Introduction
Cathepsin B (CTSB) is a cysteine protease
physiologically involved in lysosomal protein degradation (1). In cancer cells, CTSB may be translocated
to the cell surface but also secreted in the extracellular space
(2). This expression distribution
lends various protumorigenic properties to CTSB in solid
malignancies (3,4). CTSB enhances the invasiveness of cancer
cells through the degradation of extracellular matrix proteins, by
cleavage and inactivation of tissue inhibitors of
metalloproteinases, and by disruption of cell-cell junctions.
Furthermore, CTSB may affect tumor cell proliferation, migration
and apoptosis, and participates in cancer angiogenesis and
chemoresistance (5).
In breast cancer mouse models, knockout of the CTSB
gene resulted in retarded tumor growth and fewer lung metastases
(6,7).
Correspondingly, previous studies on human breast cancer have
revealed that CTSB tissue overexpression is a strong and
statistically independent adverse prognostic factor (8–12). In
previous decades, it was revealed that CTSB also participates in
immunoregulatory processes, as it is involved in the lysosomal
cleavage of cellular proteins, eventually leading to major
histocompatibility complex presentation. Cell-surface CTSB may also
protect cancer cells from cytotoxic effector molecules secreted by
tumor-suppressive immune cells (13).
Another potential immune evasion mechanism results
from the ability of CTSB to cleave and inactivate chemokines,
including the C-X-C motif chemokine receptor 3 (CXCR3) chemokine
receptor ligands C-X-C motif chemokine ligand (CXCL)9 and CXCL10
(14,15). CXCR3 is expressed by tumor-suppressive
lymphocytes such as cytotoxic T cells and natural killer cells,
mediating their infiltration into solid malignancies (16,17).
Cancer cells may therefore co-opt CTSB to cleave and inactivate
CXCR3 ligands, which may lead to a reduced number of
tumor-infiltrating lymphocytes. A similar underlying mechanism has
also been demonstrated for dipeptidyl peptidase 4, which cleaves
CXCR3 chemokines (18).
Therefore, the present study hypothesized that tumor
cells may upregulate CTSB expression upon CXCR3 activation by
employing a negative feedback mechanism, similar to the one
demonstrated for the induction of matrix metalloproteinase (MMP)-9
in colon and breast cancer (19,20). To
the best of our knowledge, the current study demonstrated for the
first time that the CXCR3 ligands CXCL9 and CXCL10 induce the
expression and secretion of CTSB in human breast cancer cells in
concert with CXCR3 overexpression.
Materials and methods
Patient characteristics
Patients with early-stage breast cancer (n=88) who
underwent primary surgical treatment between March 1987 and
December 1991 at the Department of Gynecology and Obstetrics,
Technical University of Munich (Germany) were included in the
study. The present study was approved by the local ethics
committee, and all patients provided written informed consent. The
median duration of clinical follow-up was 112 months (range,
1–329). Patient characteristics are as follows: Median age, 59
years (range, 33–84); incidence of mortality, 55/88 patients (63%).
The tumor stages wereT1 (22/88, 25%), T2 (37/88, 42%), T3 (11/88,
13%), T4 (12/88, 14%) and unknown (6/88, 7%). The nuclear grading
was as follows: G1 (4/88, 5%), G2 (38/88, 43%), G3 (25/88, 28%) and
unknown (21/88, 24%). The lymph node status was node-negative
(34/88, 39%), node-positive (47/88, 53%) and unknown (7/88, 8%).
The estrogen receptor-α statuses were as follows: Negative (13/88,
15%), positive (61/88, 69%) and unknown (14/88, 16%). No
information regarding the human epidermal growth factor receptor 2
status was recorded. Patients received adjuvant treatment according
to consensus recommendations (9).
Reagents, antibodies and cell
lines
Dulbecco's modified Eagle's medium (DMEM), fetal
calf serum (FCS), gentamycin,
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and
glutamine were obtained from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Recombinant human CXCL4/PF4 protein (CXCL4),
recombinant human CXCL9/MIG protein (CXCL9) and recombinant human
CXCL10/IP-10 protein (CXCL10) were from R&D Systems, Inc.
(Minneapolis, MN, USA) and were reconstituted in PBS supplemented
with 5% bovine serum albumin (A1470; Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany). The 7-aminoactinomycin D (7-AAD)
viability staining solution was purchased from eBioscience, Inc.
(catalog no. 00-6993-50; San Diego, CA, USA). All other chemicals
were of analytical grade and obtained from Merck Millipore.
Monoclonal antibodies to the human antigens CTSB (catalog no.
MAB965; clone 73317; rat IgG1), CXCR3 (catalog no. MAB160, clone
49801, mouse IgG1) and the IgG1 isotype control antibody (catalog
no. MAB002) were all purchased from R&D Systems, Inc.
Monoclonal mouse anti-GAPDH (catalog no. CB1001, clone 6C5) was
obtained from EMD Millipore (Billerica, MA, USA). Alexa Fluor
488-conjugated goat anti-mouse IgG (heavy and light chains) was
purchased from Thermo Fisher Scientific, Inc. (catalog no.
A-11001). Horseradish peroxidase-conjugated goat anti-mouse IgG
(Jackson Immuno Research Laboratories, Inc., Burlington, ON, USA).
The MDA-MB-231 and MCF-7 human breast cancer cell lines (American
Type Culture Collection, Manassas, VA, USA) were cultured in a
humidified 5% CO2 atmosphere at 37°C in DMEM
supplemented with glutamine, 10% FCS, 10 mM HEPES and 20 µg/ml
gentamycin.
