Introduction
Vascular cells, particularly endothelial cells
(ECs), generate reactive oxygen species (ROS) including superoxide
anions (O2·−), hydroxyl radicals
(·OH) and hydrogen peroxide
(H2O2). ROS are considered harmful to the
vasculature and may initiate pathological processes that contribute
to atherosclerosis, restenosis, hypertension and diabetic vascular
complications (1,2). However, there is also an apparent role
for ROS in the maintenance of vascular homeostasis via the
regulation of cellular events that govern cell death,
differentiation and proliferation (1,3,4). Due to its solubility in lipid and
aqueous environments, H2O2 can freely diffuse
through the cell membrane to reach remote cells prior to reacting
with particular molecular targets (5).
The modulation of ROS levels by oxygen
concentrations in lung tissue is important for control of the
pulmonary vascular system (6).
Vascular ECs are implicated in the control of blood pressure, blood
coagulation, inflammation and angiogenesis (7). H2O2 influences the
function of ECs via intricate mechanisms; for example, the ambient
production of O2·− in vasculature and the
subsequent low level generation of H2O2
affects EC growth and proliferation (2), and enhanced oxidative stress owing to
the high level of H2O2 may lead to the
apoptotic death of ECs, causing endothelial dysfunction in vascular
system (1).
Mitogen-activated protein kinases (MAPKs) are
evolutionarily preserved signaling proteins in eukaryotes that
arbitrate responses to various stimuli (8). Extracellular signal regulated kinases
(ERK1/2), the c-Jun N-terminal kinase/stress-activated protein
kinases (JNK/SAPK) and the p38 kinases are the three major MAPK
groups identified in mammals (9). The
activation of multiple MAPKs is the primary constituent of the
various signaling pathways that control cell proliferation,
survival, differentiation and cell death (10). MAPKs in ECs and smooth muscle cells
are activated by a variety of growth factors, including
platelet-derived growth factor (PDGF), vascular endothelial growth
factor (VEGF) and angiotensin II (Ang II) (11–13). MAPKs
can discern the cellular redox status and they are, in turn,
targets for ROS; for example, JNK and p38 are generally activated
by mild oxidative stress, and their activation then leads to
apoptosis (14,15). However, these two kinases
differentially affect apoptosis in pyrogallol-treated ECs; JNK
promotes survival in these cells, whereas p38 is associated with
cell death (16). In addition, ROS
can stimulate the ERK pathway via ERK phosphorylation (17). ERK activation typically produces a
pro-survival effect rather than a pro-apoptotic effect (18). Furthermore, the activity of MAPKs is
sustained by the activity of MAPK phosphatases, which are directly
regulated by H2O2 (19).
H2O2 inhibits the
phosphorylation of ERK1/2 in human umbilical vein ECs (HUVEC)
(20), whereas other studies have
demonstrated that H2O2 enhances the
phosphorylation of ERK1/2 in HUVEC (21) and bovine aortic ECs (BAEC), which is
associated with their apoptosis (22). Treatment with
H2O2 promotes p38 phosphorylation in HUVEC
(20,21) and BAEC (23). JNKs and their downstream target,
c-Jun, have been demonstrated to be involved in the apoptosis of
ECs induced by H2O2 and other stresses
(12,21,24). Thus,
the effects of H2O2 on the activities of
MAPKs, particularly mitogen-activated protein kinase kinase 1
(MEK)-ERK signaling, may differ depending on EC types and
experimental conditions, resulting in diverse cellular responses.
The action of H2O2 in aggravating endothelial
dysfunction and cell death has been extensively investigated
(25,26). However, the mechanisms underlying the
varied outcomes with respect to MAPKs remain obscure.
Using MAPK-specific inhibitors (including the
SP600125 JNK inhibitor, the PD98059 MEK inhibitor and the SB203580
p38 inhibitor), the present study addressed the function of various
MAPKs in H2O2-induced cell death and the
attenuation of cell growth. H2O2 exposure to
well-established calf pulmonary arterial ECs (CPAECs), as performed
in our previous studies (27,28), was used to analyze the effect of MAPK
inhibitors on cell growth, death, mitochondrial membrane potential
(MMP) and glutathione (GSH) levels.
Materials and methods
Cell culture
CPAECs were purchased from the Korean Cell Line Bank
(KCLB, Seoul, Korea) and were cultured in RPMI-1640 supplemented
with 10% fetal bovine serum (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and 1% penicillin-streptomycin (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). CPAECs were harvested using a
solution of trypsin-EDTA (Gibco; Thermo Fisher Scientific, Inc.)
during the exponential phase of growth. CPAECs were maintained in
100-mm plastic tissue culture dishes (Nalge Nunc International,
Penfield, NY, USA) in humidified incubator containing 5%
CO2, at 37°C.
