Introduction
Pancreatic ductal adenocarcinoma (PDA) is one of the
most aggressive types of malignant cancer, with a 5 year survival
rate of 5% (1). The majority of PDA
cases are identified at a progressed stage that is too late for
surgical intervention, due to a lack of symptoms and diagnostic
markers at earlier stages (1). Even
following surgical intervention, the 5-year survival rate is
<15% without adjuvant therapy or <25% with adjuvant
chemotherapy (1). Gemcitabine (GEM)
-based regimens or a combined regimen of 5-fluorouracil,
leucovorin, irinotecan and oxaliplatin (FOLFIRINOX) are standard,
widely used first-line treatments for patients with advanced PDA
(2). Second-line chemotherapy may be
beneficial for patients with good performance status and
tolerability to additional chemotherapy. Although a number of phase
II and III second-line chemotherapy trials have been completed,
sufficient evidence for efficacy has not been obtained (3). Paclitaxel is effective for advanced
pancreatic cancer refractory to GEM, GEM-based regimens or
FOLFIRINOX as second-line chemotherapy (4,5). In
addition, nanoparticle albumin-bound paclitaxel (nab-paclitaxel;
Abraxane) was previously used for first-line chemotherapy in the
combination with GEM in phase I and II chemotherapy trials
(6,7).
Paclitaxel binds to β-tubulin and stabilizes
microtubules that induce metaphase arrest, inducing apoptotic cell
death via the spindle assembly checkpoint pathway (8,9). Genetic
mutations in the paclitaxel-binding region of β-tubulin may modify
the sensitivity of cells to paclitaxel (10,11).
Overexpression of a neuron-specific subtype βIII-tubulin (TUBB3) is
involved in taxane resistance in malignant tumors (11–13),
including PDA (14).
Overexpression of anti-apoptotic proteins is also
associated with taxane-based therapeutics. B-cell lymphoma-2
(Bcl-2) family proteins that consist of both pro- and
anti-apoptotic members regulate mitochondrial apoptotic signals and
thus cell fate. Apoptotic signals are mediated by Bcl-2
homology-3-only proteins that induce oligomerization of
Bcl-2-associated X protein and Bcl-2 antagonist/killer, cytochrome
c release and caspase activation. The anti-apoptotic members,
including Bcl-2, B-cell lymphoma extra-large (Bcl-xL),
Bcl-2-like protein 2 (Bcl-w) and myeloid cell leukemia 1 (Mcl-1)
antagonize this activation (15).
Mcl-1 suppresses apoptosis triggered by paclitaxel or vincristine
in ovarian cancer and non-small-cell lung cancer (NSCLC) cells
(16). Loss-of-function mutations in
the F-box and WD-40 domain protein 7 gene, which encodes an E3
ubiquitin ligase for Mcl-1 degradation, stabilizes Mcl-1 protein
and contributes to taxane resistance in these tumors (17). The association between Bcl-2 family
protein expression and chemosensitivity for taxane derivatives has
not been fully elucidated in PDA. Furthermore, ABT-737 is a
small-molecule inhibitor of Bcl-2, Bcl-xL and Bcl-w
(18). Navitoclax (ABT-263) is an
orally bioavailable inhibitor with a similar binding profile that
is under evaluation in clinical trials (19–21). In
combination with taxane-based therapy, these small molecules
improved responses in a number of human malignancies (22–24).
The present study investigated the expression of
Bcl-2-associated anti-apoptotic proteins, and evaluated the
efficacy of combining ABT-737 with paclitaxel in PDA cell
lines.
Materials and methods
Cell culture
A total of 6 PDA cell lines (PK-9, PK-8, KLM-1,
PK-59, PK-45-P and PK-45-H) were obtained from the RIKEN
BioResource Centre (Tsukuba, Japan). A total of two PDA cell lines
(MIA-PaCa-2 and Panc-1) were purchased from the American Type
Culture Collection (Manassas, VA, USA). MIA-PaCa-2 cells were
maintained at 37°C under 5% CO2 in Dulbecco's modified
Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc.); other cell lines were
maintained in RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS.
Reagents and antibodies
Paclitaxel (Calbiochem; Merck KGaA, Darmstadt,
Germany) and ABT-737 (Selleck Chemicals, Houston, TX, USA) were
prepared at a stock concentration of 10 mM in dimethyl sulfoxide.
The following antibodies were used for western blotting: Monoclonal
anti-Bcl-2 (dilution, 1:250; cat. no., 05-729; EMD Millipore,
Billerica, MA, USA), monoclonal anti-Mcl-1 (dilution, 1:1,000; cat.
no., #5453), monoclonal anti-Bcl-xL (dilution, 1:1,000;
cat. no., #2764) monoclonal anti-Bcl-w (dilution, 1:1,000; cat.
no., #2764), anti-poly-(ADP-ribose) polymerase (PARP; dilution,
1:1,000; cat. no., #9542; all from Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-TUBB3 (dilution, 1:1,000; cat. no.,
MMS-435P; Covance, Inc., Princeton, NJ, USA) and mouse monoclonal
anti-α-tubulin (dilution, 1:1,000; cat. no., T5168; Sigma-Aldrich;
Merck KGaA). Horseradish peroxidase (HRP)-linked anti-mouse
immunoglobulin (Ig) G or anti-rabbit IgG (1:3,000; NA931 or NA934,
respectively; GE Healthcare Life Sciences, Chalfont, UK) were used
as a secondary antibodies for western blotting.
