Introduction
Glioblastoma multiforme (GBM) is not only the most
common type of brain tumor in adulthood, but also one of the most
malignant types of cancer, with a median survival time (World
Health Organization grade IV) of 14–15 months with maximum
treatment (1). Its infiltrative and
heterogeneous characteristics, combined with high proliferation
rates, makes treatment challenging. Standard therapy includes
surgery, adjuvant radiotherapy and chemotherapy with the alkylating
agent temozolomide (TMZ) (1). TMZ was
first identified by Stevens et al in 1984 (2) as an oral anti-cancer treatment. The
active metabolite of TMZ methylates guanine residues in the DNA,
leading to double strand breaks (3).
The O6-methylguanine-DNA methyltransferase (MGMT) gene codes for a
protein that removes the methylation performed by TMZ and abrogates
its effects (4). This gene's promoter
is methylated in ~50% of cases, which is an independent and
favorable prognostic factor for patients with GBM (4).
Previous studies have established the association
between chemotherapy with TMZ and autophagy induction (5,6).
Autophagy, from the Greek meaning ‘self-eating’, is a recycling
machinery of intracellular proteins and organelles like
mitochondria (7). Macroautophagy is
the major type of autophagy and is referred to as autophagy for the
remainder of the current study. This process comprises several
sequestration steps, beginning with a phagophore enclosing cellular
waste and forming the autophagosome by elongation (8). Fusion of the autophagosome and a
lysosome allows acidic hydrolases to degrade the inner components
of the now termed ‘autolysosome’ (8).
For the purposes of monitoring the autophagic flux,
the microtubule-associated protein light chain 3 (LC3) is one of
the most reliable proteins (8).
Pro-LC3 is cleaved by the autophagy-related-gene protease 4 (Atg4)
to form LC3-I (9). The conjugation of
LC3-I to phosphatidylethanolamine forms LC3-II, which is the
lipidated form of LC3 and is located at the autophagosome cytosolic
and intraluminal membrane (8).
Subsequently, LC3-II at the intraluminal membrane is degraded by
lysosomal hydrolyases in the final step of autophagy (8). The drug chloroquine inhibits this last
step, resulting in an accumulation of LC3-II (10).
Beclin-1, the mammalian homolog of Atg6, is required
to initiate the autophagic process (11,12).
Notably, Wei et al (13)
described an interaction between the epidermal growth factor
receptor (EGFR) and autophagy in non-small cell lung cancer (NSCLC)
cells. Active EGFR inhibited the initiation of autophagy via
phosphorylation of Beclin-1, and activation of EGFR was performed
through the addition of EGF or by transfection of cells with the
truncated EGFRvIII version (13). This mutated receptor is common in
NSCLC cells, as well as in GBM (14,15). Due
to an in-frame deletion of exon 2–7, the extracellular binding
portion is deleted, leading to the continuous activation of EGFR
(15). Wei et al (13) identified that this activation inhibits
autophagy through an interaction with Beclin-1 (13). It remains to be established whether
the same underlying mechanisms are relevant in GBM.
Materials and methods
Reagents
TMZ and chloroquine were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). EGF was purchased from PeproTech
Inc. (Rocky Hill, NJ, USA).
Cell culture and cell treatment
The LN18 human glioblastoma cell line, first
characterized in 1981 (16), was
obtained from Dr. Van Meir (University of Lausanne, Switzerland).
pGBM T1 and pGBM T12 cells were isolated from tissues that were
collected in cooperation with the Department for Neurosurgery at
Technische Universität München (Munich, Germany). Primary cell
culture was established by Dr. Andrea Schäfer as previously
described (17). The resection
(January and May 2008) and subsequent processing were performed
with patients' consent according to the Technische Universität
München medical faculty's guidelines for tissue preservation.
Primary single cell suspensions were only cultured at early
passages. The primary glioblastoma stem cell line X01 was obtained
from Dr Andreas Androutsellis-Theotokis (Carl Gustav Carus
University, Dresden, Germany).
LN18, pGBM T1 and pGBM T12 cells were maintained in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) under standard cell culture conditions in the
presence of 5% CO2 at 37°C in a humidified incubator.
When treating cells with chemotherapeutics, the concentration of
fetal calf serum (FCS; Thermo Fisher Scientific, Inc.) was reduced
from 4 to 0.1% to minimize off-target FCS effects. The primary
glioblastoma tumor stem cell line X01 was cultured in RPMI-1640
medium (Thermo Fisher Scientific, Inc.) supplemented with 1%
L-glutamine (Thermo Fisher Scientific, Inc.), 1% N1 (Thermo Fisher
Scientific, Inc.), 1% non-essential amino acids (Thermo Fisher
Scientific, Inc.), 1 ng/ml transforming growth factor β (Thermo
Fisher Scientific, Inc.), 20 ng/ml basic fibroblast growth factor
(Thermo Fisher Scientific, Inc.), 20% BIT100 (Pelo Biotech GmbH,
Planegg, Germany), 0.1% primocin (InvivoGen, San Diego, USA), and
20 ng/ml epidermal growth factor (PeproTech, Inc.) to simulate
cancer stem cell conditions. To investigate EGFR alterations, LN18
cells were transfected with plasmid DNA carrying the constitutively
active EGFRvIII variant (LN18vIII) by using
lipofectamine transfection reagent (Thermo Fisher Scientific,
Inc.). Cells were cultured and maintained in the presence of the
selective antibiotic G418 (Thermo Fisher Scientific, Inc.). Stable
expression of EGFRvIII was routinely analyzed by western
blotting using EGFR antibody (Ab)-12 (cocktail R19/48; 1:500;
catalog no. MS-400-P; Thermo Fisher Scientific, Inc.). The antibody
is able to detect extracellular and cytoplasmic domains allowing
differentiation between wild-type EGFR (170 kDa) and
EGFRvIII (145 kDa) forms of the protein.
