Introduction
Haematopoiesis relates to the production,
proliferation, self-renewal, and differentiation of blood cells
(1). In response to growth factors
such as stem cell factor glycoproteins (interleukins 1 to 7) and
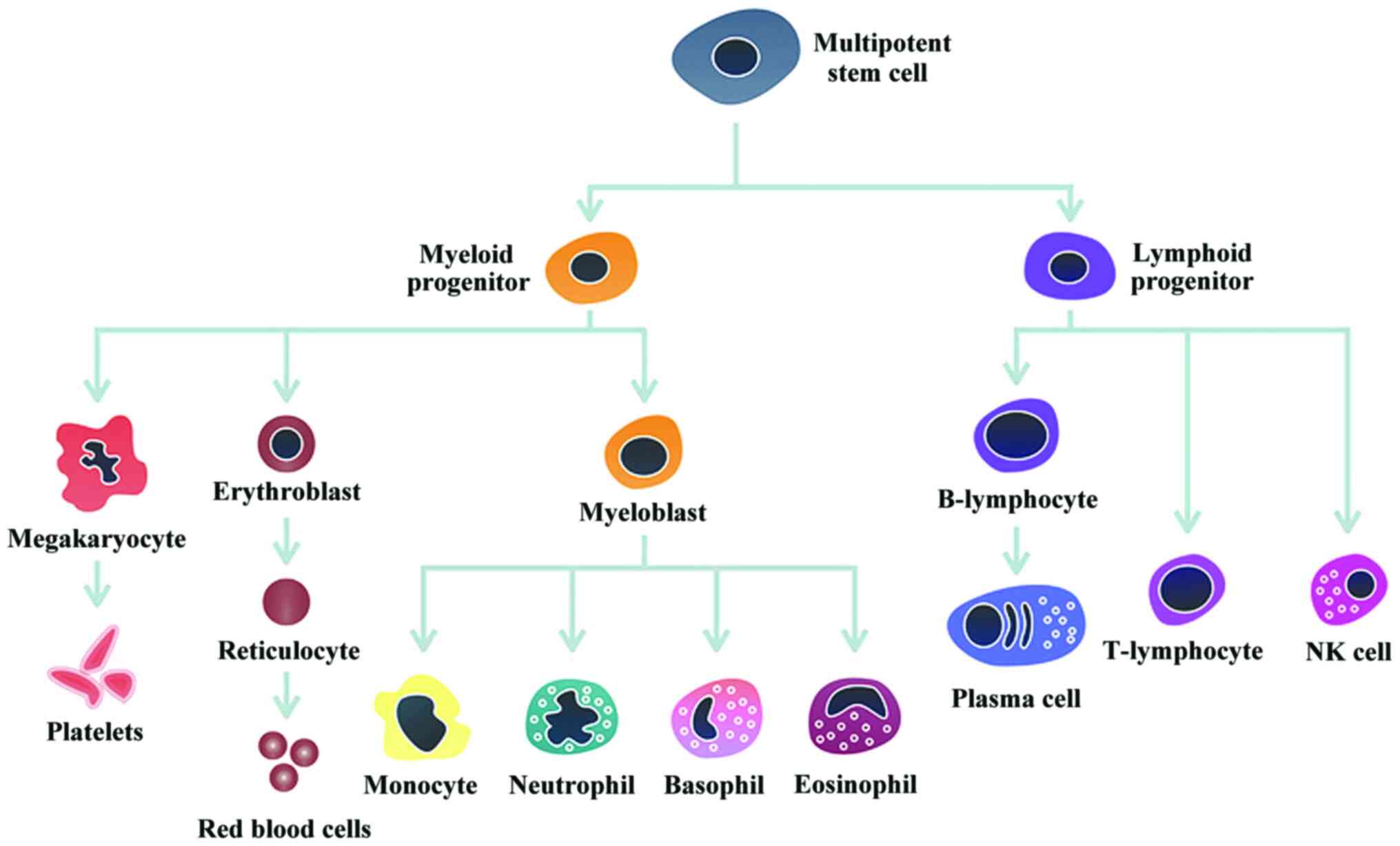
colony-stimulating factors, multipotent haematopoietic stem cells
generate and maintain all differentiated lymphoid and myeloid cells
present in the blood, bone marrow, spleen, and thymus (2). The haematopoietic stem cells produce two
progenitor cells: the common myeloid and common lymphoid cells. The
common lymphoid progenitor cell originates the natural killer, T-
and B-cells that are part of the immune system and have the key
role of controlling infections. The common myeloid progenitor cell
generates three lineages of cells: erythrocyte, megakaryocyte, and
myeloblast. The erythrocyte (red blood cell) is responsible for
carrying and delivering oxygen to the body organs and tissues. The
megakaryocyte produces the platelets or thrombocytes, responsible
for blood clotting. The myeloblast cell differentiates into four
types of cells, which have the capability of defending the body
against infection and toxins: neutrophils, eosinophils, basophils
and monocytes. Fig. 1 shows a
schematic representation of human haematopoiesis.