Cell viability assay
Cells were seeded into 96-well cell culture plates
at a density of 104 cells/well for 24 h. To examine the
dose-response association, cells were incubated with paclitaxel
(2-fold serial dilution from 1 pM to 2.5 µM) and/or ABT-737 (500
nM), or 0.1% dimethyl sulfoxide (DMSO) as a control, for 72 h at
37°C. Cell viability assays were performed using Cell Counting
Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan),
according to the manufacturer's protocol. Absorbance at 450 nm was
evaluated using a Multiskan spectrum (Thermo Fisher Scientific,
Inc.). IC50 was determined by fitting to a normal
distribution.
Western blotting
Cells at 80–90% confluence were washed twice with
cold PBS and harvested by scraping. To analyze apoptotic marker
proteins, cells were treated with 100 nM paclitaxel and/or 500 nM
ABT-737, or 0.1% DMSO as a control, for 24–72 h at 37°C. Adherent
and floating cells were harvested followed by washing with cold
PBS. Cells were collected by centrifugation (200 × g for 3 min at
4°C) and lysed with sonication (5 sec, output 4; Branson Sonifier
S-150D; Branson Ultrasonics, Danbury, CT, USA) in ice-cold RIPA
buffer (50 mM Tris pH 8.0, 150 mM NaCl, 10 mM NaF, 2 mM
Na3VO4, 1% NP-40, 0.5% sodium deoxycholate,
0.1% SDS) supplemented with Complete Protease Inhibitor cocktail
(EDTA-free; Roche Diagnostics GmbH, Mannheim, Germany) and 0.5 mM
PMSF. The insoluble fraction was removed by centrifugation (20,000
× g for 10 min at 4°C). Protein concentration was evaluated using a
BCA Protein Assay kit (Novagen, Inc., Madison, WI, USA). Protein
samples (20 µg/lane) were separated on 10% SDS-PAGE gel and then
transferred onto polyvinylidene difluoride transfer membranes (Pall
Corporation, Port Washington, NY, USA). The membranes were blocked
with 5% non-fat dried milk (cat. no. 9999; Cell Signaling
Technology, Inc.) in 0.1% Tween-20/PBS for 1 h at room temperature,
and then incubated with primary antibodies overnight at 4°C and
with HRP-conjugated secondary antibodies (GE Healthcare Life
Sciences) for 1 h at room temperature. Signals were detected with
enhanced chemiluminescence prime detection reagents (GE Healthcare
Life Sciences) and processed with ChemiDoc XRS with Quantity One
software (version 4.5.2; Bio-Rad Laboratories, Inc., Hercules, CA,
USA), with α-tubulin as the loading control.
Knockdown of Bcl-2, Bcl-xL and
TUBB3
Silencer Select Validated short interfering (si)RNA
against Bcl-2 (cat. no. s224526), Bcl-xL (cat. no.
s1920), TUBB3 (cat. no. s20297) and Negative Control siRNA No. 1
(cat. no., 4390843) were purchased from Ambion (Thermo Fisher
Scientific, Inc.). Cells were washed once with Opti-Minimum
Essential medium (Gibco; Thermo Fisher Scientific, Inc.) and then
transfected with 20 nM siRNA mixed with Lipofectamine RNAiMAX
transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Culture media were
refreshed 6 h after transfection and cells were incubated for a
further 48 h at 37°C. Cells were then replated for the cell
viability assay and western blotting as previously described.
Results
Bcl-2 family and TUBB3 expression and
sensitivity to ABT-737
The expression level of the Bcl-2 family and TUBB3
was analyzed in 8 PDA cell lines (Fig.
1A). There was strong expression of Bcl-2 in PK-59 and moderate
expression in other cell lines, but no expression in Panc-1.
Bcl-xL was detectable in all cell lines, with relatively
low expression in Panc-1 and MIA-PaCa-2. Bcl-w and Mcl-1 expression
was detectable in all cell lines with certain variations in
expression. Strong expression of TUBB3 was detected in Panc-1 and
MIA-PaCa-2 but not in PK-8, PK-9, PK-59 and PK-45-P (Fig. 1A).