CXCR3 immunohistochemistry and flow
cytometry
Immunohistochemistry was performed on 4-µm thick
sections cut from routine formalin-fixed paraffin-embedded
(FFPE)-blocks prepared from invasive breast cancer tissue specimens
(n=88) obtained from patients with breast cancer treated at the
Department of Obstetrics and Gynecology, Medical School of the
Technical University of Munich. Briefly, cut sections were
deparaffinized by treatment with xylene followed by a graded series
of ethanol (100–70%) and rehydration in distilled H2O,
and subjected to heat-induced epitope retrieval in citrate buffer
(2.1 g citric acid monohydrate for 1 liter aqua dest;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), pH 6.0).
Endogenous peroxidase activity was blocked by treatment of the
sections with 3% H2O2 in distilled
H2O for 20 min at room temperature, followed by
endogenous avidin/biotin block using a blocking kit (catalog no.
AB972; Zytomed, Berlin, Germany), according to the manufacturer's
instructions, and subsequent incubation with 5% goat serum (Dako,
Glostrup, Denmark). The sections were then incubated (1 h, room
temperature) with 0.5 µg/ml of antibody MAB160 to human CXCR3
diluted in green antibody diluent (catalog no. S2022; Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA). Subsequently, for
detection of the primary antibody binding reaction, the LSAB-kit
(Zytomed Systems, Berlin, Germany) was employed according to the
manufacturer's protocol. Sections were washed thoroughly between
incubations and cell nuclei were counterstained with Meyer's
hematoxylin. Staining of tumor cells for CXCR3 protein expression
was assessed semi-quantitatively as absent (0), weak (1+), moderate
(2+) or intense (3+) cytoplasmic staining. As tumors generally
demonstrated diffuse staining of varying intensity and no
considerable intra-tumoral heterogeneity, the number of positive
cells was not included into the scoring algorithm. As an internal
positive reference, normal fallopian tube epithelium, previously
classified as 2+ positivity for CXCR3 protein expression, was
additionally stained.
To assess breast cancer cell lines for cell surface
expression of CXCR3, MCF-7 and MDA-MB-231 cells were incubated with
monoclonal antibody MAB160 to human CXCR3 (dilution, 1:6.25 in 50
µl; 200 µg/ml in 0.5% FCS; 0.01% NaN3; 1 h on ice).
Monoclonal antibody MAB002 served as the isotype control antibody
in equivalent concentrations (dilution, 1:6.25 in 50 µl; 200 µg/ml
in 0.5% FCS; 0.01% NaN3; 1 h on ice). Subsequently,
cells were incubated with Alexa Fluor 488-conjugated antibody
A-11001 (dilution, 1:350; 2.85 µg/ml in 0.5% FCS; 0.01%
NaN3; 30 min on ice) to visualize binding of MAB160.
Dead cells were excluded by simultaneous staining with propidium
iodide. Fluorescence as a measure of antibody binding or 7-AAD
reaction with cell nuclei was recorded using the FACSCalibur
(Becton-Dickinson, Heidelberg, Germany) and histograms were
evaluated and plotted using Flowing Software 2 (version 2.5.1;
Turku Center of Biotechnology, Turku, Finland).
Western blot analysis
Fresh tumor samples of human breast cancer tissue
were obtained during surgery, examined by a pathologist and stored
in liquid nitrogen until further use. Tumor tissue homogenates and
extracts were prepared as described previously (21). Protein concentrations of the tissue
extracts were determined by applying the BCA Protein Assay kit
(Pierce; Thermo Fisher Scientific, Inc.).
MCF-7 or MDA-MB-231 cells were seeded on 6 cm Ø
culture plates and grown to 70% confluency. Following washing in
PBS, the cells were starved for 24 h in serum-free medium, which
was renewed 30 min prior to the addition of the indicated
stimulants (100 ng/ml CXCL4, CXCL9 or CXCL10). The cells were
incubated for 48 h and then washed in ice-cold PBS to be lysed in
SDS-PAGE sample buffer (pH 6.0) containing 50 mM
Na2HPO4/NaH2PO4, 0.2 M
NaCl, 5 mM EDTA and 1% Triton X-100. The lysates were chilled on
ice for 20 min and then subjected to ultrasound treatment (2×10
sec, 4°C) prior to storage at −20°C until further use. The BCA
Protein Assay kit (Pierce; Thermo Fisher Scientific, Inc.) was
applied for assessment of the protein concentration of the
homogenates according to the manufacturer's protocol. Equal amounts
of protein (30 µg) were applied to 10% SDS-PAGE as described
previously (22). Blotting of the
separated protein bands to a nitrocellulose membrane (Schleicher
& Schuell, Dassel, Germany) was performed by semi-dry transfer
technology (Biometra, Göttingen, Germany). Blots were blocked (1 h,
room temperature) with PBS/0.5% Tween 20 (w/v) (PBST) containing 5%
skimmed milk powder (Sigma-Aldrich; Merck Millipore), washed 3
times with PBST and then incubated overnight at 4°C with the
following antibodies: MAB160 to CXCR3 (0.67 µg/ml), MAB965 to CTSB
(0.5 µg/ml) in 5% bovine serum albumin/PBST, 6C5 to GAPDH (0.1
µg/ml) in 5% skimmed milk/PBST. Subsequently, the blots were washed
with PBST and then incubated with horseradish peroxidase-coupled
anti-mouse (for MAB160, 6C5) or anti-rat IgG (for MAB965) at room
temperature for 1 h. The blots were washed with PBST prior to
antibody reaction visualization using enhanced chemiluminescence
detection (GE Healthcare Life Sciences, Uppsala, Sweden). For this,
the blots were exposed to CEA RP-new (Agfa HealthCare NV, Mortsel,
Belgium) and developed by Cawomat 2000 IR (Cawo, Schrobenhausen,
Germany). For CXCR3 western blotting, Tris-buffered saline
containing 0.1% Tween 20 was used instead of PBST.