Leukaemic transformation of a progenitor
haematopoietic cell involves a disruption in the course of normal
proliferation and differentiation process, resistance to apoptotic
signals, and increased self-renewal. The prevalent theory of
leukaemogenesis is that a single haematopoietic cell suffers
mutation and goes into an unlimited process of self-renewal
resulting in malignant, poorly differentiated haematopoietic cells
(clonal origin of leukaemic cell) (3). Leukaemia cells behave differently than
normal haematopoietic precursors, with slower cell division and
longer time to produce DNA. Yet, these cells accumulate
persistently in the bone marrow of leukaemic patients and
progressively replace haematopoietic cells. Furthermore, this
causes bone marrow failure that is associated with severe anemia,
bleeding, and infections.
Diagnosis and classification
Previously, morphologic analysis was used to
classify leukaemia into myeloid or lymphoid (4). Currently, advanced diagnostic techniques
such as flow cytometry, conventional and molecular genetics as well
as next-generation sequencing- based multi-gene mutation profiling
provide precise diagnosis and classification of leukaemias. Some
tests are useful not only for diagnosis, but also to evaluate if
therapy has been effective or modifications of the initial
treatment is required. The common tests used to determine initial
response to therapy include the fluorescence in situ
hybridization test, flow cytometry and polymerase chain reaction
(PCR) (5). The diagnosis of leukaemia
often performed by analysis of the peripheral blood. However, bone
marrow examination is also required because up to 20% of patients
might not present with blasts in the peripheral blood at the time
of clinical presentation (6). In
addition, the morphology of leukaemia cells in the blood might
differ from the cells in the bone marrow.
Clinical features
The clinical features of acute leukaemia are
secondary to the accumulation of malignant cells with consequent
bone marrow failure. Symptoms observed or recorded are often
non-specific. These include, easy or spontaneous bruising, fever,
lethargy, pallor and infection. Bone pain and/or limping are more
common symptoms. Physical examination might also show
lymphadenopathy, hepatomegaly and weight loss.
Childhood cancer is a rare disease. Yet, the numbers
of affected children are on continuous rise as the confirmed cases
are rapidly increasing worldwide (7).
With current population growth and decrease in childhood mortality
rates (mostly due to lower mortality from infectious diseases), the
incidence of childhood cancer is expected to increase by 30% by
2020 (8). About 70% of new cases are
predicted to occur in low- and middle-income countries, where more
than 80% of paediatric cancer deaths currently occur (9).
Leukaemia is the most common type of childhood
malignancy, representing a third of all paediatric cancers and
about 10% of malignancies in adolescents in the developed world.
Acute lymphoblastic leukaemia (ALL) accounts for approximately
75–80% of all childhood leukaemias with an annual incidence rate of
approximately 4/100,000 persons per year in the United States and
Europe (7).