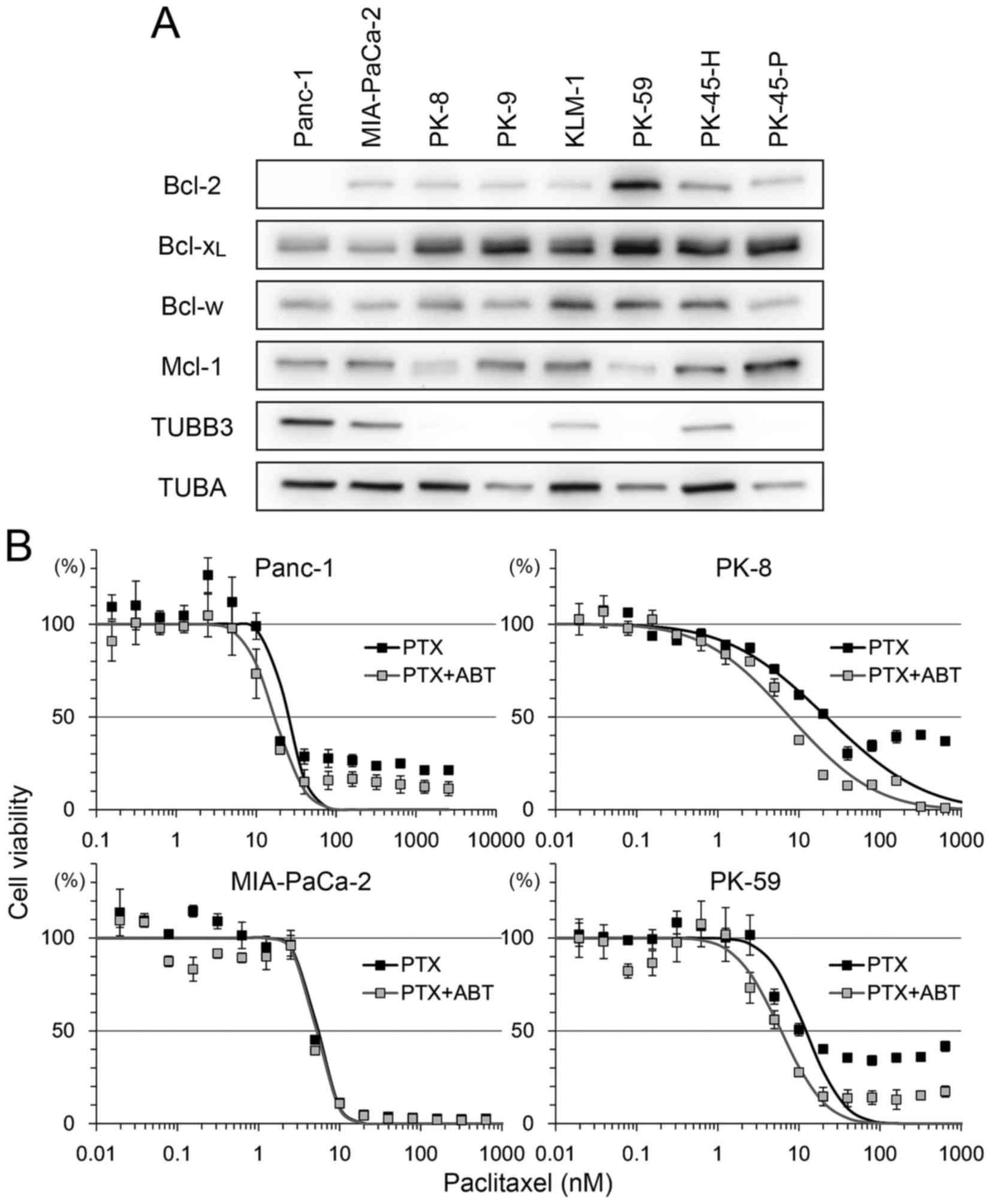 | Figure 1.Bcl-2 family protein expression
levels in PDA and sensitivity to PTX/ABT. (A) A total of eight PDA
cells lines were subjected to western blotting to detect Bcl-2,
Bcl-xL, Bcl-w, Mcl-1, TUBB3 and TUBA. (B) A total of
four PDA cell lines were seeded into 96-well plates and treated
with 2-fold serial dilution of PTX with or without 500 nM ABT for
72 h. Cell viability was determined using Cell Counting Kit-8.
Viability was normalized to control samples (treated with vehicle
or ABT alone) and expressed as the mean ± standard deviation (n=5).
Mortality of shifting points were fitted to a normal distribution
and used to calculate the half maximal inhibitory concentration.
PDA, pancreatic ductal adenocarcinoma; PTX, paclitaxel; ABT,
ABT-737; Bcl-2, B-cell lymphoma-2; Bcl-xL, B-cell
lymphoma extra-large; Bcl-w, Bcl-2-like protein 2; Mcl-1, myeloid
cell leukemia 1; TUBB3, βIII-tubulin; TUBA, α-tubulin. |
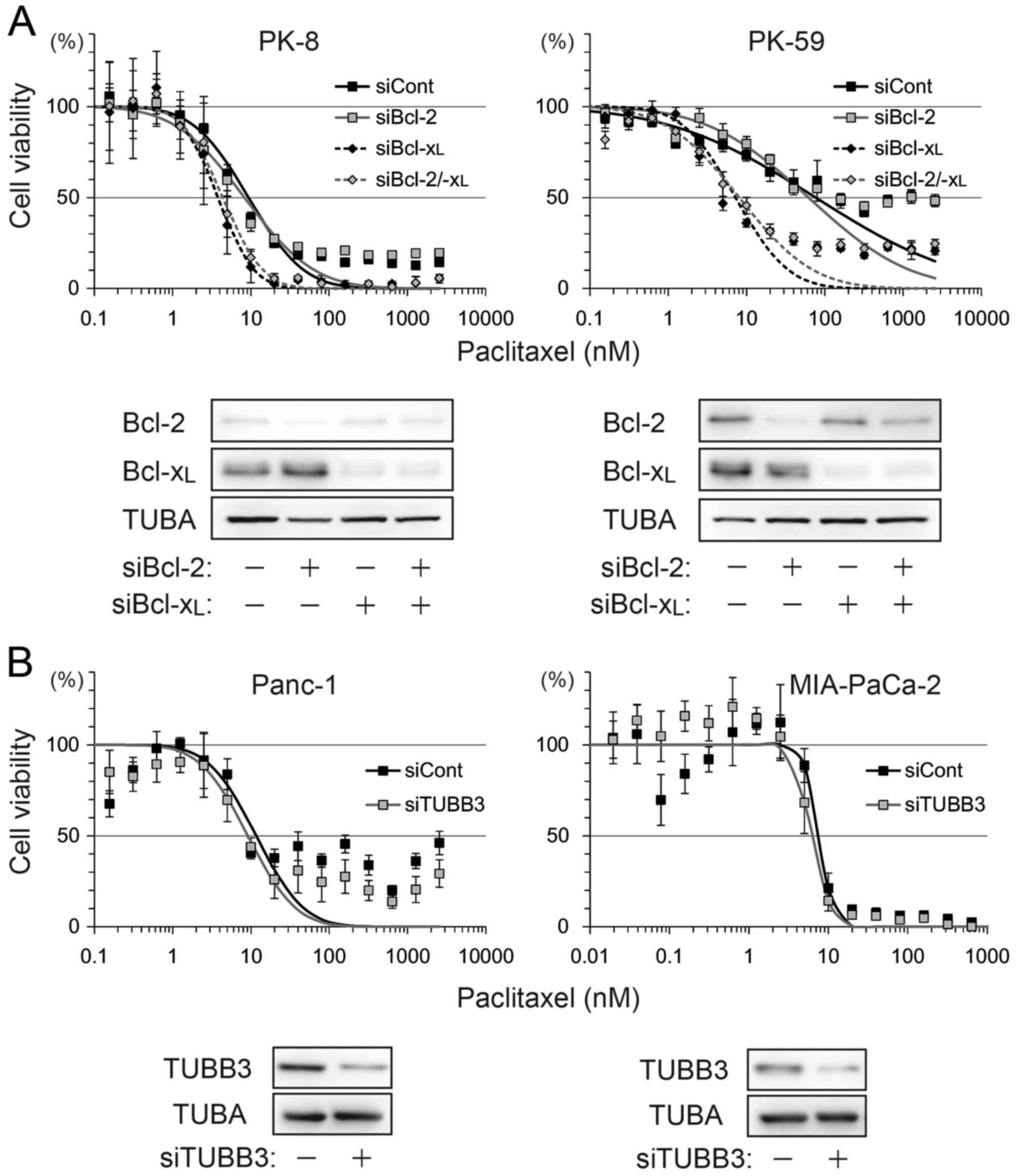
To determine the association between these factors
and sensitivity to paclitaxel and ABT-737, four cell lines, Panc-1
(Bcl-2-negative, Bcl-xL-low, TUBB3-high), MIA-PaCa-2
(Bcl-2/Bcl-xL-low, TUBB3-high), PK-8 (Bcl-2-low,
Bcl-xL-high, TUBB3-negative) and PK-59
(Bcl-2/Bcl-xL -high, TUBB3-negative), were selected for
use in viability assays following treatment with paclitaxel and/or
ABT-737. Treatment with paclitaxel decreased the viability of all