Reagents
H2O2 was purchased from
Sigma-Aldrich (Merck KGaA). The JNK inhibitor (SP600125), MEK
inhibitor (PD98059) and p38 inhibitor (SB203580) were obtained from
Calbiochem (Merck KGaA). All reagents were dissolved in dimethyl
sulfoxide (Sigma-Aldrich; Merck KGaA) to 10 mM. Cells were
pretreated with each MAPK inhibitor for 30 min prior to treatment
with H2O2 in the conditions previously
described. A dose of 10 µM of each MAPK inhibitor was applied in
all experiments.
Cell growth assay
The effect of drugs on the growth of CPAECs was
determined by evaluating the MTT (Sigma-Aldrich; Merck KGaA) dye
absorbance, according to a previously described method (29). Cells were exposed to 30 µM
H2O2 with or without 10 µM JNK inhibitor, MEK
inhibitor or p38 inhibitor for 24 h in the conditions previously
described.
Cell cycle analysis
Sub-G1 cells were assessed using
propidium iodide (Sigma-Aldrich; Merck KGaA) staining, as per a
previously described method (30).
Cells were exposed to 30 µM H2O2 in the
presence or absence of 10 µM JNK inhibitor, MEK inhibitor or p38
inhibitor for 24 h in the conditions previously described. Cell DNA
content was assessed using a BD FACStar™ flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) and CellQuest Pro software
(version 5.1; BD Biosciences).
Annexin V staining for the detection
of apoptosis
Apoptotic cell death was verified by measuring cells
stained with Annexin V-fluorescein isothiocyanate (FITC;
Invitrogen; Thermo Fisher Scientific, Inc.), as per a previously
described method (31). Cells were
exposed to 30 µM H2O2 with or without 10 µM
JNK inhibitor, MEK inhibitor or p38 inhibitor for 24 h in the
conditions previously described. Annexin V staining was analyzed
with a BD FACStar flow cytometer, as aforementioned.
Measurement of MMP
MMP was measured using a rhodamine 123 fluorescent
dye (Sigma-Aldrich; Merck KGaA), as previously described (32). Cells were exposed to 30 µM
H2O2 in the presence or absence of 10 µM JNK
inhibitor, MEK inhibitor or p38 inhibitor for 24 h in the
conditions previously described. Rhodamine 123 staining intensity
was assessed by a BD FACStar flow cytometer as aforementioned. The
absence of rhodamine 123 in cells designated the loss of MMP in
CPAECs. MMP levels in the cells were expressed as mean fluorescence
intensity (MFI), which was calculated using CellQuest™ Pro
software, as aforementioned.
Measurement of intracellular ROS
levels
Intracellular ROS levels were measured with
2′,7′-dichlorodihydrofluorescein diacetate (DCF; Invitrogen; Thermo
Fisher Scientific, Inc.), and O2·− levels
were evaluated using dihydroethidium (DHE, Invitrogen; Thermo
Fisher Scientific, Inc.) fluorescent dyes. Cells were exposed to 30
µM H2O2 with or without 10 µM JNK inhibitor,
MEK inhibitor or p38 inhibitor for 24 h in the previously described
conditions. Cells were then incubated with 20 µM H2DCFDA
or DHE at 37°C for a further 30 min. DCF and DHE fluorescence
levels were measured using the BD FACStar flow cytometer. ROS and
O2·− levels were stated as MFI.
Detection of the intracellular
glutathione (GSH)
The GSH level was analyzed with a
5-chloromethylfluorescein diacetate dye (CMF; Invitrogen; Thermo
Fisher Scientific, Inc.). Cells were incubated with 30 µM
H2O2 in the presence or absence of 10 µM JNK
inhibitor, MEK inhibitor or p38 inhibitor for 24 h in the
previously described conditions. Cells were then incubated with 5
µM CMF at 37°C for a further 30 min. CMF fluorescence intensity was
measured using the BD FACStar flow cytometer as previously
described. GSH depletion was indicated with negative CMF staining.
CMF levels in cells were expressed as MFI.
Statistical analysis
The data are presented as the mean ± standard
deviation of ≥2 independent experiments. The data were analyzed
using GraphPad Prism software version 5 (GraphPad Software, Inc.,
La Jolla, CA, USA). The Student's t-test and one-way analysis of
variance followed by Tukey's multiple comparison test were utilized
for parametric data. P<0.05 was considered to indicate a
statistically significance difference.
Results
MAPK inhibitors affect cell growth and
death in H2O2-treated CPAECs
The effects of MAPK inhibitors (including a JNK
inhibitor, MEK inhibitor and p38 inhibitor) on the growth of
H2O2-treated CPAECs were examined using MTT
assays. The inhibitors were selected based on those used in our
prior studies (33–36). According to another previous study
(25), the IC50 of
H2O2 in CPAECs is ~20 µM at 24 h. Therefore,
30 µM of H2O2 was selected for use in the
present study.
