-Cazaña J:
Intracellular EP2 prostanoid receptor promotes cancer-related
phenotypes in PC3 cells. Cell Mol Life Sci. 72:3355–3373. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato T, Konomi K, Fujii R, Aono H, Aratani
S, Yagishita N, Araya N, Yudoh K, Beppu M, Yamano Y, et al:
Prostaglandin EP2 receptor signalling inhibits the expression of
matrix metalloproteinase 13 in human osteoarthritic chondrocytes.
Ann Rheum Dis. 70:221–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brosius J: Waste not, want not-transcript
excess in multicellular eukaryotes. Trends Genet. 21:287–288. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pollard KS, Salama SR, King B, Kern AD,
Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch
R, et al: Forces shaping the fastest evolving regions in the human
genome. PLoS Genet. 2:e1682006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seok J, Warren HS, Cuenca AG, Mindrinos
MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy
L, et al: Genomic responses in mouse models poorly mimic human
inflammatory diseases. Proc Natl Acad Sci USA. 110:3507–3512. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bastien L, Sawyer N, Grygorczyk R, Metters
KM and Adam M: Cloning, functional expression, and characterization
of the human prostaglandin E2 receptor EP2 subtype. J Biol Chem.
269:11873–11877. 1994.PubMed/NCBI
|