four cell lines in a dose-dependent manner (Fig. 1B). Compared with paclitaxel alone,
combination treatment with ABT-737 shifted the survival curve to a
lower paclitaxel concentration in Panc-1, PK-8 and PK-59 cells.
ABT-737 alone did not decrease the viability of PDA cell lines
(data not shown), a result similar to that obtained by a previous
study in melanoma cell lines (25).
The IC50 of paclitaxel vs. paclitaxel/ABT-737
combination in each cell line was as follows: Panc-1 (25.05 vs.
17.14 nM), MIA-PaCa-2 (5.36 vs. 5.17 nM), PK-8 (21.87 vs. 7.75 nM)
and PK-59 (12.16 vs. 6.02 nM). In addition, small populations of
Panc-1, PK-8 and PK-59 cells survived following the treatment with
paclitaxel at saturating concentrations (>100 nM). However, the
survival population was decreased by combined treatment with
paclitaxel and ABT-737 (Fig. 1B).
ABT-737 abrogates the Bcl-xL dependent
anti-apoptotic effect
ABT-737 lowered paclitaxel-induced IC50
by >2-fold in PK-8 and PK-59 cells, but <2-fold in Panc-1 and
MIA-PaCa-2 cells. To determine which factor contributed to ABT-737
sensitivity, Bcl-2, Bcl-xL and TUBB3 were depleted by
siRNA transfection and subjected to a viability assay following
treatment with paclitaxel alone. Compared with control siRNA, the
IC50 was shifted by Bcl-xL knockdown or
Bcl-2/Bcl-xL double knockdown, but not by Bcl-2
knockdown in PK-8 and PK-59 cell lines (Fig. 2A). TUBB3 knockdown in Panc-1 and
MIA-PaCa-2 slightly shifted IC50 by <2-fold (Fig. 2B).