LN18, pGBM T1 and pGBM T12 were exposed to 50 µM
chloroquine for 2 h. LN18 cells were treated with 20 ng EGF for 30
min. The concentration of TMZ treatment varied from 100–500 µM for
2–72 h due to different responses in established and primary cell
lines.
Hypoxic treatment of cells
pGMB X01 cells were placed in the hypoxic incubating
chamber at 37°C and O2 was gradually replaced by
nitrogen within 11 cycles. Following incubation for 24 h at 1%
O2, the cells were lysed at the same time as the
normoxic pGBM X01 control cells.
Immunoblotting
Collected and washed LN18, LN18vIII, pGBM
T1 and pGBM T12 proteins were resuspended in freshly prepared lysis
buffer (20% L-Buffer, 2% phenylmethanesulfonyl fluoride; Cell
Signaling Technology, Inc., Danvers, MA, USA) and incubated
rotating at 4°C for 10 min. Protein quantification was measured
using a Bradford protein assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Following quantification, equal amounts of
proteins (20 µg) were unfolded and charged by the detergent sodium
dodecyl sulfate. SDS-PAGE gels were prepared with the permeability
(7–12%) adjusted to the size of the proteins. Following separation
through the gel matrix, proteins were transferred to an
immobilizing polyvinylidene difluoride membrane. Blocking of
nonspecific binding sites was performed with 5% milk for 1 h at
room temperature. Cells were incubated with the following primary
antibodies by rotating at 4°C overnight: Beclin-1 (1:1,000; catalog
no. 4445S; Cell Signaling Technology, Inc., Danvers, MA, USA),
light chain 3 B-II (LC3B; 1:1,000; catalog no. 4445S; Cell
Signaling Technology, Inc.), MGMT (1:1,000; catalog no. 2739; Cell
Signaling Technology, Inc.), EGFR Ab-12 cocktail R19/48 (1:500;
catalog no. MS-400-P; Thermo Fisher Scientific, Inc.) and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000; catalog
no. G8795; Sigma-Aldrich, Merck KGaA). Blots were incubated with
horseradish peroxidase-conjugated anti-mouse or anti-rabbit
secondary immunoglobulin for 1 h at room temperature (1:10,000;
catalog nos. 7076 and 7074, respectively; Cell Signaling
Technology, Inc.). Every step was followed by three 5 min washes
with phosphate-buffered saline (PBS; Thermo Fisher Scientific,
Inc.). To visualize the binding sites, enhanced chemiluminescence
solution (Thermo Fisher Scientific, Inc.) was added to the blots,
and the chemiluminescence reaction was detected using an X-ray
film.
Immunofluorescence
LN18 and LN18vIII cells were cultured on
gelatin-coated glass slips for 48 h prior to TMZ (500 µM)
application. At 48 h following treatment, cells were fixed with 4%
paraformaldehyde at room temperature for 30 min and washed 3 times
with PBS. To allow access to intracellular antigens, cells were
exposed to the surfactant 0.1% Triton X-100 (Carl Roth GmbH+Co. KG,
Karlsruhe, Germany)/PBS for 10 min. Blocking of unspecific binding
sites was conducted with an antibody blocking buffer containing 1%
bovine serum albumin (Bio-Rad Laboratories), 0.01% Tween-20 (Carl
Roth GmbH+Co. KG) and 2.5% goat-serum (Thermo Fisher Scientific,
Inc.) for 30 min at room temperature. The cells were incubated with
the primary antibodies against EGFR Ab-12 cocktail R19/48 (1:200;
catalog no. MS-400-P; Thermo Fisher Scientific, Inc.) and Beclin-1
(1:200; catalog no. SC-11427; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) for 2 h at room temperature. Following washing
with PBS, the cells were exposed to Alexa-568-conjugated anti-mouse
(1:500; catalog no. A-11004) and Alexa-488-conjugated anti-rabbit
secondary antibodies (1:500; catalog no. A-11008; Thermo Fisher
Scientific, Inc.). The cells were subsequently incubated for 45 min
at room temperature in the dark. The slides were analyzed with the
Axio Imager 1 microscope (Zeiss AG, Oberkochen, Germany).
Co-immunoprecipitation
To detect protein-protein interactions, a Pierce
Classic immunoprecipitation kit (Thermo Fisher Scientific, Inc.)
was used according to the manufacturer's protocol. The cells were
lysed with ice-cold immunoprecipitation lysis buffer and purified
by using control agarose resin. The flow-through was prepared in
order to form the immune complex with the primary antibody
overnight. The complex was captured in a spin column containing
resin slurry by gently mixing for 1 h. Following three washing
steps, the immune complex was eluted by incubation in 2X
non-reducing sample buffer with dithiothreitol (20 mM) and
collected by centrifugation at 1,000 × g at 4°C for 45 sec. The
antibodies against Beclin-1 (immobilization, 1:100; immunoblotting,
1:1,000; catalog no. 3495; Cell Signaling Technology, Inc.) and
EGFR (immobilization, 1:100; immunoblotting 1:1,000; catalog no.
2232; Cell Signaling Technology, Inc.) were used. A protein complex
can be pulled down with one or two antibodies depending on whether
only the interaction is a matter of interest, or the expression of
the two individual proteins is being examined (18). In this case, Beclin-1 and EGFR are
established as being expressed in LN18 cells (19,20). For
that reason, co-immunoprecipitation was performed using Beclin-1
alone for pull-down.
Statistical analysis
ImageJ (version 1.51; National Institutes of Health,
Bethesda, USA) and Microsoft Excel 2007 (Microsoft Corporation,
Redmond, WA, USA) were used for analyzing western blots and
immunofluorescence. Relative normalization control values of
respective GAPDH lanes were used for normalizing the protein of
interest. Statistical analysis of Student's t-test and Pearson's
correlation analysis was performed with R Studio (version 3.2.3; R
Studio, Boston, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Chloroquine application increases
LC3B-II levels
To assess whether the cell lines in the present
study are responsive to autophagy, chloroquine was applied to the
established cell line LN18 and the primary lines pGBM T1 and pGBM
T12. Fig. 1A and the corresponding
analysis (Fig. 1B) indicates the
significant increase of LC3B-II levels in chloroquine-treated cells
(P<0.05), suggesting a block of autophagy. Beclin-1 was
ubiquitously expressed, but its expression levels were not affected
by chloroquine treatment.