Acute myeloid leukaemia (AML) comprises about 15–20%
of all paediatric leukaemias, with an annual incidence rate of
0.8/100,000 persons per year in the United States. Of those
diagnosed with this malignancy, approximately 33% are adolescents
and 50% are adults in the developed countries (10). There is also substantial geographical
variation in the incidence of AML, with the highest incidence rates
reported in China, and Japan, and among the Maori population in New
Zealand intermediate rates are reported. Although AML is a neoplasm
more frequent in older people, this disease can occur at any age
and remains the leading cause of cancer deaths among patients aged
≤39 years (11). In contrast to ALL,
there is no evidence of a significant increase of incidence of AML
among children, adolescents and young adults in the United States
and Europe over the last few decades.
Risk stratification, treatment and
prognosis
The recognition that ALL and AML are heterogeneous
disease has guided risk-directed therapy aimed at improving
survival as well as the quality of life (12). The identification of patients with
high risk of relapse invites intensive treatment; hence excessive
toxic effects among cases with low-risk disease are prevented
(13). Cytogenetic and molecular
characteristics as well as assessment of minimal residual disease
have been substituting many conventional prognostic factors in both
ALL and AML. The risk stratification of acute leukaemias and
several risk adapted-treatments are described in the following
sections (14).
Acute lymphoblastic leukaemia
Specific treatment approaches for ALL might differ
depending on the disease presentation, but they regularly include
remission-induction, intensification or consolidation treatment,
and maintenance therapy to eradicate leukaemia cells. Patients with
ALL generally need long maintenance treatment in order to prevent
relapse. The mechanism by which lower dose chemotherapy regimen
eradicates the residual leukaemic cells is poorly understood. When
continuation treatment was given during a shorter period of time
(18 months or less) for children and adults with ALL, survival was
lower than that of conventional treatment. While approximately 65%
of young patients may be cured with only 12 months of chemotherapy,
it is not possible to identify these cases with certainty and the
current recommendation is to treat these patients for two years or
more; common drugs used for the treatment of ALL include
corticosteroids, asparaginase and vincristine (15).
Currently, early response to therapy is evaluated by
measurement of residual disease at specific time points in the
peripheral blood and bone marrow, respectively. Minimal residual
disease detected by PCR or by flow cytometry, is recognized as an
important prognostic factor for survival in patients with ALL. This
suggested that continuous minimal residual disease monitoring might
be useful to identify patients with high or intermediate risk of
relapse (those with a somewhat slow early response to therapy) and
guide therapy. However, clinicians should be aware that some
patients with persistent minimal residual disease may be cured,
while some patients with minimal residual disease negative
(undetectable) at remission can still present leukaemia recurrence
(16). This emphasises the
fundamental role of maintenance treatment for patients with ALL.
Treatment directed at the central nervous system is of key
importance and should be initiated early in the course of
treatment. Several factors are taken into account when selecting
the intensity of therapy, such as risk of relapse and the quantity
of leukaemic cells in the cerebral spinal fluid. It has been
established that cranial irradiation could cause various acute and
late adverse effects such as secondary malignancies, neurocognitive
disorders and endocrinopathies. Consequently, in many centres,
intrathecal and systemic chemotherapy have replaced cranial
irradiation (17).
Induction failure is rare in children, and could be
defined as the presence of leukaemic cells in the peripheral blood,
bone marrow or extra medullary location after 4–6 weeks of
induction therapy. The children with ALL who have induction failure
are considered very high-risk patients and haematopoietic stem-cell
transplantation is recommended (18).
Further, a study in the recent past confirmed higher survival time
in children who had T-cell leukaemia with transplantation in
comparison with the children who had undergone chemotherapy with
transplantation (19). Detailed
information on adolescents and young adults is comparatively scarce
because of the smaller number of incident cases in this age group,
as well as lower enrolment of these patients in clinical trials
compared with children and older adults (20). Consequently, adolescents are commonly
examined together with children aged 10–15 years in paediatric
trials or with patients aged 20–30 years in adult trials. There is
evidence that substantial decline in survival is observed beyond 15
years of age at diagnosis (21). At
present, evidence suggests that intensified treatment protocols
might reduce or eliminate the influence of some prognostic factors,
such as male, black race and Down syndrome, on survival (22).