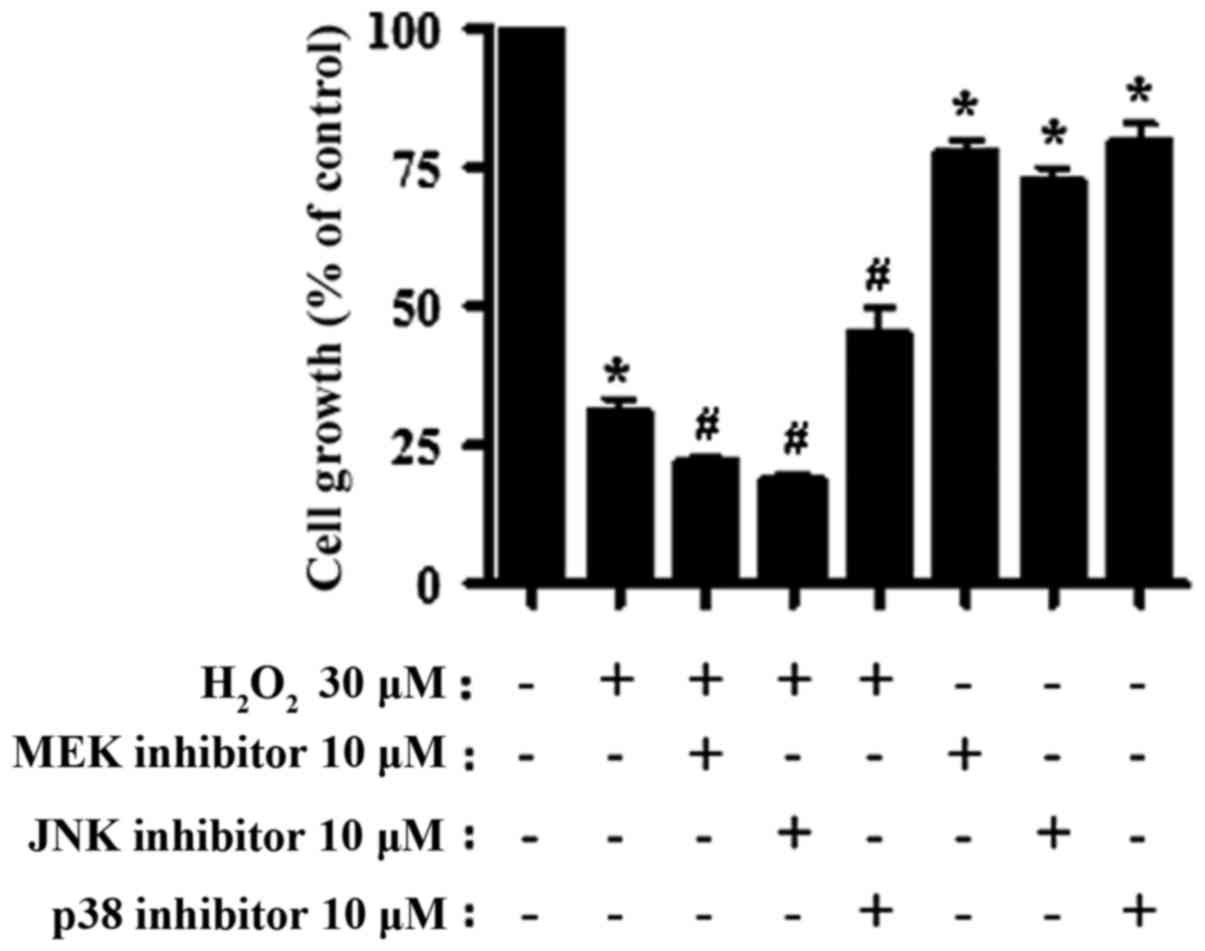
H2O2 treatment caused ~70%
growth inhibition of CPAECs within 24 h (Fig. 1; P<0.05, compared with no
treatment). The addition of the MEK or JNK inhibitors further
stalled cell growth (P<0.05, compared with the
H2O2-only group). The JNK inhibitor was the
most potent in augmenting the negative effect of
H2O2 on cell growth, although it was not
statistically different from the MEK inhibitor (P=0.168; Fig. 1). On the other hand, treatment with
the p38 inhibitor partially attenuated the effect of
H2O2 on CPAEC growth (P<0.05; Fig. 1). All inhibitors also diminished the
growth of the CPAECs when administered without
H2O2 (P<0.05; Fig. 1).
The percentages of the sub-G1 cells in
CPAECs were measured in a similar manner to a number of previous
studies (37–39). Treatment with
H2O2 alone increased the percentage of the
sub-G1 cells by ~20% compared with the
H2O2-untreated CPAEC control group (Fig. 2A and B). MEK inhibitor treatment
exhibited a trend towards increasing the number of
sub-G1 cells in H2O2-treated
CPAECs (Fig. 2A and B). Treatment
with the JNK inhibitor significantly increased, whereas the p38
inhibitor significantly decreased the number of sub-G1
cells in H2O2-treated CPAECs (both P<0.05;
Fig. 2A and B). In addition,
H2O2 treatment also increased the percentage
of Annexin V-FITC stained CPAECs, indicating that the death of
CPAECs subsequent to H2O2 treatment may occur
via apoptosis (P<0.05; Fig. 2C).