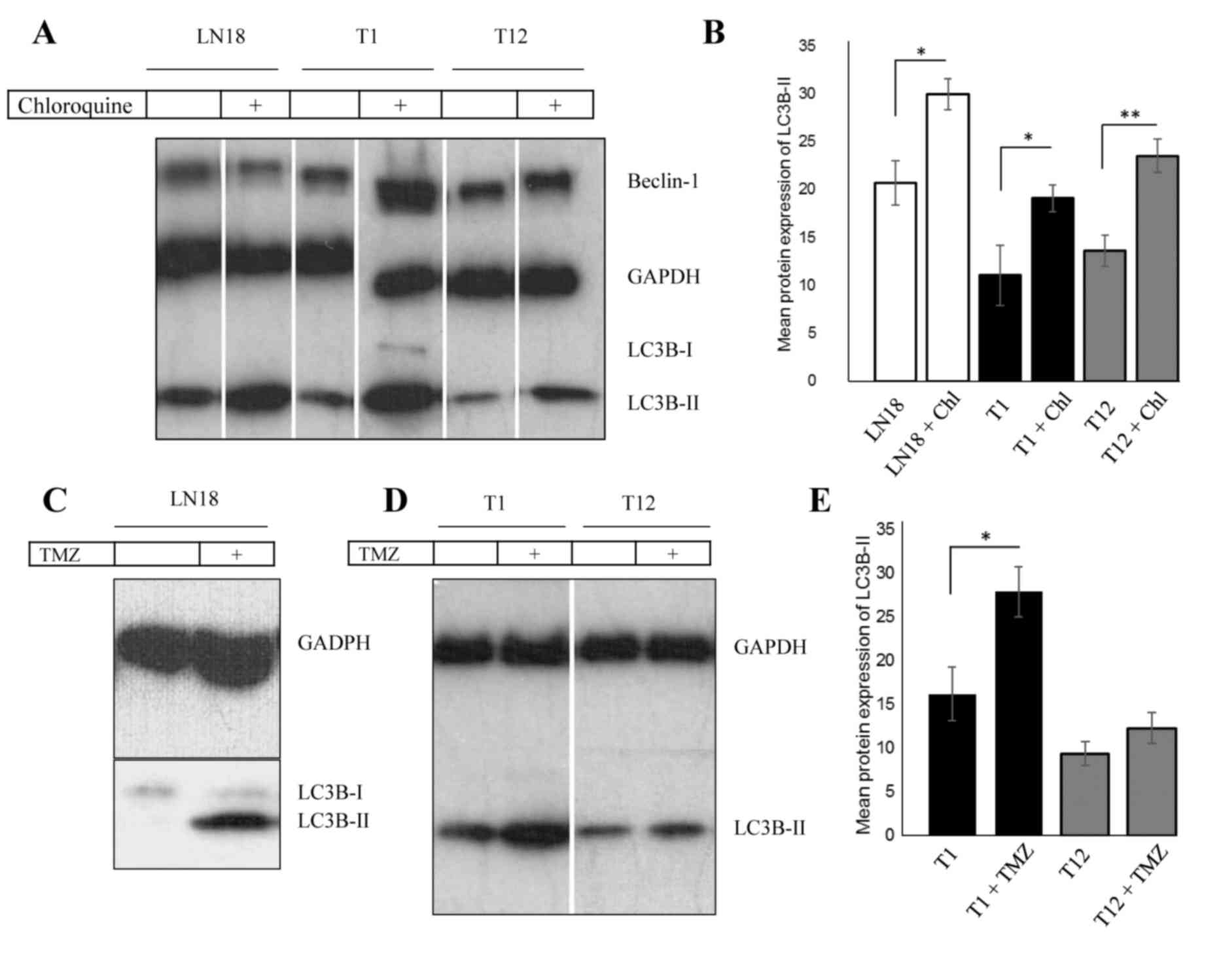 | Figure 1.TMZ upregulates LC3B-II levels in
primary and established GBM cells. (A) Western blot analysis
revealed increased levels of LC3B-II following treatment with 50 µM
chloroquine for 2 h in GBM LN18, pGBM T1 and pGBM T12 cells. (B)
Analysis of chloroquine treatment, with LC3B-II upregulation
normalized to GAPDH. (C) LN18 exhibits an upregulation of LC3B-II
following long-term TMZ treatment. TMZ was applied for 72 h at a
concentration of 500 µM. (D) Autophagy levels were increased in
pGBM T1 following short-term application of low-dose TMZ. In pGBM
T1 cells, the level of LC3B-II protein was increased following TMZ
treatment (200 µM, 2 h). In pGBM T12 cells, the LC3B-II protein
levels were not notably modified by short-term TMZ treatment. (E)
Densitometric analysis of protein expression following TMZ
treatment. *P<0.05, **P<0.01. Error bars indicate the mean
densitometric value ± standard deviation. Chl, chloroquine; TMZ,
temozolomide; LC3B-II, light chain 3 B-II; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma
multiforme; LN18vIII, constitutively active
EGFRvIII variant. |
TMZ induces autophagy in primary and
established glioblastoma cells
LN18 cells were treated with 100–200 µM TMZ for 2 h
each, which did not result in an increase in LC3B-II protein levels
(data not shown). Conversely, TMZ treatment (500 µM) for 72 h
resulted in a arked upregulation of LC3B-II (Fig. 1C). To investigate these findings with
primary cells, pGBM T1 and pGBM T12 cells were exposed to TMZ (200
µM), which resulted in a significant increase of LC3B-II in pGBM T1
cells (Fig. 1D and E; P=0.0168). The
primary cell line pGBM T12 exhibited only a minor modification of
LC3B-II levels.