Measurement of CTSB using ELISA
Tumor cell stimulation experiments with CXCL9,
CXCL10 and CXCL4 were performed as described previously (23). Briefly, MDA-MB-231 cells were plated
on 12-well culture plates and grown to 70% confluency, washed with
PBS and then starved for 24 h in serum-free medium. The serum-free
medium was renewed 30 min prior to addition of the relevant
stimulant. Supernatants were collected after 48 h and stored at
−20°C until further use. In each experiment, 8 wells were
stimulated. Secretion levels of CTSB were determined by using the
human CTSB Duo set ELISA kit (DY2176, R&D Systems, Inc.),
according to the manufacturer's protocol. CTSB content values
determined in the respective breast cancer tumor tissues were
retrieved from the tumor tissue data bank of the Department of
Obstetrics and Gynecology of the Technical University of
Munich.
Statistical analyses
Univariate survival analyses were performed and
plotted for overall survival according to the Kaplan-Meier method
(24). For multivariable survival
analyses, a Cox proportional hazards model was used. Results of the
cell culture stimulation experiments were assessed using mean
values taken from at least three independent experiments and
analyzed and employing the Mann-Whitney test (SPSS version 21.0;
IBM SPSS, Armonk, NY, USA). Results are presented as the mean ±
standard error of the mean. Statistical significance was defined as
P≤0.05, P≤0.005 or P≤0.001. A minimum threshold of P<0.05 was
considered to indicate a statistically significant difference.
Results
CXCR3 expression in human breast
cancer is associated with an adverse clinical outcome
As a prerequisite for the present study, the protein
expression levels of the chemokine receptor CXCR3 were determined
using immunohistochemical staining of 88 human primary invasive
breast cancer FFPE-tissue sections. In these malignant tissues,
CXCR3 was located in the region of tumor cells in adjacent
non-malignant duct cells, lymphocytes and endothelial cells
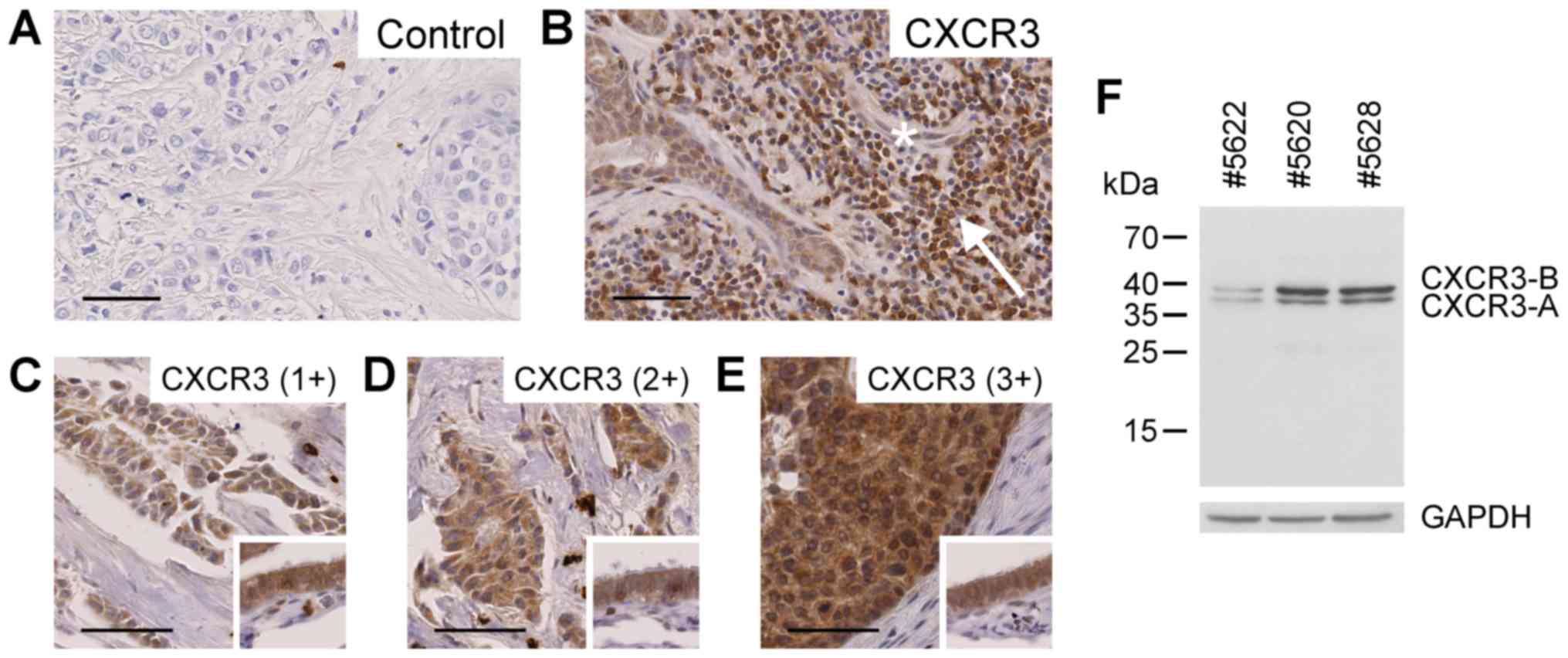
(Fig. 1A and B), confirming earlier
findings by Datta et al (25)
and Ma et al (26). Tumor cell
CXCR3 protein expression levels were assessed semi-quantitatively
as absent (0, 7/88), weak (1+, 24/88), moderate (2+, 25/88) or
intense (3+, 32/88) based on cytoplasmic staining (Fig. 1C-E). The majority of tissue samples
exhibited moderate to intense staining in tumor cells. Immunoblot
analysis of breast cancer tissue extracts employing the same
antibody as engaged in the immunohistochemical study resulted in
recognition of the two splice variants CXCR3-A and CXCR3-B
(Fig. 1F).