None of the MAPK inhibitors significantly affected the Annexin
V-FITC positive cell number in H2O2-treated
CPAECs (Fig. 2C); however, treatment
with the JNK inhibitor alone increased the number of Annexin V-FITC
positive cells in CPAECs in the absence of
H2O2 (P<0.05; Fig. 2C).
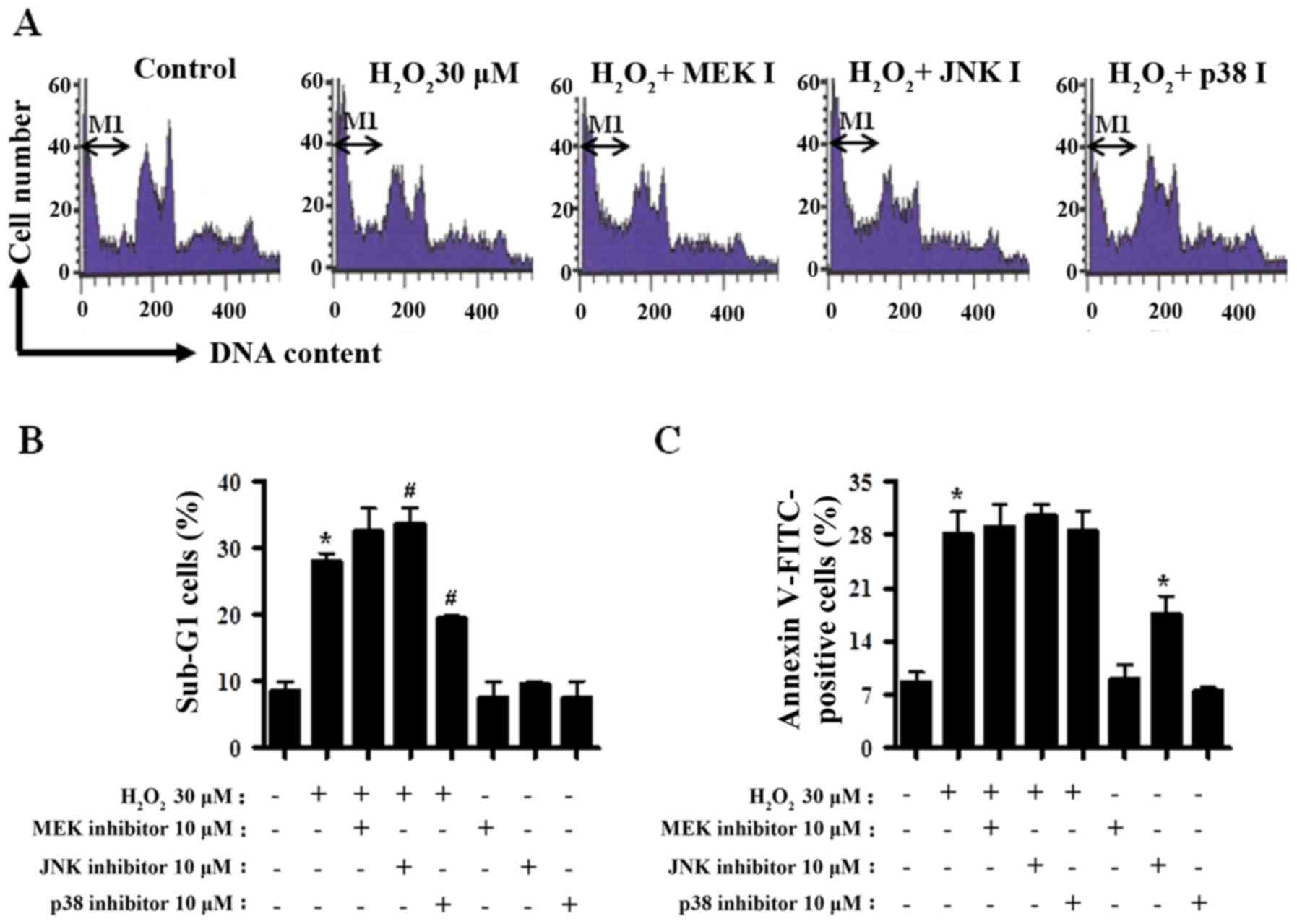 | Figure 2.Analysis of apoptosis in MAPK
inhibitor- and H2O2-treated CPAECs. Following
a 30-min pre-incubation with each MAPK inhibitor, CPAECs in the
exponential growth phase were treated with
H2O2 for 24 h, and subsequently analyzed for
the sub-G1 and apoptotic population using flow
cytometry. (A) Representative of DNA content histograms of cells.