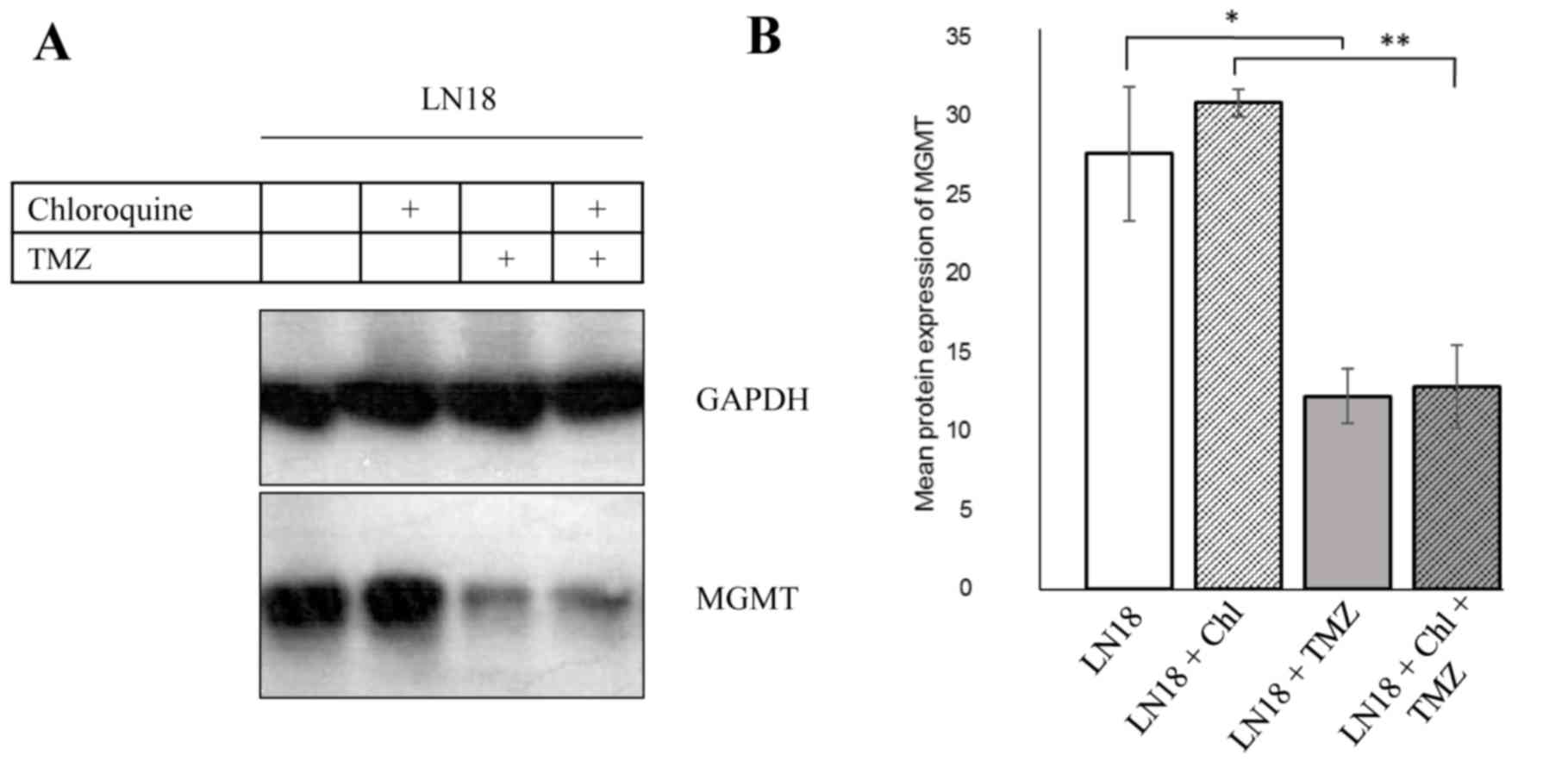
TMZ application decreases MGMT protein
levels
Methylation of the promoter of the MGMT gene and its
associated lowered protein levels results in a higher
susceptibility towards TMZ treatment (21). LN18 cells expressed high levels of
MGMT protein (Fig. 2A). By contrast,
pGBM T1 and pGBM T12 did not express MGMT protein (data not shown).
The high levels of MGMT in LN18 cells decreased following the
application of TMZ (Fig. 2A and B,
P<0.05), indicating a reduction in MGMT protein.