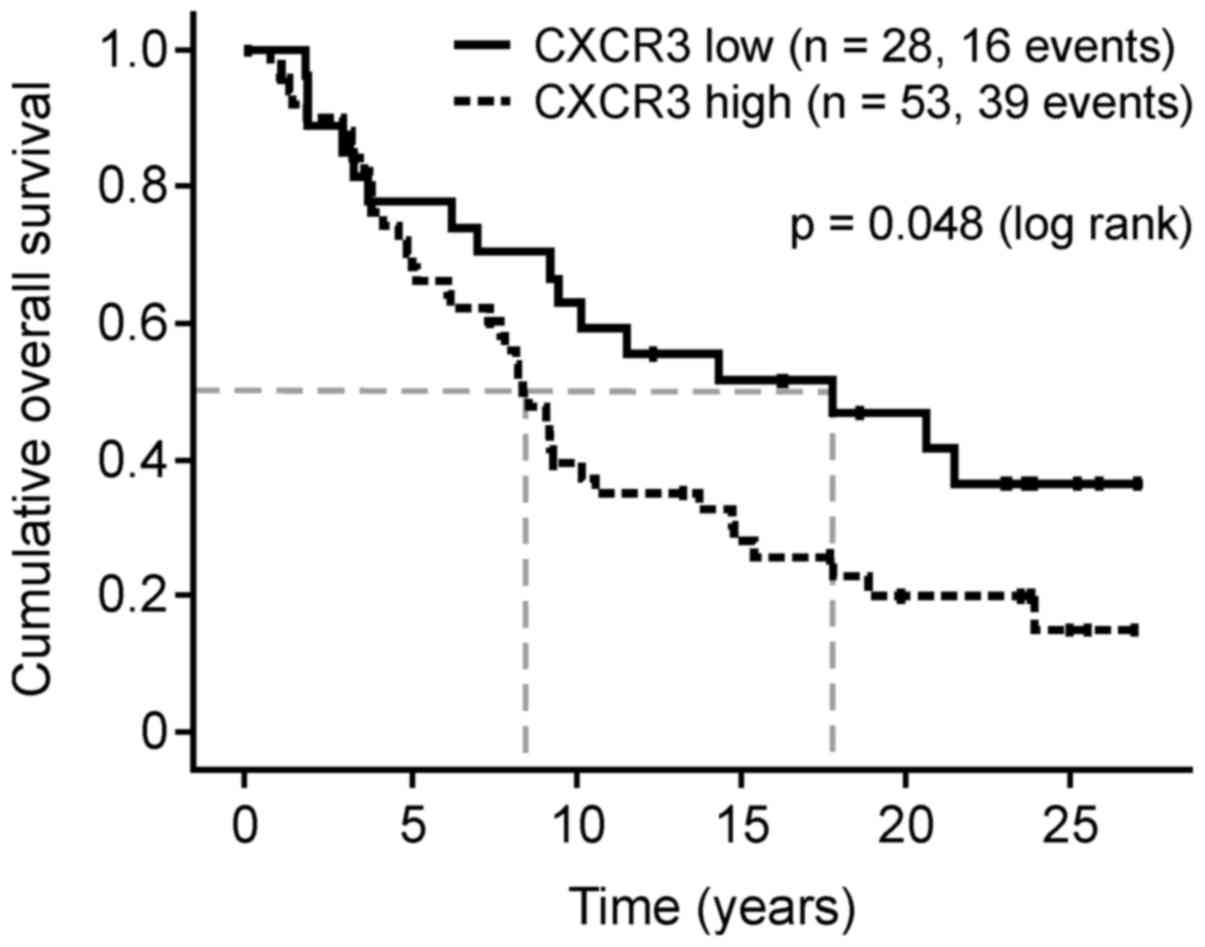
The CXCR3 protein expression levels in FFPE breast
cancer tumor cells were divided into groups with low (0, 1+; n=31)
and high expression (2+, 3+; n=57). Patients with breast cancer
with high CXCR3 protein expression levels exhibited a significantly
reduced median overall survival compared with the low-level group
(102 vs. 217 months, hazard ratio (HR), 1.79; 95% confidence
interval (CI), 1.00–3.23; P=0.048; Fig.
2). The median disease-free-survival time was also reduced (120
vs. 227 months; HR, 3.10; 95% CI, 0.88–10.96; P=0.08).