Moreover, although children with Down syndrome have
an increased risk of developing ALL and lower survival, outcomes
are comparable in children with and without Down syndrome after
adjustment for favourable and unfavourable cytogenetic lesions
(23). Clinical trials have
identified several factors predictive of outcomes after ALL
(24). The few particular subgroups
of high-risk patients with ALL that should be treated with a
risk-adapted protocol are described below. Due to the extremely
poor prognosis in the past, children Philadelphia
chromosome-positive ALL are currently treated with intensive
chemotherapy plus imatininb or desatinib, tyrosine kinase
inhibitors. Haematopoietic stem cell transplantation might be
recommended in case of relapse.
Adolescent and young adult acute
lymphoblastic leukaemia
The adolescents and young adults with ALL have
poorer outcomes compared with children. In the mid-2000s,
collaborative trials began to treat these patients with paediatric
protocols and several studies have shown excellent results
(25). Between 2007 and 2012, a large
prospective adult intergroup trial (C10403) (26) in the United States, investigated the
adoption of a successful protocol used by the Children's Oncology
Group (ALL0232) for treatment of patients aged 16–39 years with ALL
(27). The significant improvement in
outcomes of B- or T-cell ALL patients supported the use of
paediatric protocols by adult haematologists to treat adolescents
and young adults with this neoplasm. However, despite improvement,
there is evidence that these patients continue to be treated with
low-intensity chemotherapy regimens in many centres (28). An explanation might be that some
clinicians are not yet convinced about the paediatric approach
superiority compared to the conventional treatment and may await
more evidence from a randomized phase III study. Another
possibility is that adult haematologists might not feel as familiar
as paediatricians in managing the treatment-related toxicity
secondary to the intensive paediatric protocol. Some complications
of treatment such as pancreatitis, osteonecrosis, hyperglycemia,
and infection seem to occur more often in older patients (>10
years old). This might cause adult clinicians to change prescribed
drug dosage and schedule (29,30).
The observation of ALL relapse leading to deaths of
pediatric patients still raises the risk of death in spite of the
dramatic improvement in survival. The main determinants of survival
are time to relapse, site of relapse, leukaemia immunophenotype,
and more recently, minimal residual disease. Patients who present
relapse within 36 months of diagnosis have a dismal 5-year overall
survival of approximately 15% (31).
Salvage chemotherapy or even haematopoietic stem
cell transplantation for both adults and children with relapsed or
refractory ALL have not improved outcome, and intensive research
continues to be done in order to find new therapeutic agents able
to improve survival in these patients. New monoclonal antibodies
such as cluster of differentiation (CD)19, CD20, CD22, and CD52
have been developed. The rationale for the use of monoclonal
antibodies is that lymphoblasts express various cell-surface
antigens that may be favourable targets for this therapy. For
instance, over 95% of B-cell ALL and more than 90% of lymphoblasts
express CD19 and CD22, respectively (32). Monoclonal antibody therapy has been
recently used in clinical trials to treat children and adults with
relapsed or refractory ALL. The initial results have been
favourable with good tolerability and high levels of negative
minimal residual disease. However, longer follow-up time is
necessary to assess toxicity and long-term outcome.
Acute myeloid leukaemia
Similar to ALL, risk-adapted therapy has become
critical for AML. AML has been risk stratified in two major groups.
The low risk group (about 25% of the cases) includes patients with
core-binding factor (CBF) AML [t(8;21), inv(16), t(15;17)], infant AML, AML with Down
syndrome, AML with CEBPA and NPM1 mutations (non-FLT3-ITD) or
megakaryoblastic AML with the t(1;22) abnormality, and minimal
residual disease negative. The high-risk group (about 25% of the
cases) includes patients with unfavourable cytogenetic alterations
(monosomies 5 and 7), FLT3-ITD and TP53 mutations, secondary AML,
AML associated with myelodisplastic syndrome, and minimal residual
disease positive. In approximately 40–50% of cases there is not a
good genetic or molecular marker to determine the disease prognosis
and clinicians use minimal residual disease assessment to guide
treatment (33). In general, the
treatment of AML is performed using four to five intensive courses
of cytarabine and anthracyclines chemotherapy. Maintenance therapy
appears not to have any advantage in AML as it occurs for ALL.