EGFR and EGFRvIII do not
phosphorylate Beclin-1 independently of TMZ or EGF application
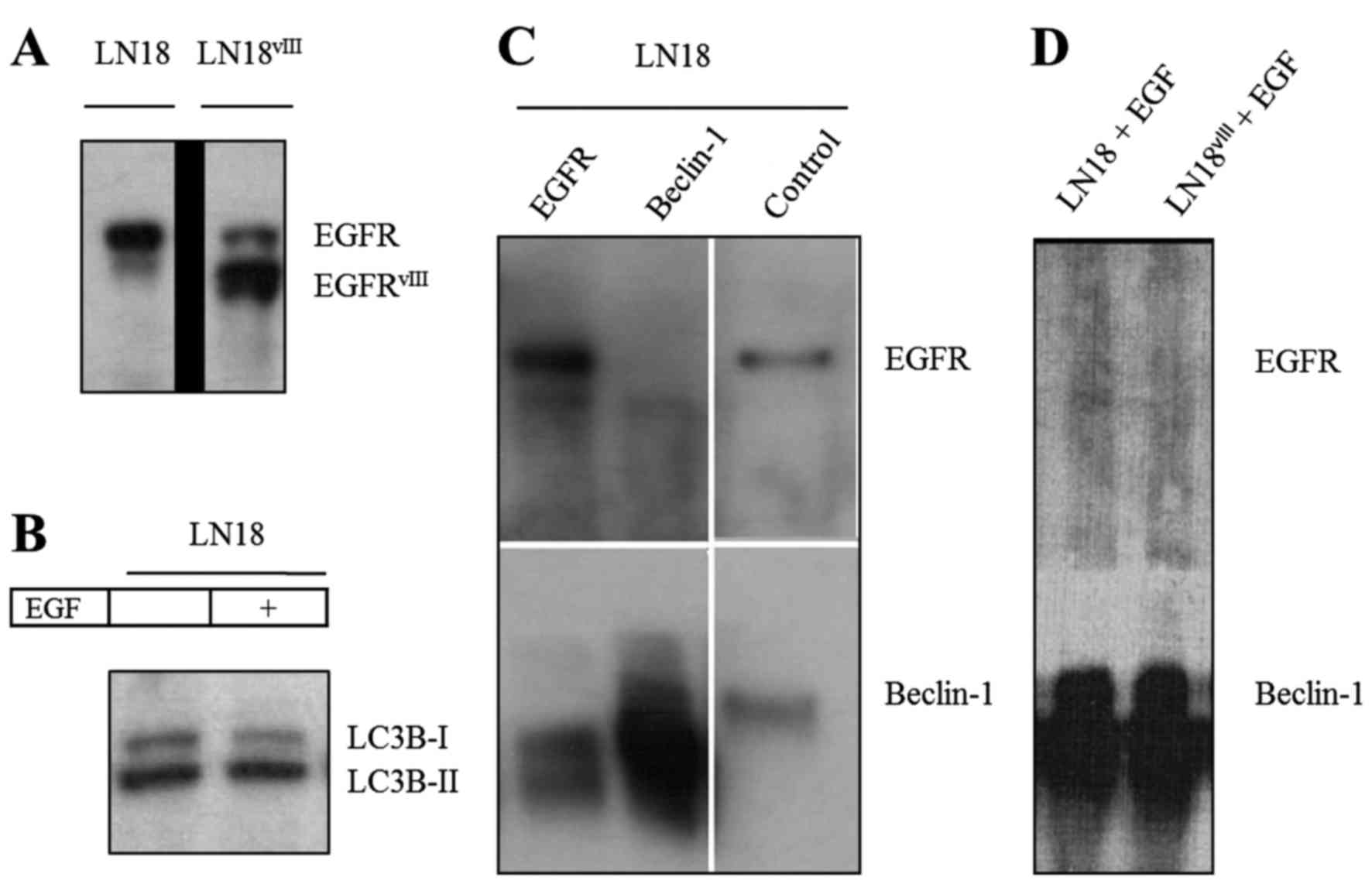
In the present study, several techniques were
applied to monitor the potential interactions of Beclin-1 and EGFR.
Immunoblotting revealed the phosphorylation status of Beclin-1,
demonstrating that, in LN18 cells, Beclin-1 was not phosphorylated
independently of TMZ application. To minimize the effects of
inactive EGFR, LN18 cells transfected with the constitutively
active vIII mutant were cultured (Fig.
3A). In addition, LN18 was incubated with EGF (20 ng, 30 min).
Beclin-1 phosphorylation status was not divergent from the controls
following EGF application or in LN18vIII. LC3B-II levels
were not altered in LN18 by stimulation with EGF (Fig. 3B).
EGFR and EGFRvIII do not
co-immunoprecipitate with Beclin-1 independently of TMZ or EGF
application
In the GBM cell lines LN18 and LN18vIII,
Beclin-1 and EGFR did not co-immunoprecipitate (Fig. 3C and D). The EGFR-Beclin-1 interaction
was not promoted by treatment with TMZ (500 µM, 24 h) or by
stimulation with EGF (20 ng, 30 min; Fig.
3D). These results were further confirmed by
co-immunoprecipitation of the primary GBM cell line X01. To
investigate whether hypoxia, as is present in the center of tumor
masses, interferes with the EGFR-Beclin-1 association, pGBM X01 was
incubated under hypoxic cell culture conditions for 24 h. Formation
of the EGFR-Beclin-1 complex was not induced by hypoxia for 24 h in
X01 cells (data not shown).
Immunofluorescence detects varied
localizations for EGFR and Beclin-1 independent of TMZ
application
Immunofluorescence is a method enabling the
visualization of co-localized proteins, suggesting their
interaction. As presented in Fig. 4,
the majority of EGFR spots could be identified in
LN18vIII cells due to the antibody detection of
wild-type EGFR, as well as the aberrant form vIII. EGFR and
Beclin-1 were not adjacent in LN18 and LN18vIII. This
result was not altered by treatment with high-dose TMZ. Compared
with LN18vIII expressing the truncated and
constitutively active EGFR, TMZ treatment appeared to restrict
growth more in wild-type LN18 cells.