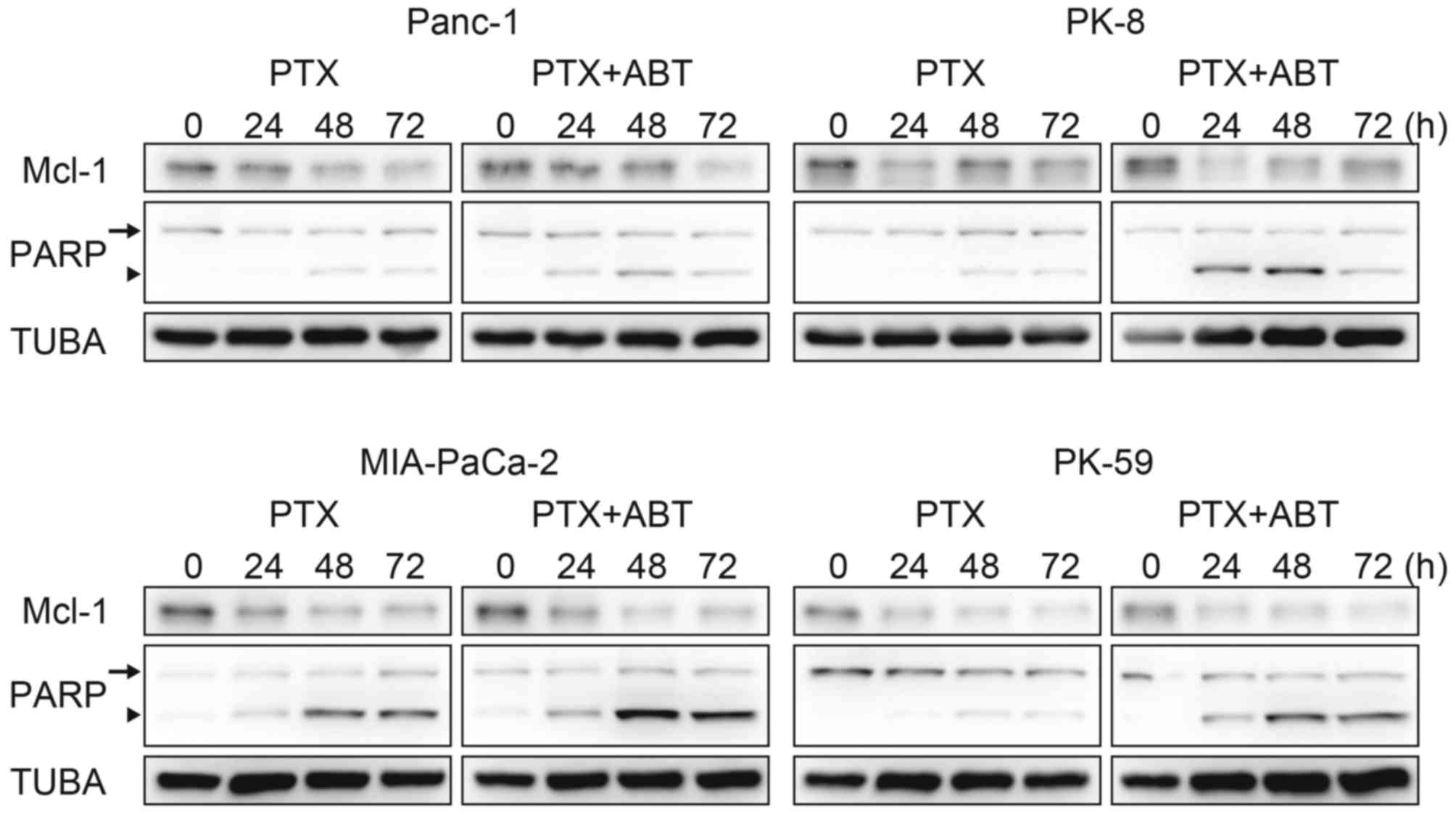
ABT-737 accelerates paclitaxel-induced
cell death
The survival population at the saturating
concentration of paclitaxel was decreased by combination treatment
with ABT-737 in Panc-1, PK-8 and PK-59 cells (Fig. 1A), and by Bcl-xL knockdown
in PK-8 and PK-59 cell lines (Fig.
2A). Although Mcl-1 degradation during mitotic arrest was
observed in all four cell lines, ABT-737 did not affect the Mcl-1
degradation kinetics (Fig. 3).
Subsequently, cleavage of PARP by caspase-3 was detected as an
apoptotic marker. In Panc-1, PK-8 and PK-59, cleaved PARP was
detected from 48 to 72 h following treatment with paclitaxel alone.
In contrast, cleaved PARP was detected from 24 h following combined
treatment with paclitaxel and ABT-737. In addition, ABT-737
combination treatment increased the amount of cleaved PARP compared
with treatment with paclitaxel alone (Fig. 3). Acceleration of apoptosis onset by
paclitaxel/ABT-737 combination treatment was confirmed by knockdown
of Bcl-2 and/or Bcl-xL. In the PK-8 and PK-59 cells,
knockdown of Bcl-xL was sufficient to increase the
amount of cleaved PARP 24 h following treatment with paclitaxel
alone, whereas knockdown of Bcl-2 did not affect PARP cleavage
compared with the control siRNA (Fig.
4).