The trend in overall survival difference was greater
in the node-negative disease cases (median survival, 251
(CXCR3low) vs. 100 months (CXCR3high);
P=0.207), compared with the node-positive cases [median survival,
112 (CXCR3low) vs. 111 months (CXCR3high);
P=0.088], which supports initial data from Ma et al
(26). Using multivariate analysis,
the negative prognostic impact of CXCR3 overexpression remained
independent of lymph node status, tumor size and nuclear grading
(Table I). The 88 breast cancer FFPE
tumor tissue specimens are part of the tissue collective assessed
by Thomssen et al (9) for CTSB
protein expression using ELISA. However, no significant correlation
between CXCR3 protein expression levels in tumor cells assessed by
immunohistochemistry and CTSB protein levels determined by ELISA
was observed in the current study (data not shown).
 | Table I.Cox multivariate analysis of
clinicopathological factors as well as CXCR3 expression for overall
survival. |
Table I.
Cox multivariate analysis of
clinicopathological factors as well as CXCR3 expression for overall
survival.
| Variable | Hazard ratio | 95% CI | P-value |
|---|
| CXCR3 expression
(high vs. low) | 1.99 | 1.00–3.00 | 0.050 |
| Nodal status
(positive vs. negative) | 0.89 | 0.46–1.46 | 0.741 |
| Tumor size (T3/4
vs. T1/2) | 2.88 | 1.53–5.53 | 0.001 |
| Nuclear grading (G3
vs. G1/2) | 1.44 | 0.75–2.75 | 0.272 |
CXCR3-directed chemokines CXCL9 and
CXCL10 induce CTSB in human breast cancer cells
For demonstration of the interaction of
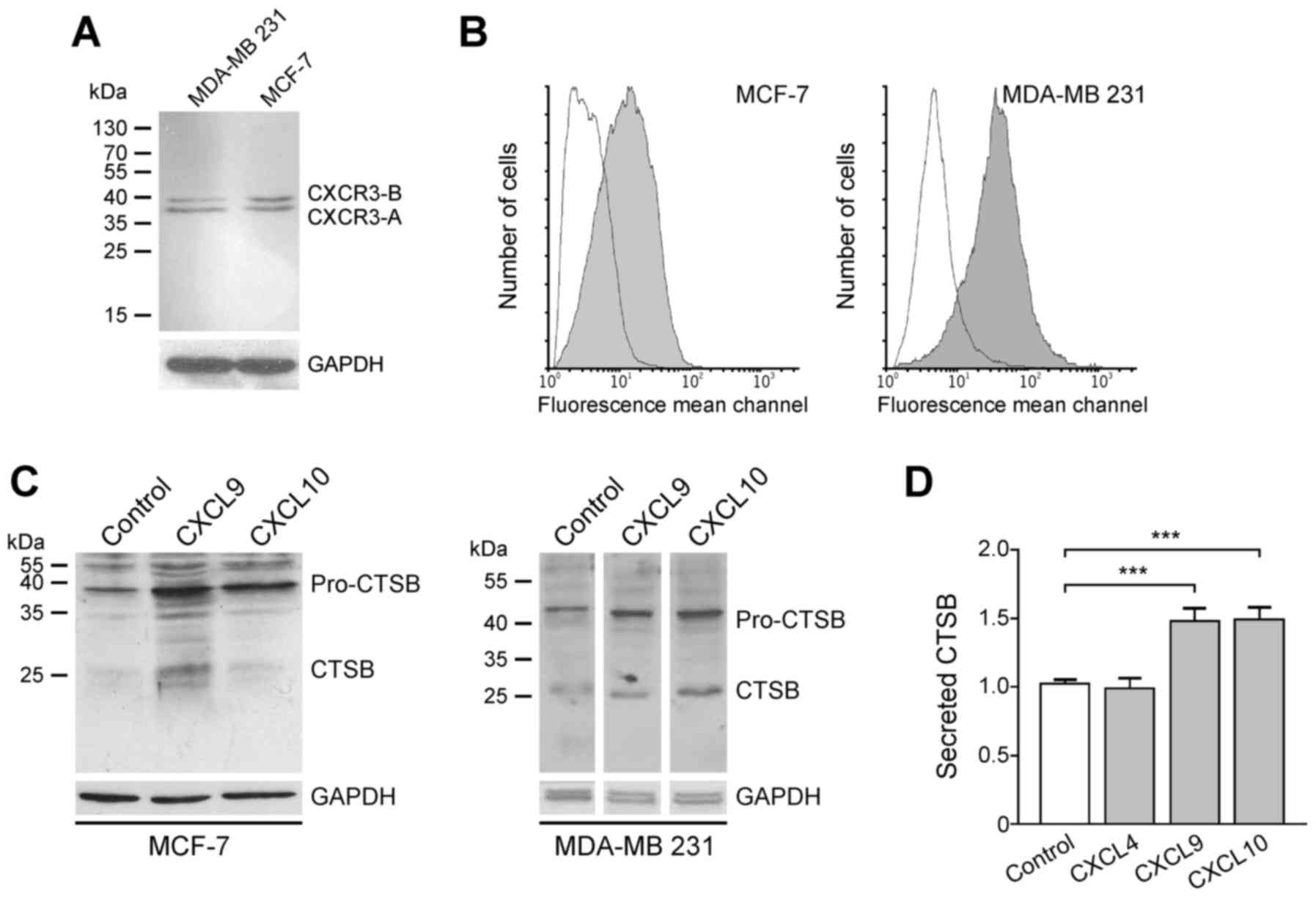
CXCR3-directed chemokines CXCL9 and CXCL10, CXCR3 protein was
initially assessed in the human breast cancer cell lines MCF-7 and
MDA-MB-231. Western blot analysis revealed that the two cell lines
express the splice variants CXCR3-A and CXCR3-B (Fig. 3). Although CXCR3 was identified in the
cytosol of tumor cells (Fig. 1), flow
cytometry analysis revealed that CXCR3 protein is also located on
the cell surface of both cell lines, a prerequisite for stimulation
of tumor cells by chemokines via the membrane-associated CXCR3
receptor (Fig. 3).