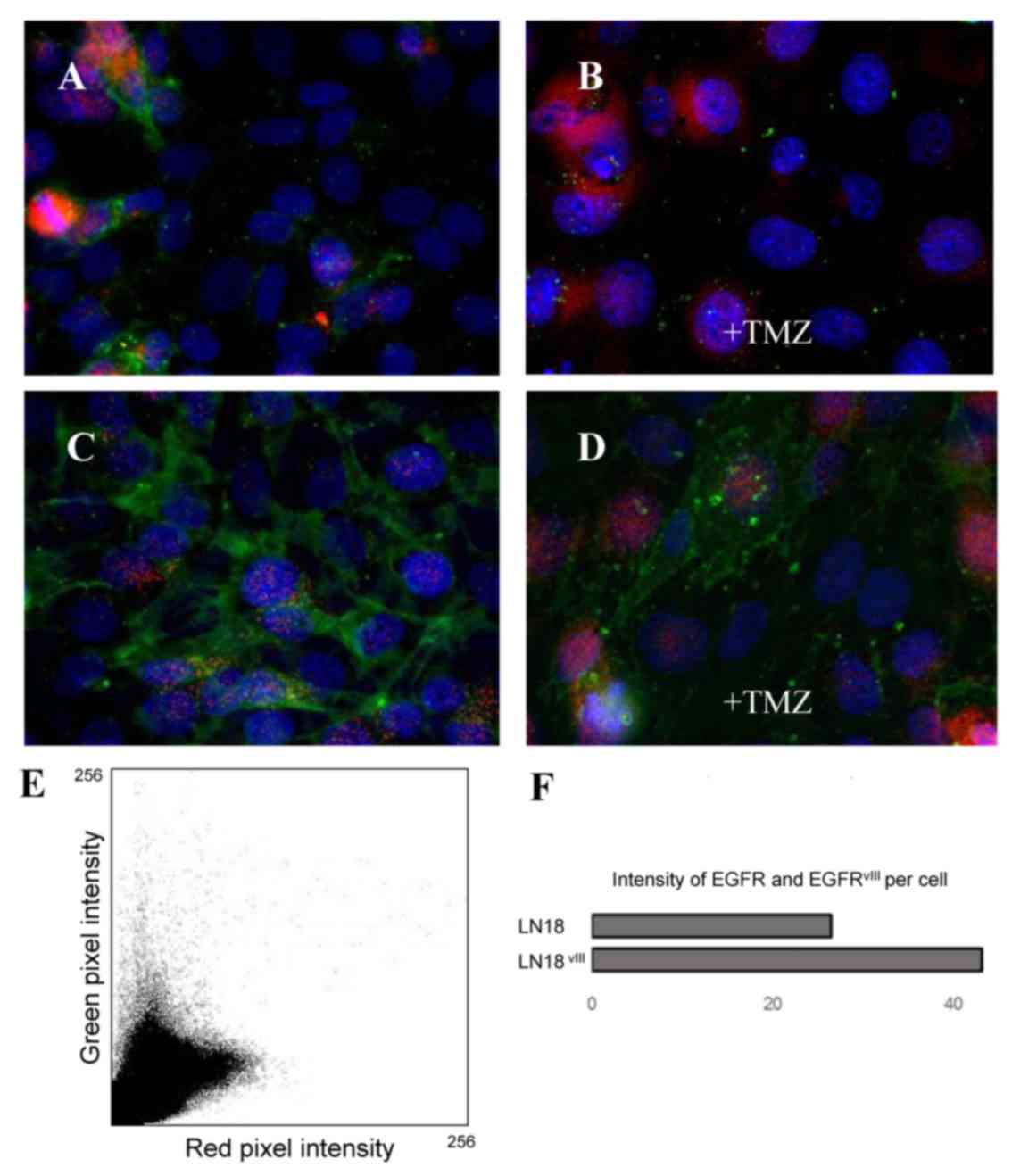 | Figure 4.Immunofluorescence double-labeling of
Beclin-1 (red) and EGFR (green). (A) GBM cell line LN18, (B) GBM
cell line LN18+TMZ, (C) GBM cell line LN18vIII and (D)
GBM cell line LN18vIII+TMZ. Magnification, ×63.
Microphotographs reveal Beclin-1 protein expression at similar
levels in the two cell lines, whereas LN18vIII expresses
higher levels of EGFR. EGFR and Beclin-1 are not directly adjacent
to one another. (E) Intensity of green and red pixels were
identified per cell and analyzed for colocalization with the plugin
‘colocalization colormap’ in ImageJ. The scatterplot reflects the
lack of correlation by the two separate groups of points. This was
verified for each cell line and each condition. Pearson's
correlation coefficient (pixel-by-pixel covariance): 0.39 (LN18),
0.00 (LN18+TMZ), 0.01 (LN18vIII) and −0.06
(LN18vIII+TMZ). (F) Results of an analysis of 6
different immunofluorescence images of each cell line of one
experiment. EGFR, epidermal growth factor receptor;
LN18vIII, constitutively active EGFRvIII
variant, TMZ, temozolomide. |
Discussion
Previous studies on autophagy have provided insight
into its underlying mechanisms at a molecular level (22). However, there remain challenges to our
understanding, including the investigation of interacting signaling
pathways and their impact on the deleterious or protective
character of autophagy. With an interdisciplinary effort, the
elucidation of the interaction between autophagy proteins and other
significant cellular pathways may aid the treatment of malignant
cancer, including GBM.
GBM is the most malignant type of brain tumor in
adulthood and poses a major challenge in terms of therapy options
(23). At present, the alkylating
drug TMZ is used as the first-line treatment for primary GBM
(1). The heterogeneous character of
GBM cells complicates the evaluation of interference of TMZ with
various cellular signaling pathways (24). However, the primary influences of TMZ
on cellular signaling pathways require elucidation to analyze
adverse side effects as well as potential accompanying therapeutic
approaches. One affected pathway appears to be the catabolic
process of autophagy (5). The
conversion of LC3 has now become the most widely used method to
monitor autophagy (8). An increase of
LC3B-II, as presented in Fig. 1A, may
be induced in primary and established GBM cell lines by autophagy
regulation using chloroquine. This drug inhibits the fusion of
lysosomes with autophagosomes, resulting in an accumulation of
LC3B-II (10). Chloroquine has been
used in several studies to suppress tumor growth, including in lung
cancer cells (25). In GBM, the
effects of chloroquine were promising and led to a significant
extension of overall patient survival time (26).
Primary cell culture, which is an improved mimicry
of relevant in vivo conditions, was compared to established
GBM cell lines, as they are hypothesized to vary in
dedifferentiation (27). The
investigated primary cell lines pGBM T1 and pGBM T12 did not
express MGMT, in contrast to LN18. The findings of the current
study indicate that autophagy is increased in primary cell lines,
predominantly in pGBM T1, by a lower concentration of TMZ and
reduced treatment time compared with the established cell line
LN18. The positive regulation of TMZ on autophagy may be
interpreted as a response to adverse cellular conditions (5). The LN18 cells were not affected by low
dose and short-term treatment with TMZ in the present study, which
indicates TMZ-resistant characteristics. However due to adverse
side effects, the high dose of TMZ required for autophagy induction
in LN18 is not feasible for use in patients with resistant GBM.