M1 regions indicate the portion of sub-G1 cells. (B)
Percentage of sub-G1 cells (M1 regions in A). (C)
Percentage of Annexin V-FITC positive cells, indicative of
apoptosis. *P<0.05, compared with the control (no treatment)
group. #P<0.05, compared with cells treated with
H2O2 only. MAPK, mitogen-activated protein
kinase; CPAEC, calf pulmonary arterial endothelial cell; MEK,
mitogen-activated protein kinase kinase 1; JNK, c-Jun N-terminal
kinase; FITC, fluorescein isothiocyanate. |
MAPK inhibitors influence MMP in
H2O2-treated CPAECs
Cell death is closely associated with the loss of
MMP (40). Thus, MMP in
H2O2-treated CPAECs was determined using a
rhodamine 123 dye at 24 h of treatment. There was a significant
loss of MMP in H2O2-treated cells (P<0.05;
Fig. 3A and B). Treatment with the
MEK inhibitor did not influence the MMP level in
H2O2-treated CPAECs (Fig. 3A and B). JNK inhibitor boosted,
whereas p38 inhibitor decreased, the loss of MMP in
H2O2-treated CPAECs (both P<0.05; Fig. 3A and B). Treatment with JNK inhibitor
alone triggered a significant loss of MMP in the control CPAECs
(Fig. 3A and B). When disregarding
rhodamine 123-negative cells, treatment with
H2O2 non-significantly increased the MMP
level in CPAECs (Fig. 3A and C).
Treatment with the MEK or JNK inhibitor reduced the MMP level in
H2O2-treated CPAECs (P<0.05; Fig. 3A and C), whereas treatment with the
p38 inhibitor did not alter the level (Fig. 3A and C). Whilst treatment with the MEK
or JNK inhibitor reduced the MMP level in
H2O2-untreated control CPAECs, treatment with
the p38 inhibitor augmented the level (P<0.05; Fig. 3A and C).
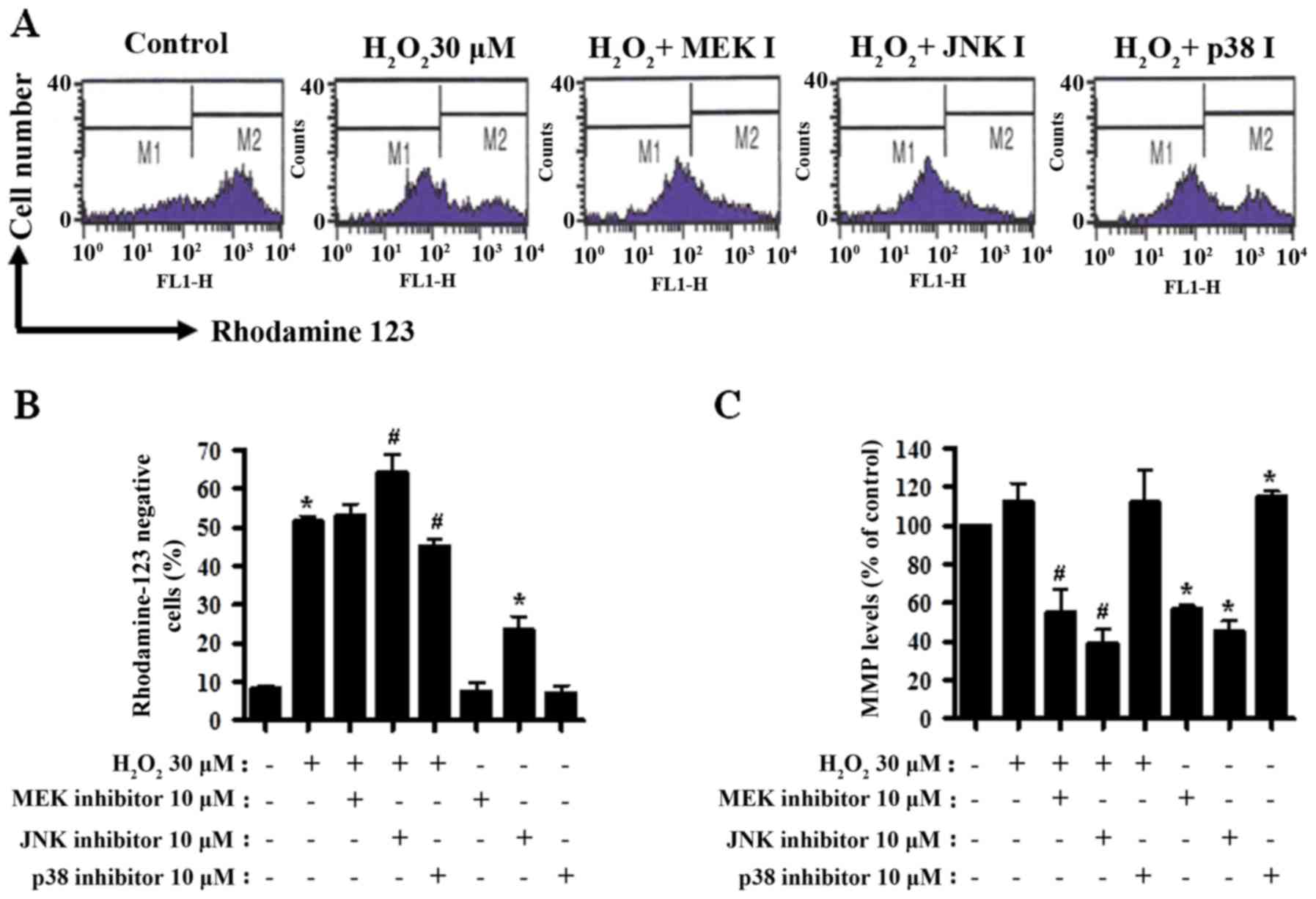 | Figure 3.Assessment of MMP in
H2O2-treated CPAECs in the presence and
absence of MAPK inhibitors. Following a 30-min pre-incubation with
each MAPK inhibitor, CPAECs in the exponential growth phase were
treated with H2O2 for 24 h. MMP in CPAECs was
measured using rhodamine 123 intensity with flow cytometry. (A)
Representative rhodamine 123 stained cell histograms. M1 regions
contain rhodamine 123-negative cells, i.e., cells where MMP is
reduced; M2 regions contain rhodamine 123-positive cells. (B)
Percentage of rhodamine 123-negative cells (M1 regions in A). (C)
Percentage of rhodamine 123-positive cells (M2 regions in A).