Subsequently, whether CXCR3 chemokines are able to
induce CTSB expression in breast cancer cells was assessed, as this
trend has been described for MMP-2 and MMP-9 in MDA-MB-231 cells
and the colon cancer cell lines SW480, SW620, KM12C and KM12SM
(19,20). Therefore, MCF-7 and MDA-MB-231 cells
were incubated with 100 ng/ml recombinant CXCL9 and CXCL10,
respectively. After 24 h (MCF-7) or 48 h (MDA-MB-231), cells were
harvested and CTSB protein expression was evaluated by immunoblot
analysis. In the two cell lines, cellular pro-CTSB (~46 kDa) and
CTSB (~25 kDa) were upregulated followingCXCR3-directed chemokine
exposure (Fig. 3). In this scenario,
the chemokines CXCL9 and CXCL10 (100 ng/ml) significantly increased
the secretion of CTSB by ~1.5 fold following 48 h of incubation
(Fig. 3). The CXCR3-B-selective
chemokine CXCL4, however, did not effect such a release, indicating
that the CXCR3-A splice variant accounts for CTSB induction upon
CXCL9 or CXCL10 stimulation.
Discussion
In solid malignancies, the CXCR3 chemokine receptor
is a double-edged sword, as on the one hand it mediates the
recruitment of tumor-suppressive lymphocytes (TILs) into the tumor
microenvironment, leading to reduced tumor growth and metastatic
spread associated with improved survival, but in tumor cells, CXCR3
over expression may promote cell proliferation, migration and
invasion, resulting in poor clinical outcomes for patients
(27,28).
In breast cancer, elevated intratumoral
concentrations of CXCR3 ligands are associated with enhanced
numbers of TILs and improved patient survival (29–31). In
syngeneic murine breast cancer models, overexpression of CXCL9 or
CXCL10 leads to reduced tumor growth and lowered metastatic spread
(32,33). However, forced overexpression of CXCR3
in tumor cells augments pulmonary metastasis with reduced survival
in these mouse models (26,34). During the mammary tumorigenesis
process in mice, CXCR3 is among the most upregulated genes;
however, its underlying mechanisms have not yet been fully
elucidated (35).
In the present breast cancer study, CXCR3 was
located in tumor cells, tumor-infiltrating lymphocytes and
endothelial cells, which is concordant with previous reports
(25). The present findings, which
reveal that CXCR3 overexpression in tumor cells is associated with
significantly reduced overall survival, supports results described
by Ma et al (26). In this
previous study, the association of CXCR3 protein expression with
unfavorable patient outcome was significant only in the
node-negative subgroup. The current results are concordant with
this observation; however, this effect was also seen in the
node-positive subgroup. Hilborn et al (36) reported that CXCR3 protein expression
in breast cancer tissue is able to predict the tamoxifen treatment
response of patients, and that CXCR3 protein overexpression is
correlated with reduced survival.
In the present study, to the best of our knowledge,
the induction of a cysteine-type cathepsin family member, CTSB, by
CXCR3 ligands in tumor cells was demonstrated for the first time.
Induction of other proteases by CXCR3 ligands has been demonstrated
in MDA-MB-231 breast cancer cells for the matrix metalloproteases
MMP-2 and MMP-9 (19) and in the
colon cancer cells SW480, SW620, KM12C and KM12SM for MMP-9
(20). In general, overexpression of
proteases is linked to the enhanced invasion of tumor cells through
the degradation of extracellular matrix proteins (37). Nevertheless, MMP-2 and MMP-9 may also
cleave CXCR3 ligands (38,39). Previously, this feature was
demonstrated for CTSB and other cathepsin family members (15). Induction of these proteases,
therefore, may represent a negative feedback mechanism through
which cells recruited by CXCR3 ligands start to degrade the
chemokine gradient immediately upon arrival at the site.
Physiologically, this may limit an otherwise unregulated
inflammatory reaction. Cancer cells may retain such a mechanism to
increase tumor cell spread, impair CXCR3 ligands and reduce
lymphocyte infiltration. Proteolytic cleavage of CXCR3 ligands
therefore may represent a protective evasion mechanism. Such a
mechanism has been described for dipeptidyl peptidase IV, which
also inactivates CXCL10 (18).
Induction of cysteine proteases such as CTSB by
CXCR3 may explain the unfavorable clinical value of CXCR3
overexpression in breast cancer. Notably, the tumor promoting
effect of CXCR3 in a syngeneic murine breast cancer model depended
on IFN-γ (40), which is one of the
strongest inducers of CXCR3 ligands in breast cancer cells
(23). However, the present study did
not find a correlation between CXCR3 expression levels determined
immunohistochemically and intratumoral CTSB concentrations as
measured by ELISA by Thomssen et al (9). This may also be explained by other
mechanisms contributing to CTSB expression in breast cancer.
In conclusion, CTSB is induced by CXCR3 ligands in
human breast cancer cells, which represents another underlying
mechanism contributing to the negative prognostic impact of CXCR3
in breast cancer cells. The present findings suggest that breast
cancer cells may exploit originally tumor-suppressive CXCR3
chemokines to enhance their invasiveness and reduce immune
infiltration through the degradation of these chemokines.