Taken together, this process of hindering cells from escaping
adverse cellular conditions using chloroquine may be a promising
addition to TMZ treatment.
Another established underlying mechanism to
circumvent TMZ-induced damages is MGMT, which removes methylated
DNA adducts (4). The expression of
MGMT is a negative predictive factor for survival time in patients
with GBM (28). In the current study,
LN18 cells expressed MGMT, but the protein levels were markedly
decreased by TMZ (Fig. 2). This
indicates that the cellular supply of MGMT is exhausted when
repairing methylated TMZ lesions, concordant with the findings of
Wick et al (29). The primary
cell lines pGBM T1 and pGBM T12 did not express MGMT, which may be
interpreted as favorable regarding TMZ treatment.
The transmembrane signaling module EGFR serves a
significant role in numerous cellular signaling pathways and its
amplification or mutations are encountered in numerous cases of
GBM, the most common of which is EGFRvIII (15). Lacking its outer regulative portion,
EGFRvIII provides constant signaling for the tumor cell
(14). This signaling not only
enhances phosphoinositide 3-kinase/protein kinase B activation, but
also appears to possess divergent characteristics compared with
wild-type EGFR signaling (30,31). In
the present study, the expression of the truncated vIII form
appeared to be linked to higher resistance towards high-dose
application of TMZ in LN18, as detected by immunofluorescence
(Fig. 4). Notably, the co-expression
of wild-type and truncated EGFR, as with LN18vIII, is
hypothesized to result in an antagonistic connection between the
two receptors (32). This may induce
malignancy in GBM (32). However, the
prognostic role of the truncated version of EGFR remains
controversial. Shinojima et al (33) identified a poor prognostic outcome in
patients with GBM due to EGFRvIII in combination with
EGFR amplification. By contrast, Montano et al (34) revealed a prolonged survival for
patients with GBM expressing EGFRvIII (34). The altered signaling function of
EGFRvIII and its implication in GBM growth and therapy
resistance must be investigated in detail in the future.
Wei et al (13)
suggested an important role for EGFR in the inhibition of autophagy
initiation (13). This previous study
described the phosphorylation of Beclin-1 by active EGFR in NSCLC
cells (13). If EGFR in GBM cells
also regulates Beclin-1, this is of note as EGFR is frequently
amplified or mutated in GBM (35). In
the current study, Beclin-1 was not phosphorylated by inactive or
active EGFR independently of TMZ treatment in LN18 and
LN18vIII. GBM cells did not exhibit modification of
autophagy monitored by wild-type EGFR, application of EGF or
EGFRvIII. Beclin-1 did not bind to EGFR or
EGFRvIII in control cells or TMZ-treated cells in
established and primary GBM. The interaction between Beclin-1 and
EGFR remained unaffected by hypoxic conditions for 24 h.
Immunofluorescence revealed that Beclin-1 locations are not
directly adjacent to EGFR locations in LN18 or LN18vIII,
which did not vary upon TMZ application. These data demonstrate
that Beclin-1 does not interact directly with EGFR in the GBM cell
lines used in the present study and suggests varied regulation,
particularly for primary GBM culture. This is concordant with Zhu
and Shah (36), suggesting other
regulative pathways for autophagy than EGFR in GBM.
Further studies may focus on the interaction between
inactive EGFR and autophagy. Tan et al (37) demonstrated that a knockdown of EGFR
inhibited autophagy in different cell lines, but this study did not
include brain tumor cells (37).
However, these findings are in favor of an important stimulus of
autophagy by inactive EGFR in certain tumor entities, and may be
evaluated in GBM cells.
Acknowledgements
The authors would like to thank Dr Andreas
Androutsellis-Theotokis (Carl Gustav Carus University, Dresden,
Germany) for the cell line X01, Dr Daniela Schilling (Department of
Radiooncology Technische Universität, Munich, Germany) for
providing the hypoxia-incubating chamber and Mrs. Sandra Baur
(Department of Neuropathology, Technische Universität, Munich,
Germany) for technical support in the laboratory. This work was
supported by the Deutsche Forschungsgemeinschaft (SFB 824: ‘Imaging
for the Selection, Monitoring, and Individualization of Cancer
Therapies,’ Project B6).
Glossary
Abbreviations
Abbreviations:
|
Atg
|
autophagy-related gene
|
|
GBM
|
glioblastoma multiforme
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
pGBM
|
primary glioblastoma multiforme cell
line
|
|
TMZ
|
temozolomide
|
References
|
1
|
Koshy M, Villano JL, Dolecek TA, Howard A,
Mahmood U, Chmura SJ, Weichselbaum RR and McCarthy BJ: Improved
survival time trends for glioblastoma using the SEER 17
population-based registries. J Neurooncol. 107:207–212. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stevens MF, Hickman JA, Stone R, Gibson
NW, Baig GU, Lunt E and Newton CG: Antitumor imidazotetrazines. 1.
Synthesis and chemistry of
8-carbamoyl-3-(2-chloroethyl)imidazo[5,1-d]-1,2,3, 5-tetrazin-4(3
H)-one, a novel broad-spectrum antitumor agent. J Med Chem.
27:196–201. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Agarwala SS and Kirkwood JM: Temozolomide
a novel alkylating agent with activity in the central nervous
system, may improve the treatment of advanced metastatic melanoma.
Oncologist. 5:144–151. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hegi ME, Diseren AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 325:997–1003. 2005. View Article : Google Scholar
|
|
5
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Natsumeda M, Aoki H, Miyahara H, Yajima N,
Uzuka T, Toyoshima Y, Kakita A, Takahashi H and Fujii Y: Induction
of autophagy in temozolomide treated malignant gliomas.