Central nervous system directed therapy with triple agents is also
recommended for AML. Haematopoietic stem cell transplantation is
performed more often among young patients with AML than for ALL
(about 30 vs. 5%).
Core-binding factor AML
CBF AML is associated with chromosomal
rearrangements between chromosomes 8 and 21 (t8;21) and within
chromosome 16 [inv(16)]. This
subtype accounts for about 25% of all childhood AML cases and its
prevalence decreases with advancing age. CBF AML prevalence is
approximately 10–15% in adults aged 60 years or younger and 5% in
patients older than 60 years (34).
With intensive chemotherapy regimens with three to four drugs, CBF
AML has become a group of good prognosis with 3-year overall
survival of approximately 90% in children and adolescents and about
69% for young adults. Furthermore, haematopoietic stem cell
tansplantation is not recommended for children in complete
remission, but might be indicated for those who relapse.
Down syndrome effects
The patients diagnosed with both AML and Down
syndrome have a better prognosis than non-Down syndrome patients
with AML. Therefore, the current treatment approach is to reduce
chemotherapy agents in order to avoid complications of treatment,
particularly cardiotoxicity.
Neonatal and infant acute myeloid
leukaemia
Because neonates with AML may have spontaneous
remission, some clinicians may choose to observe them rather than
begin chemotherapy immediately. When chemotherapy is necessary,
careful dose adjustments should be done in order to avoid toxicity.
Neonates tend to have worse clinical course due to complications of
treatment and disease resistance than children. On the contrary,
infants with AML often have similar prognosis as older children
provided that they receive intensive chemotherapy regimen.
Haematopoietic stem cell transplantation seems not to improve
outcome in these patients and can cause serious adverse
effects.
AML with altered genes (FLT3-ITD, NPM and CEBPA
mutations) FLT3-ITD mutations are associated with poor prognosis in
children (5-year overall survival <35%) (35). The use of haematopoietic stem cell
transplantation is still controversial, being usually reserved for
high-risk patients. In contrast, patients with FTL3 point mutation
have a better outcome and are often treated with chemotherapy only.
Patients with NPM and CEBPA mutations have a favourable prognosis;
therefore haematopoietic stem cell transplantation is not usually
recommended for these patients. AML with MLL-rearrangements is a
heterogeneous disease and prognosis may vary from 22% for patients
with t(6;11) to 100% for those with t(1;11). The role of
haematopoietic stem cell transplantation remains controversial in
this subtype of disease (36).
Conclusion
The prognosis of acute leukaemia has improved
substantially in the last few decades, mainly for ALL and some
subtypes of AML. This improvement was possible due to national and
international collaborative clinical trials that investigate the
association of various factors on outcome. Risk-adapted therapy
based on patient clinical and genetic features and minimal residual
disease assessment have largely contributed to treatment success in
both ALL and AML treatments. However, for specific types of
disease, prognosis is still poor and new treatment approaches are
warranted.
References
|
1
|
Li Z, Zhang P, Yan A, Guo Z, Ban Y, Li J,
Chen S, Yang H, He Y, Li J, et al: ASXL1 interacts with the cohesin
complex to maintain chromatid separation and gene expression for
normal hematopoiesis. Sci Adv. 3:e16016022017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Szilvassy SJ: The biology of hematopoietic
stem cells. Arch Med Res. 34:446–460. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pui CH: Acute lymphoblastic
leukemiaChildhood Leukemias. 3rd. Cambridge University Press;
Cambridge: pp. 332–366. 2012, View Article : Google Scholar
|
|
4
|
Collins L, Beaumont L, Cranston A, Savoie
S, Nayiager T and Barr R: Anthropometry in long-term survivors of
acute lymphoblastic leukemia in childhood and adolescence. J
Adolesc Young Adult Oncol. Jan 24–2017.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
El-Rifai W, Ruutu T, Elonen E, Volin L and
Knuutila S: Prognostic value of metaphase-fluorescence in situ
hybridization in follow-up of patients with acute myeloid leukemia
in remission. Blood. 89:3330–3334. 1997.PubMed/NCBI
|
|
6
|
Onciu M and Pui CH: Diagnosis and
classificationChildhood Leukemias. Pui CH: 3rd. Cambridge
University Press; Cambridge: pp. 21–48. 2012, View Article : Google Scholar
|
|
7
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodriguez-Galindo C, Friedrich P,
Morrissey L and Frazier L: Global challenges in pediatric oncology.