Acknowledgements
The authors would like to thank Joachim Hornung,
Elisabeth Schüren and Alexandra Stöckel (Department of Gynecology
and Obstetrics, Technical University of Munich) for excellent
technical assistance. This study was supported by grants from the
Wilhelm Sander-Stiftung (grant nos. 2011.006.1 and 2011.006.2) to
Holger Bronger, Stefanie Avril and Manfred Schmitt. Stefanie Avril
is supported by the Clinical and Translational Science
Collaborative of Cleveland (grant no. KL2TR000440) from the
National Center for Advancing Translational Sciences component of
the National Institutes of Health and NIH roadmap for Medical
Research.
References
|
1
|
Yan S and Sloane BF: Molecular regulation
of human cathepsin B: Implication in pathologies. Biol Chem.
384:845–854. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roshy S, Sloane BF and Moin K:
Pericellular cathepsin B and malignant progression. Cancer
Metastasis Rev. 22:271–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aggarwal N and Sloane BF: Cathepsin B:
Multiple roles in cancer. Proteomics Clin Appl. 8:427–437. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olson OC and Joyce JA: Cysteine cathepsin
proteases: Regulators of cancer progression and therapeutic
response. Nat Rev Cancer. 15:712–729. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kos J, Mitrović A and Mirković B: The
current stage of cathepsin B inhibitors as potential anticancer
agents. Future Med Chem. 6:1355–1371. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasiljeva O, Korovin M, Gajda M, Brodoefel
H, Bojic L, Krüger A, Schurigt U, Sevenich L, Turk B, Peters C and
Reinheckel T: Reduced tumour cell proliferation and delayed
development of high-grade mammary carcinomas in cathepsin
B-deficient mice. Oncogene. 27:4191–4199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vasiljeva O, Papazoglou A, Krüger A,
Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt
K, Bogyo M, et al: Tumor cell-derived and macrophage-derived
cathepsin B promotes progression and lung metastasis of mammary
cancer. Cancer Res. 66:5242–5250. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lah TT, Cercek M, Blejec A, Kos J,
Gorodetsky E, Somers R and Daskal I: Cathepsin B, a prognostic
indicator in lymph node-negative breast carcinoma patients:
Comparison with cathepsin Dcathepsin L, and other clinical
indicators. Clin Cancer Res. 6:578–584. 2000.PubMed/NCBI
|
|
9
|
Thomssen C, Schmitt M, Goretzki L, Oppelt
P, Pache L, Dettmar P, Jänicke F and Graeff H: Prognostic value of
the cysteine proteases cathepsins B and cathepsin L in human breast
cancer. Clin Cancer Res. 1:741–746. 1995.PubMed/NCBI
|
|
10
|
Maguire TM, Shering SG, Duggan CM,
McDermott EW, O'Higgins NJ and Duffy MJ: High levels of cathepsin B
predict poor outcome in patients with breast cancer. Int J Biol
Markers. 13:139–144. 1998.PubMed/NCBI
|
|
11
|
Foekens JA, Kos J, Peters HA, Krasovec M,
Look MP, Cimerman N, Meijer-van Gelder ME, Henzen-Logmans SC, van
Putten WL and Klijn JG: Prognostic significance of cathepsins B and
L in primary human breast cancer. J Clin Oncol. 16:1013–1021. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Harbeck N, Alt U, Berger U, Krüger A,
Thomssen C, Jänicke F, Höfler H, Kates RE and Schmitt M: Prognostic
impact of proteolytic factors (urokinase-type plasminogen
activator, plasminogen activator inhibitor 1, and cathepsins B, D,
and L) in primary breast cancer reflects effects of adjuvant
systemic therapy. Clin Cancer Res. 7:2757–2764. 2001.PubMed/NCBI
|
|
13
|
Balaji KN, Schaschke N, Machleidt W,
Catalfamo M and Henkart PA: Surface cathepsin B protects cytotoxic
lymphocytes from self-destruction after degranulation. J Exp Med.
196:493–503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hasan L, Mazzucchelli L, Liebi M, Lis M,
Hunger RE, Tester A, Overall CM and Wolf M: Function of liver
activation-regulated chemokine/CC chemokine ligand 20 is
differently affected by cathepsin B and cathepsin D processing. J
Immunol. 176:6512–6522. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Repnik U, Starr AE, Overall CM and Turk B:
Cysteine cathepsins activate ELR chemokines and inactivate non-ELR
chemokines. J Biol Chem. 290:13800–13811. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen DS and Mellman I: Oncology meets
immunology: The cancer-immunity cycle. Immunity. 39:1–10. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wendel M, Galani IE, Suri-Payer E and
Cerwenka A: Natural killer cell accumulation in tumors is dependent
on IFN-gamma and CXCR3 ligands. Cancer Res. 68:8437–8445. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
da Silva R Barreira, Laird ME, Yatim N,
Fiette L, Ingersoll MA and Albert ML: Dipeptidylpeptidase 4
inhibition enhances lymphocyte trafficking, improving both
naturally occurring tumor immunity and immunotherapy. Nat Immunol.
16:850–858. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin SY, Nam JS, Lim Y and Lee YH:
TNFα-exposed bone marrow-derived mesenchymal stem cells promote
locomotion of MDA-MB-231 breast cancer cells through
transcriptional activation of CXCR3 ligand chemokines. J Biol Chem.