Neuropathology. 31:486–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N, Yoshimorim T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fujita N, Hayashi-Nishino M, Fukumoto H,
Omori H, Yamamoto A, Noda T and Yoshimori T: An Atg4B mutant
hampers the lipidation of LC3 paralogues and causes defects in
autophagosome closure. Mol Biol Cell. 19:4651–4659. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA
and Koh JY: Induction of lysosomal dilatation, arrested autophagy
and cell death by chloroquine in cultured ARPE-19 cells. Invest
Ophthalmol Vis Sci. 51:6030–6037. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng Y, Ren X, Hait WN and Yang JM:
Therapeutic targeting of autophagy in disease: Biology and
pharmacology. Pharmacol Rev. 65:1162–1197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sinha S and Levine B: The autophagy
effector Beclin 1: A novel BH3-only protein. Oncogene. 27 Suppl
1:S137–S148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei Y, Zou Z, Becker N, Anderson M,
Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et
al: EGFR-mediated Beclin 1 phosphorylation in autophagy
suppression, tumor progression, and tumor chemoresistance. Cell.
154:1269–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okamoto I, Kenyon LC, Emlet DR, Mori T,
Sasaki J, Hirosako S, Ichikawa Y, Kishi H, Godwin AK, Yoshioka M,
et al: Expression of constitutively activated EGFRvIII in non-small
cell lung cancer. Cancer Sci. 94:50–56. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gan HK, Kaye AH and Luwor RB: The EGFRvIII
variant in glioblastoma multiforme. J Clin Neurosci. 16:748–754.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ishii N, Maier D, Merlo A, Tada M,
Sawamura Y, Diserens AC and Van Meir EG: Frequent co-alterations of
TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human
glioma cell lines. Brain Pathol. 9:469–479. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schäfer A, Teufel J, Ringel F, Bettstetter
M, Hoepner I, Rasper M, Gempt J, Koeritzer J, Schmidt-Graf F, Meyer
B, et al: Aldehyde dehydrogenase 1A1-a new mediator of resistance
to temozolomide in glioblastoma. Neuro Oncol. 14:1452–1464. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berezowska S, Diermeier-Daucher S,
Brockhoff G, Busch R, Duyster J, Grosu AL and Schlegel J: Effect of
additional inhibition of human epidermal growth factor receptor 2
with the bispecific tyrosine kinase inhibitor AEE788 on the
resistance to specific EGFR inhibition in glioma cells. Int J Mol
Med. 26:713–721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ciechomska IA, Przanowski P, Jackl J,
Wojtas B and Kaminska B: BIX01294, an inhibitor of histone
methyltransferase, induces autophagy-dependent differentiation of
glioma stem-like cells. Sci Rep. 6:387232016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rappl A, Piontek G and Schlegel J:
EGFR-dependent migration of glial cells is mediated by
reorganisation of N-cadherin. J Cell Sci. 121:4089–4097. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bobola MS, Tseng SH, Blank A, Berger MS
and Silber JR: Role of O6-methylguanine-DNA methyltransferase in
resistance of human brain tumor cell lines to the clinically
relevant methylating agents temozolomide and streptozotocin. Clin
Cancer Res. 2:735–741. 1996.PubMed/NCBI
|
|
22
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Arozena A Acevedo, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (III edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weller M, Cloughesy T, Perry J and Wick W:
Standards of care for treatment of recurrent glioblastoma-are we
there yet? Neuro Oncol. 15:4–27. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soeda1 A, Hara A, Kunisada T, Yoshimura S,
Iwama1 T and Park D: The evidence of glioblastoma heterogeneity.
Sci Rep. 5:79792015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Briceño E, Calderon A and Sotelo J:
Institutional experience with chloroquine as an adjuvant to the
therapy for glioblastoma multiforme. Surg Neurol. 67:388–391. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Seidel S, Garvalov BK and Acker T:
Isolation and culture of primary glioblastoma cells from human
tumor specimens. Methods Mol Biol. 1235:263–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thon N, Kreth S and Kreth FW: Personalized
treatment strategies in glioblastoma: MGMT promoter methylation
status. Onco Targets Ther. 6:1363–1372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wick W, Platten M and Weller M: New
(alternative) temozolomide regimens for the treatment of glioma.
Neuro Oncol. 11:69–79. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bleeker FE, Molenaar RJ and Leenstra S:
Recent advances in the molecular understanding of glioblastomas. J
Neurooncol. 108:11–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eskilsson E, Røsland G, Talasila K, Jahedi
R, Leiss L, Saed H, Keunen O, Foerster S, Euskirchen P, Hossain J,
et al: Distinct EGFR signaling in Glioblastoma: Wild-type EGFR
promotes invasion while EGFRvIII drives prototypical SFK c-SRC
activation to foster angiogenesis. Neuro Oncol. 16 Suppl 5:v32014.
View Article : Google Scholar
|
|
32
|
Li L, Puliyappadamba VT, Chakraborty S,
Rehman A, Vemireddy V, Saha D, Souza RF, Hatanpaa KJ, Koduru P,
Burma S, et al: EGFR wild type antagonizes EGFRvIII-mediated
activation of Met in glioblastomas. Oncogene. 34:129–134. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shinojima N, Tada K, Shiraishi S, Kamiryo
T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, et
al: Prognostic value of epidermal growth factor receptor in
patients with glioblastoma multiforme. Cancer Res. 63:6962–6970.
2003.PubMed/NCBI
|
|
34
|
Montano N, Cenci T, Martini M,
D'Alessandris QG, Pelacchi F, Ricci-Vitiani L, Maira G, de Maria R,
Larocca LM and Pallini R: Expression of EGFRvIII in glioblastoma:
Prognostic significance revisited. Neoplasia. 12:1113–1121. 2011.
View Article : Google Scholar
|
|
35
|
Padfield E, Ellis HP and Kurian KM:
Current therapeutic advances targeting EGFR and EGFRvIII in
glioblastoma. Front Oncol. 5:52015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu Y and Shah K: Multiple lesions in
receptor tyrosine kinase pathway determine glioblastoma response to
pan-ERBB inhibitor PF-00299804 and PI3K/mTOR dual inhibitor
PF-05212384. Cancer Biol Ther. 15:815–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|