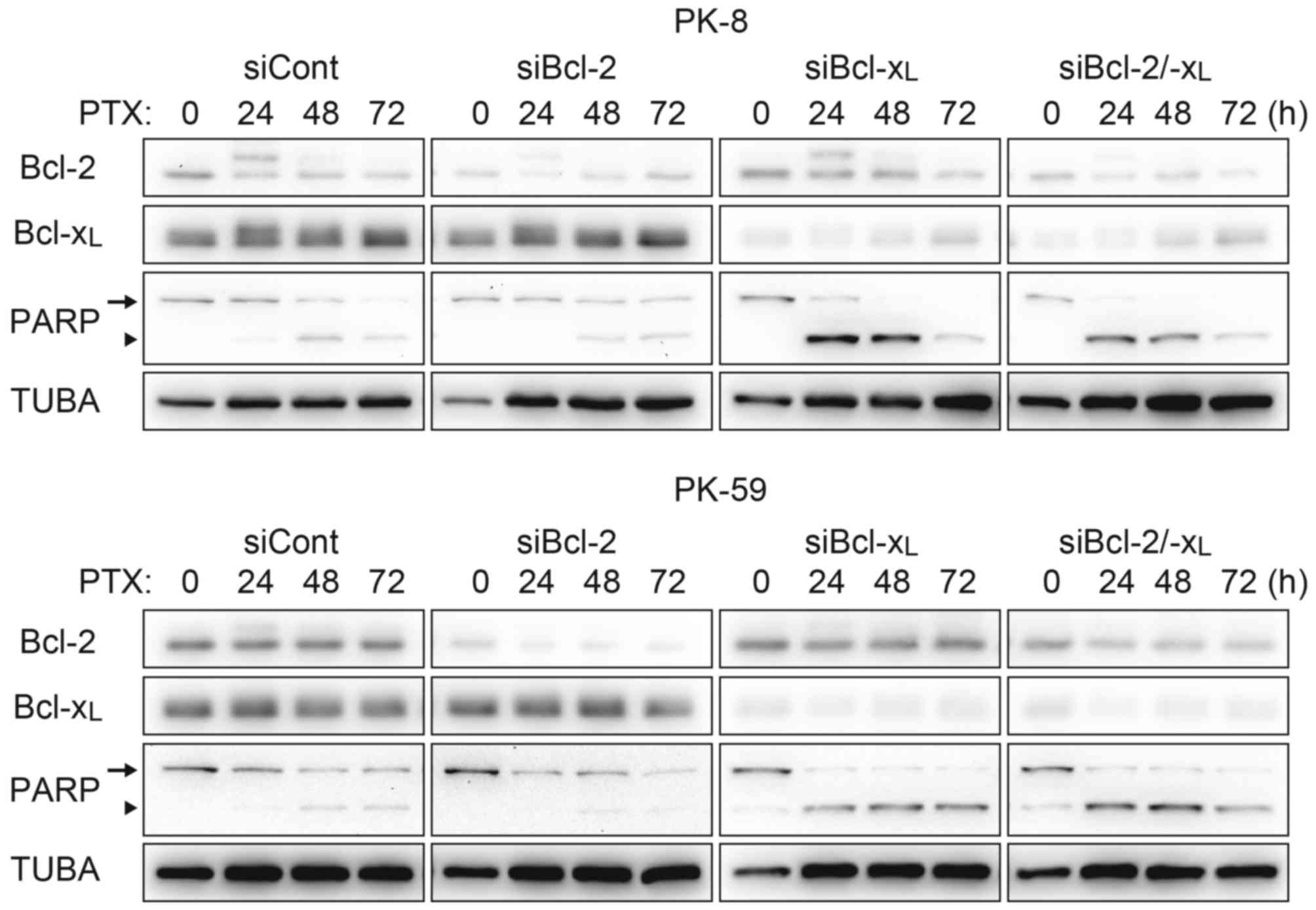 | Figure 4.Acceleration of the apoptotic pathway
by Bcl-xL knockdown. PK-8 and PK-59 cells were
transfected with siRNA against Bcl-2 and/or Bcl-xL and
incubated for 48 h. Cells were re-plated in a culture dish and
incubated for 24 h, and then treated with 100 nM PTX for 24, 48 or
72 h. Adherent and floating cells were subjected to western
blotting to detect Bcl-2, Bcl-xL, full length PARP
(arrow) and its cleaved form (arrowhead). Bcl-xL, B-cell
lymphoma extra-large; siRNA/si, short interfering RNA; Bcl-2,
B-cell lymphoma-2; PTX, paclitaxel; PARP, Poly-(ADP-ribose)
polymerase; TUBA, α-tubulin; cont, control. |
Discussion
To the best of our knowledge, the present study is
the first to evaluate the efficacy of combining ABT-737 with
paclitaxel in PDA. Although ABT-737 targets Bcl-2,
Bcl-xL and Bcl-w, the sensitization by ABT-737 was
dependent on Bcl-xL alone in the PDA cell lines
investigated in the present study. In spite of other factors
associated with taxane resistance, including Bcl-2 (15) and TUBB3 (11–13)
expression levels, the abundance of Bcl-xL protein was
the key determinant of paclitaxel sensitivity in PDA based on the
results of the present study. In addition, paclitaxel-induced Mcl-1
degradation was comparable in PDA cell lines evaluated, and was
independent of ABT-737 sensitivity. Abundant Bcl-xL
expression contributed to an anti-apoptotic effect that raised the
IC50 of paclitaxel and delayed the onset of apoptotic
events. In the case of chemotherapy treatment for human
malignancies, physiological concentrations of paclitaxel (26) and ABT-737 (27) were decreased by metabolic kinetics.