285:30731–30740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zipin-Roitman A, Meshel T, Sagi-Assif O,
Shalmon B, Avivi C, Pfeffer RM, Witz IP and Ben-Baruch A: CXCL10
promotes invasion-related properties in human colorectal carcinoma
cells. Cancer Res. 67:3396–3405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jänicke F, Pache L, Schmitt M, Ulm K,
Thomssen C, Prechtl A and Graeff H: Both the cytosols and detergent
extracts of breast cancer tissues are suited to evaluate the
prognostic impact of the urokinase-type plasminogen activator and
its inhibitor, plasminogen activator inhibitor type 1. Cancer Res.
54:2527–2530. 1994.PubMed/NCBI
|
|
22
|
Bronger H, König J, Kopplow K, Steiner HH,
Ahmadi R, Herold-Mende C, Keppler D and Nies AT: ABCC drug efflux
pumps and organic anion uptake transporters in human gliomas and
the blood-tumor barrier. Cancer Res. 65:11419–11428. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bronger H, Kraeft S, Schwarz-Boeger U,
Cerny C, Stöckel A, Avril S, Kiechle M and Schmitt M: Modulation of
CXCR3 ligand secretion by prostaglandin E2 and cyclooxygenase
inhibitors in human breast cancer. Breast Cancer Res. 14:R302012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaplan EL and Meier P: Nonparametric
estimation from incomplete observations. J Amer Statist Assn.
53:457–481. 1958. View Article : Google Scholar
|
|
25
|
Datta D, Flaxenburg JA, Laxmanan S, Geehan
C, Grimm M, Waaga-Gasser AM, Briscoe DM and Pal S: Ras-induced
modulation of CXCL10 and its receptor splice variant CXCR3-B in
MDA-MB-435 and MCF-7 cells: Relevance for the development of human
breast cancer. Cancer Res. 66:9509–9518. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma X, Norsworthy K, Kundu N, Rodgers WH,
Gimotty PA, Goloubeva O, Lipsky M, Li Y, Holt D and Fulton A: CXCR3
expression is associated with poor survival in breast cancer and
promotes metastasis in a murine model. Mol Cancer Ther. 8:490–498.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cerny C, Bronger H, Davoodi M, Sharma S,
Zhu L, Obana S, Sharma J, Ebrahimi R, St John M, Lee JM, et al: The
role of CXCR3/ligand axis in cancer. International Trends in
Immunity. 3:46–52. 2015. View Article : Google Scholar
|
|
28
|
Ma B, Khazali A and Wells A: CXCR3 in
carcinoma progression. Histol Histopathol. 30:781–792.
2015.PubMed/NCBI
|
|
29
|
Denkert C, Loibl S, Noske A, Roller M,
Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R,
Hanusch C, et al: Tumor-associated lymphocytes as an independent
predictor of response to neoadjuvant chemotherapy in breast cancer.
J Clin Oncol. 28:105–113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Denkert C, von Minckwitz G, Brase JC, Sinn
BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD,
et al: Tumor-infiltrating lymphocytes and response to neoadjuvant
chemotherapy with or without Carboplatin in human epidermal growth
factor receptor 2-positive and triple-negative primary breast
cancers. J Clin Oncol. 33:983–991. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Specht K, Harbeck N, Smida J, Annecke K,
Reich U, Naehrig J, Langer R, Mages J, Busch R, Kruse E, et al:
Expression profiling identifies genes that predict recurrence of
breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer
Res Treat. 118:45–56. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dorsey R, Kundu N, Yang Q, Tannenbaum CS,
Sun H, Hamilton TA and Fulton AM: Immunotherapy with interleukin-10
depends on the CXC chemokines inducible protein-10 and monokine
induced by IFN-gamma. Cancer Res. 62:2606–2610. 2002.PubMed/NCBI
|
|
33
|
Walser TC, Ma X, Kundu N, Dorsey R,
Goloubeva O and Fulton AM: Immune-mediated modulation of breast
cancer growth and metastasis by the chemokine Mig (CXCL9) in a
murine model. J Immunother. 30:490–498. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Walser TC, Rifat S, Ma X, Kundu N, Ward C,
Goloubeva O, Johnson MG, Medina JC, Collins TL and Fulton AM:
Antagonism of CXCR3 inhibits lung metastasis in a murine model of
metastatic breast cancer. Cancer Res. 66:7701–7707. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hilborn E, Sivik T, Fornander T, Stål O,
Nordenskjöld B and Jansson A: C-X-C ligand 10 and C-X-C receptor 3
status can predict tamoxifen treatment response in breast cancer
patients. Breast Cancer Res Treat. 145:73–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wolf K and Friedl P: Mapping proteolytic
cancer cell-extracellular matrix interfaces. Clin Exp Metastasis.
26:289–298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Van den Steen PE, Husson SJ, Proost P, Van
Damme J and Opdenakker G: Carboxyterminal cleavage of the
chemokines MIG and IP-10 by gelatinase B and neutrophil
collagenase. Biochem Biophys Res Commun. 310:889–896. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Denney H, Clench MR and Woodroofe MN:
Cleavage of chemokines CCL2 and CXCL10 by matrix
metalloproteinases-2 and −9: Implications for chemotaxis. Biochem
Biophys Res Commun. 382:341–347. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu G, Yan HH, Pang Y, Jian J, Achyut BR,
Liang X, Weiss JM, Wiltrout RH, Hollander MC and Yang L: CXCR3 as a
molecular target in breast cancer metastasis: Inhibition of tumor
cell migration and promotion of host anti-tumor immunity.
Oncotarget. 6:43408–43419. 2015.PubMed/NCBI
|