*P<0.05, compared with the control (no treatment) group.
#P<0.05, compared with cells treated with
H2O2 only. MMP, mitochondrial membrane
potential; CPAEC, calf pulmonary arterial endothelial cell; MAPK,
mitogen-activated protein kinase; MEK, mitogen-activated protein
kinase kinase 1; JNK, c-Jun N-terminal kinase. |
MAPK inhibitors alter ROS, including
O2·−, levels in
H2O2-treated CPAECs
Alterations to ROS levels were assessed in
H2O2- and MAPK inhibitor-treated CPAECs. As
presented in Fig. 4A, the ROS levels
(including H2O2) significantly decreased in
CPAECs treated with H2O2 at 24 h (P<0.05).
None of the MAPK inhibitors significantly altered ROS levels in the
H2O2-treated CPAECs (Fig. 4A). By contrast, all MAPK inhibitors,
particularly the p38 inhibitor, increased the ROS levels in the
control CPAECs (P<0.05; Fig. 4A).
When O2·− levels in
H2O2-treated CPAECs were measured, the DHE
MFI, reflecting intracellular O2·−, decreased
(P<0.05; Fig. 4B). None of the
MAPK inhibitors significantly altered the DHE MFI level of
H2O2-treated CPAECs (Fig. 4B). MEK and JNK inhibitors enhanced
O2·− levels in the control CPAECs (P<0.05;
Fig. 4B).
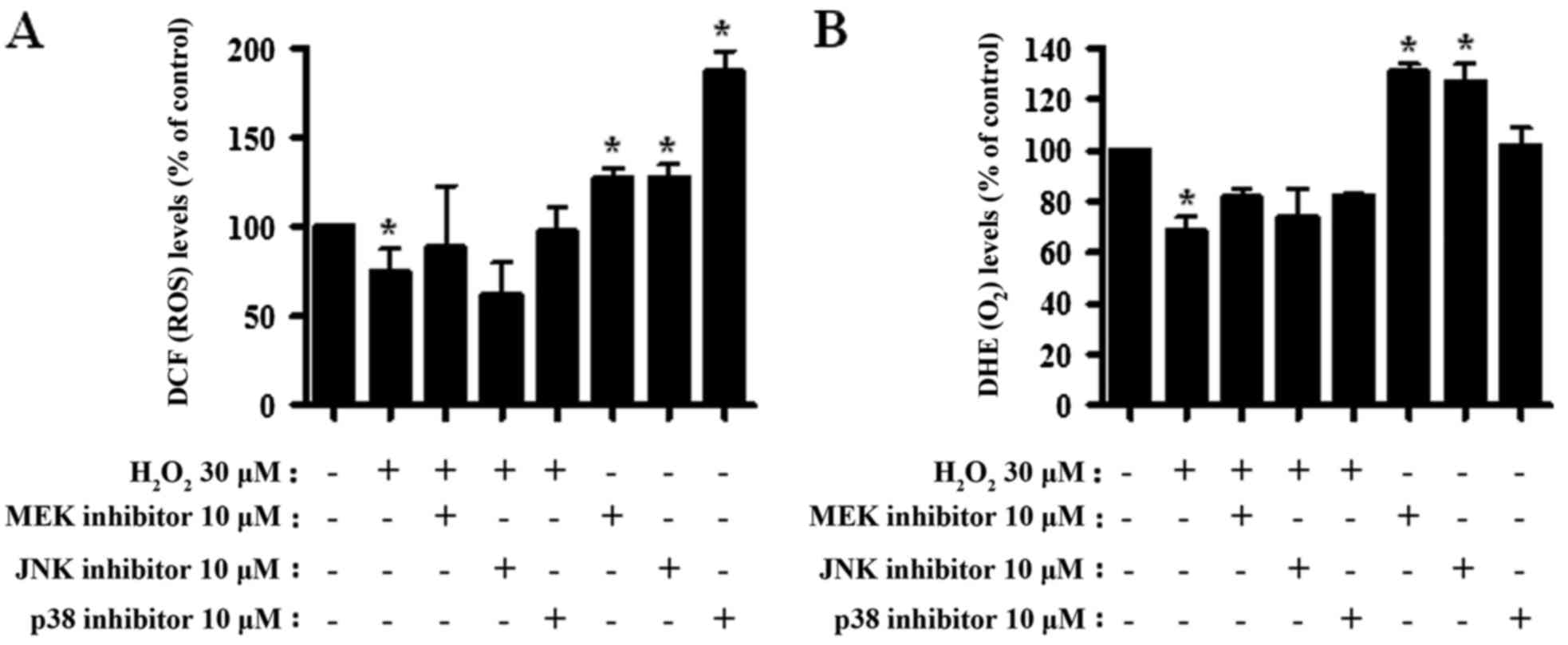 | Figure 4.Assessment of ROS levels following
treatment with MAPK inhibitors and H2O2 in
CPAECs. CPAECs in the exponential growth phase were treated with
H2O2 for 24 h following a 30-min MAPK
inhibitor pre-treatment. ROS levels in CPAECs were assessed using
DCF and DHE dyes in flow cytometry. Graphs indicate the percentage
of (A) DCF (ROS) and (B) DHE (O2·−) levels
compared with the control (no treatment) CPAECs. *P<0.05,
compared with the control (no treatment) group.