Curr Opin Pediatr. 25:3–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bray F, Jemal A, Grey N, Ferlay J and
Forman D: Global cancer transitions according to the Human
Development Index (2008–2030): a population-based study. Lancet
Oncol. 13:790–801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Creutzig U, Büchner T, Sauerland MC,
Zimmermann M, Reinhardt D, Döhner H and Schlenk RF: Significance of
age in acute myeloid leukemia patients younger than 30 years: a
common analysis of the pediatric trials AML-BFM 93/98 and the adult
trials AMLCG 92/99 and AMLSG HD93/98A. Cancer. 112:562–571. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deschler B and Lübbert M: Acute myeloid
leukemia: epidemiology and etiology. Cancer. 107:2099–2107. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pui CH, Robison LL and Look AT: Acute
lymphoblastic leukaemia. Lancet. 371:1030–1043. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pui CH: Acute lymphoblastic leukemia:
introduction. Semin Hematol. 46:1–2. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pui CH, Carroll WL, Meshinchi S and Arceci
RJ: Biology, risk stratification, and therapy of pediatric acute
leukemias: an update. J Clin Oncol. 29:551–565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pui CH, Mullighan CG, Evans WE and Relling
MV: Pediatric acute lymphoblastic leukemia: where are we going and
how do we get there? Blood. 120:1165–1174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goulden NJ, Knechtli CJ, Garland RJ,
Langlands K, Hancock JP, Potter MN, Steward CG and Oakhill A:
Minimal residual disease analysis for the prediction of relapse in
children with standard-risk acute lymphoblastic leukaemia. Br J
Haematol. 100:235–244. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Veerman AJ, Kamps WA, van den Berg H, van
den Berg E, Bökkerink JP, Bruin MC, van den Heuvel-Eibrink MM,
Korbijn CM, Korthof ET, van der Pal K, et al: Dutch Childhood
Oncology Group: Dexamethasone-based therapy for childhood acute
lymphoblastic leukaemia: results of the prospective Dutch Childhood
Oncology Group (DCOG) protocol ALL-9 (1997–2004). Lancet Oncol.
10:957–966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schrappe M, Hunger SP, Pui CH, Saha V,
Gaynon PS, Baruchel A, Conter V, Otten J, Ohara A, Versluys AB, et
al: Outcomes after induction failure in childhood acute
lymphoblastic leukemia. N Engl J Med. 366:1371–1381. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Balduzzi A, Valsecchi MG, Uderzo C, de
Lorenzo P, Klingebiel T, Peters C, Stary J, Felice MS, Magyarosy E,
Conter V, et al: Chemotherapy versus allogeneic transplantation for
very-high-risk childhood acute lymphoblastic leukaemia in first
complete remission: comparison by genetic randomisation in an
international prospective study. Lancet. 366:635–642. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bleyer A, Budd T and Montello M:
Adolescents and young adults with cancer: the scope of the problem
and criticality of clinical trials. Cancer. 107 Suppl:1645–1655.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nachman J: Clinical characteristics,
biologic features and outcome for young adult patients with acute
lymphoblastic leukaemia. Br J Haematol. 130:166–173. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bhojwani D, Pei D, Sandlund JT, Jeha S,
Ribeiro RC, Rubnitz JE, Raimondi SC, Shurtleff S, Onciu M, Cheng C,
et al: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia:
improved outcome with contemporary therapy. Leukemia. 26:265–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maloney KW, Carroll WL, Carroll AJ,
Devidas M, Borowitz MJ, Martin PL, Pullen J, Whitlock JA, Willman
CL, Winick NJ, et al: Down syndrome childhood acute lymphoblastic
leukemia has a unique spectrum of sentinel cytogenetic lesions that
influences treatment outcome: a report from the Childrens Oncology
Group. Blood. 116:1045–1050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hunger SP, Loh ML, Whitlock JA, Winick NJ,
Carroll WL, Devidas M and Raetz EA: COG Acute Lymphoblastic
Leukemia Committee: Childrens Oncology Groups 2013 blueprint for
research: acute lymphoblastic leukemia. Pediatr Blood Cancer.