Since ABT-737 accelerates the apoptotic signaling pathway and
induces a decrease in paclitaxel dosage as observed in the present
study, combination treatment with paclitaxel or other
chemotherapeutics may be beneficial for the treatment of PDA. The
abundance of Bcl-xL in biopsies or resected tumors may
be a potential biomarker to predict the effect of administration of
ABT-737.
Previously, Tan et al (28) revealed that combination treatment of
NSCLC and PDA with ABT-263 and the mitogen-activated protein kinase
(MEK) inhibitor G-963. ABT-263/G-963 combination treatment was
revealed to be efficient in NSCLC cell lines with Kirsten rat
sarcoma viral oncogene homolog mutations that constitutively
activated the MEK signaling pathway. Wong et al (22) also investigated navitoclax therapy
combined with paclitaxel or GEM in ovarian cancer cell lines. These
combinations resulted in synergistic growth inhibition in the
majority of the cell lines analyzed (22). A GEM-based regimen is considered to be
the gold standard for first-line treatment of patients with
advanced PDA, thus navitoclax may be a promising agent for the
management of these patients.
A few unique trials targeting secreted protein
acidic and rich in cysteine (SPARC) have been performed in PDA
(29–31). SPARC is an extracellular matrix
protein that functions as a stromal chaperone and is a target of
nab-paclitaxel (29,30). Abundant SPARC expression in pancreatic
cancer is associated with poor prognosis, invasion and metastasis
(29,31). In addition to the cell-directed effect
of nab-paclitaxel on SPARC-rich PDA, nab-paclitaxel facilitates the
delivery of GEM (29). A combination
of nab-paclitaxel and GEM treatment improved the median survival
rate of patients with advanced disease in phase I and II studies
(31,32). The results of the present study
support the use of navitoclax in combination with
nab-paclitaxel/GEM, as the combination may improve disease outcome
in advanced PDA.
In conclusion, the present study analyzed the
abundance of Bcl-2, Bcl-xL, Mcl-1 and TUBB3 in PDA cell
lines. Combination treatment with ABT-737 and paclitaxel induced
apoptotic cell death more efficiently compared with paclitaxel
treatment alone in PDA cells with high Bcl-xL expression
level. Knockdown experiments indicated that Bcl-xL, but
not Bcl-2 or TUBB3, counteracted paclitaxel-induced cell death.
ABT-737 is potential candidate for combination chemotherapy to
treat PDA with high Bcl-xL expression levels.
Glossary
Abbreviations
Abbreviations:
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bcl-xL
|
B-cell lymphoma extra-large
|
|
GEM
|
gemcitabine
|
|
IC50
|
half maximal inhibitory
concentration
|
|
Mcl-1
|
myeloid cell leukemia 1
|
|
nab-paclitaxel
|
nanoparticle albumin-bound
paclitaxel
|
|
NSCLC
|
non-small cell lung carcinoma
|
|
PARP
|
Poly-(ADP-ribose) polymerase
|
|
PDA
|
pancreatic ductal adenocarcinoma
|
|
SPARC
|
secreted protein acidic and rich in
cysteine
|
|
TUBB3
|
βIII-tubulin
|
References
|
1
|
Warshaw AL and Fernández-del Castillo C:
Pancreatic Carcinoma. N Engl J Med. 326:455–465. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gourgou-Bourgade S, Bascoul-Mollevi C,
Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A,
Raoul JL, Boige V, et al: Impact of FOLFIRINOX compared with
gemcitabine on quality of life in patients with metastatic
pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized
trial. J Clin Oncol. 31:23–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shukuya T, Yasui H, Boku N, Onozawa Y,
Fukutomi A, Yamazaki K, Taku K, Kojima T and Machida N: Weekly
Paclitaxel after failure of gemcitabine in pancreatic cancer
patients with malignant ascites: A retrospective study. Jpn J Clin
Oncol. 40:1135–1138. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maeda S, Motoi F, Onogawa T, Morikawa T,
Shigeru O, Sakata N, Takadate T, Naitoh T, Rikiyama T, Katayose Y,
et al: Paclitaxel as second-line chemotherapy in patients with
gemcitabine-refractory pancreatic cancer: A retrospective study.
Int J Clin Oncol. 16:539–545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saif MW: Advancements in the management of
pancreatic cancer: 2013. JOP. 14:112–118. 2013.PubMed/NCBI
|
|
7
|
Zhang DS, Wang DS, Wang ZQ, Wang FH, Luo
HY, Qiu MZ, Wang F, Li YH and Xu RH: Phase I/II study of
albumin-bound nab-paclitaxel plus gemcitabine administered to
Chinese patients with advanced pancreatic cancer. Cancer Chemother
Pharmacol. 71:1065–1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blagosklonny MV: Mitotic arrest and cell
fate: Why and how mitotic inhibition of transcription drives
mutually exclusive events. Cell Cycle. 6:70–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rowinsky EK: The development and clinical
utility of the taxane class of antimicrotubule chemotherapy agents.