#P<0.05, compared with cells treated with
H2O2 only. ROS, reactive oxygen species;
MAPK, mitogen-activated protein kinase; CPAEC, calf pulmonary
arterial endothelial cell; DCF, 2′,7′-dichlorodihydrofluorescein
diacetate; DHE, dihydroethidium; MEK, mitogen-activated protein
kinase kinase 1; JNK, c-Jun N-terminal kinase. |
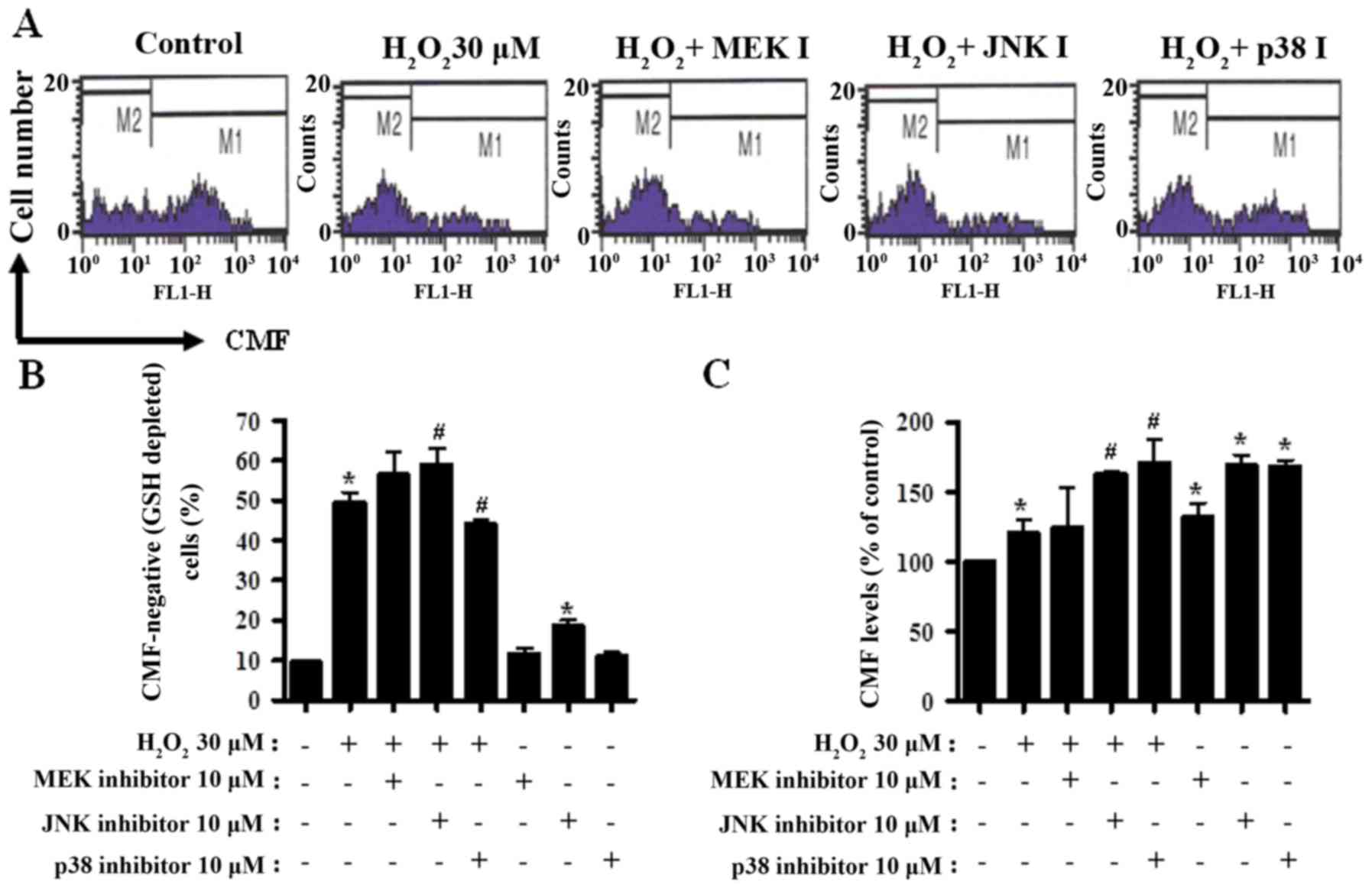
MAPK inhibitors change GSH levels in
H2O2-treated CPAECs
GSH levels in CPAECs were analyzed by CMF
fluorescence. In Fig. 5A, the M1
regions indicate CMF-positive cells, whereas the M2 regions
indicate CMF-negative (GSH-depleted) cells.