60:957–963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nachman JB, La MK, Hunger SP, Heerema NA,
Gaynon PS, Hastings C, Mattano LA Jr, Sather H, Devidas M, Freyer
DR, et al: Young adults with acute lymphoblastic leukemia have an
excellent outcome with chemotherapy alone and benefit from
intensive postinduction treatment: a report from the Childrens
Oncology Group. J Clin Oncol. 27:5189–5194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stock W, Luger S, Advani A, Geyer S,
Harvey R, Mullighan C, Willman CL, Malnassy G, Parker E, Laumann
KM, et al: Favorable outcomes for older adolescents and young
adults (AYA) with acute lymphoblastic leukemia (ALL): early results
of US intergroup trial C10403. Blood. 124:7962014.
|
|
27
|
Larsen EC, Salzer W, Nachman J, Devidas M,
Freyer DR, Raetz EA, Winick N, Hunger SP and Carroll WL: Treatment
toxicity in adolescents and young adult (AYA) patients compared
with younger patients treated for high risk B-precursor acute
lymphoblastic leukemia (HR-ALL): a report from the Childrens
Oncology Group study AALL0232. Blood. 118:15102011.
|
|
28
|
Carlson RH: Renewed calls for adolescents
and young adults with ALL to be treated with pediatric protocols.
Oncol Times. 37:20–21. 2015. View Article : Google Scholar
|
|
29
|
Advani AS, Hunger SP and Burnett AK: Acute
leukemia in adolescents and young adults. Semin Oncol. 36:213–226.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gramatges MM and Rabin KR: The adolescent
and young adult with cancer: state of the art - acute leukemias.
Curr Oncol Rep. 15:317–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nguyen K, Devidas M, Cheng SC, La M, Raetz
EA, Carroll WL, Winick NJ, Hunger SP, Gaynon PS and Loh ML;
Childrens Oncology Group, : Factors influencing survival after
relapse from acute lymphoblastic leukemia: a Childrens Oncology
Group study. Leukemia. 22:2142–2150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
DeAngelo DJ: The use of novel monoclonal
antibodies in the treatment of acute lymphoblastic leukemia.
Hematology Am Soc Hematol Educ Program. 2015:400–405.
2015.PubMed/NCBI
|
|
33
|
Rubnitz JE, Inaba H, Dahl G, Ribeiro RC,
Bowman WP, Taub J, Pounds S, Razzouk BI, Lacayo NJ, Cao X, et al:
Minimal residual disease-directed therapy for childhood acute
myeloid leukaemia: results of the AML02 multicentre trial. Lancet
Oncol. 11:543–552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mullighan C, Hunger SP and Meshinchi S:
Molecular genetics in children, adolescents and young adults with
acute lymphoblastic leukemia and acute myeloid
leukemiaHematological Malignancies in Children, Adolescents and
Young Adults. Cairo MS and Perkins SL: World Scientific Publishing
Co. Pte. Ltd.; Singapore: pp. 121–142. 2012, View Article : Google Scholar
|
|
35
|
Pratz KW, Sato T, Murphy KM, Stine A,
Rajkhowa T and Levis M: FLT3-mutant allelic burden and clinical
status are predictive of response to FLT3 inhibitors in AML. Blood.
115:1425–1432. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Balgobind BV, Raimondi SC, Harbott J,
Zimmermann M, Alonzo TA, Auvrignon A, Beverloo HB, Chang M,
Creutzig U, Dworzak MN, et al: Novel prognostic subgroups in
childhood 11q23/MLL-rearranged acute myeloid leukemia: results of
an international retrospective study. Blood. 114:2489–2496. 2009.
View Article : Google Scholar : PubMed/NCBI
|