Annu Rev Med. 48:353–374. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hari M, Loganzo F, Annable T, Tan X, Musto
S, Morilla DB, Nettles JH, Snyder JP and Greenberger LM:
Paclitaxel-resistant cells have a mutation in the
paclitaxel-binding region of beta-tubulin (Asp26Glu) and less
stable microtubules. Mol Cancer Ther. 5:270–278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mozzetti S, Ferlini C, Concolino P,
Filippetti F, Raspaglio G, Prislei S, Gallo D, Martinelli E,
Ranelletti FO, Ferrandina G and Scambia G: Class III beta-tubulin
overexpression is a prominent mechanism of paclitaxel resistance in
ovarian cancer patients. Clin Cancer Res. 11:298–305.
2005.PubMed/NCBI
|
|
12
|
Derry WB, Wilson L, Khan IA, Luduena RF
and Jordan MA: Taxol differentially modulates the dynamics of
microtubules assembled from unfractionated and purified
beta-tubulin isotypes. Biochemistry. 36:3554–3562. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kavallaris M, Kuo DY, Burkhart CA, Regl
DL, Norris MD, Haber M and Horwitz SB: Taxol-resistant epithelial
ovarian tumors are associated with altered expression of specific
beta-tubulin isotypes. J Clin Invest. 100:1282–1293. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee KM, Cao D, Itami A, Pour PM, Hruban
RH, Maitra A and Ouellette MM: Class III beta-tubulin, a marker of
resistance to paclitaxel, is overexpressed in pancreatic ductal
adenocarcinoma and intraepithelial neoplasia. Histopathology.
51:539–546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inuzuka H, Shaik S, Onoyama I, Gao D,
Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al: SCF
(FBW7) regulates cellular apoptosis by targeting MCL1 for
ubiquitylation and destruction. Nature. 471:104–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilson WH, O'Connor OA, Czuczman MS,
LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong
H, Chiu YL, et al: Safety, pharmacokinetics, pharmacodynamics, and
activity of navitoclax, a targeted high affinity inhibitor of
BCL-2, in lymphoid malignancies. Lancet Oncol. 11:1149–1159. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tse C, Shoemaker AR, Adickes J, Anderson
MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et
al: ABT-263: A potent and orally bioavailable Bcl-2 family
inhibitor. Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gandhi L, Camidge DR, de Oliveira M
Ribeiro, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM,
Litvinovich E, Hemken PM, et al: Phase I study of Navitoclax
(ABT-263), a novel Bcl-2 family inhibitor, in patients with
small-cell lung cancer and other solid tumors. J Clin Oncol.
29:909–916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wong M, Tan N, Zha J, Peale FV, Yue P,
Fairbrother WJ and Belmont LD: Navitoclax (ABT-263) reduces
Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol
Cancer Ther. 11:1026–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tan N, Malek M, Zha J, Yue P, Kassees R,
Berry L, Fairbrother WJ, Sampath D and Belmont LD: Navitoclax
enhances the efficacy of taxanes in non-small cell lung cancer
models. Clin Cancer Res. 17:1394–1404. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stamelos V, Robinson E, Redman CW and
Richardson A: Navitoclax augments the activity of carboplatin and
paclitaxel combinations in ovarian cancer cells. Gynecol Oncol.
128:377–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Watanabe A, Yasuhira S, Inoue T, Kasai S,
Shibazaki M, Takahashi K, Akasaka T, Masuda T and Maesawa C: BCL2
and BCLxL are key determinants of resistance to antitubulin
chemotherapeutics in melanoma cells. Exp Dermatol. 22:518–523.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sonnichsen DS and Relling MV: Clinical
pharmacokinetics of paclitaxel. Clin Pharmacokinet. 27:256–269.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jain HV, Richardson A, Meyer-Hermann M and
Byrne HM: Exploiting the synergy between carboplatin and ABT-737 in
the treatment of ovarian carcinomas. PLoS One. 9:e815822014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan N, Wong M, Nannini MA, Hong R, Lee LB,
Price S, Williams K, Savy PP, Yue P, Sampath D, et al: Bcl-2/Bcl-xL
inhibition increases the efficacy of MEK inhibition alone and in
combination with PI3 kinase inhibition in lung and pancreatic tumor
models. Mol Cancer Ther. 12:853–864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Infante JR, Matsubayashi H, Sato N,
Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C and
Goggins M: Peritumoral fibroblast SPARC expression and patient
outcome with resectable pancreatic adenocarcinoma. J Clin Oncol.
25:319–325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hidalgo M and Von Hoff DD: Translational
therapeutic opportunities in ductal adenocarcinoma of the pancreas.
Clin Cancer Res. 18:4249–4256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oikonomopoulos GM, Syrigos KN and Saif MW:
Prognostic factors in pancreatic cancer. JOP. 14:322–324.
2013.PubMed/NCBI
|
|
32
|
von Hoff DD, Ramanathan RK, Borad MJ,
Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias
JL, et al: Gemcitabine plus nab-paclitaxel is an active regimen in
patients with advanced pancreatic cancer: A phase I/II trial. J
Clin Oncol. 29:4548–4554. 2011. View Article : Google Scholar : PubMed/NCBI
|