H2O2 treatment resulted in an ~40% increase
in the number of GSH-depleted CPAECs (M2 region), compared with in
non-treated control cells (P<0.05; Fig. 5A and B). MEK and JNK inhibitors
appeared to increase the number of GSH-depleted cells in
H2O2-treated CPAECs; this increase was
significant for JNK inhibitor treatment (P<0.05; Fig. 5A and B). Unlike with the MEK and JNK
inhibitors, treatment with the p38 inhibitor decreased the number
of GSH-depleted cells in H2O2-treated CPAECs
(P<0.05; Fig. 5A and B). JNK
inhibitor treatment alone increased the number of GSH-depleted
cells in H2O2-untreated CPAECs (Fig. 5A and B). Furthermore, when the GSH
levels in CPAECs, without considering CMF-negative cells, were
measured, the GSH level increased in
H2O2-treated CPAECs (P<0.05; Fig. 5A and C). While treatment with the MEK
inhibitor did not alter the level of GSH in
H2O2-treated CPAECs, treatment with the JNK
or p38 inhibitors increased the levels in these cells (P<0.05;
Fig. 5A and C). All MAPK inhibitors
promoted an increase in GSH levels in the control CPAECs; the
effect was more pronounced following treatment with JNK or p38
(Fig. 5A and C).
Discussion
A variety of MAPKs occur in the vasculature,
activated by diverse growth factors, including Ang II, PDGF and
VEGF (11–13). ROS regulate MAPKs in ECs (12,20–24). Since
H2O2 inhibits the growth of CPAECs and
induces their death, the present study focused on evaluating the
effects of MAPK inhibitors on cell growth and death, and GSH in
H2O2-treated CPAECs. ERK activation typically
has a pro-survival role rather than a pro-apoptotic role (18). Treatment with the MEK inhibitor
enhanced growth inhibition in H2O2-treated
CPAECs and slightly increased the proportion of the
sub-G1 cell population. Thus, H2O2
treatment may have inactivated ERK proteins in CPAECs, resulting in
growth inhibition and cell death.
The activity of JNK and p38 can be stimulated by
ROS or an oxidative alteration to the intracellular thiol/disulfide
redox state, leading to apoptosis (14,15).
H2O2 promotes p38 phosphorylation in HUVEC
(20,21) and BAEC (23). JNKs and their downstream target,
c-Jun, have been demonstrated to be involved the apoptosis of ECs
triggered by H2O2 and other stresses
(12,21,24).
According to data from the present study, treatment with the JNK
inhibitor augmented growth inhibition and death in
H2O2-treated CPAECs, whereas treatment with
the p38 inhibitor decreased the relative extent of growth
inhibition and death in these cells. Therefore, JNK has pro-growth
and survival effects, and p38 has anti-growth and pro-death effects
in H2O2-treated CPAECs. In addition, our
previous study demonstrated that JNK inhibitor treatment increased
the rate of apoptosis in pyrogallol-treated CPAECs, whereas p38
inhibitor treatment decreased the level of apoptosis (16). This suggests that the JNK and p38
signal transduction pathways differentially affect the growth and
death of CPAECs treated with H2O2 or
pyrogallol. However, Machino et al (41) previously identified that
H2O2 promoted the phosphorylation of JNK and
p38 in human pulmonary vascular ECs. Thus, the effect of
H2O2 on JNK activity appears to be EC-type
specific; for example, it may differ in artery vs. vein, large
vessels vs. small vessels, coronary vs. pulmonary, human vs. other
species. Additionally, all the MAPK inhibitors used in the present
study reduced the growth of the control CPAECs, indicating that
individual MAPK signaling pathways may differentially affect the
growth of CPAECs in the presence or absence of
H2O2.
Treatment with 30 µM H2O2
increased the proportion of Annexin V-FITC positive cells in
CPAECs. Our prior study demonstrated that treatment with the
pan-caspase inhibitor Z-VAD significantly prohibited cell death in
H2O2-treated CPAECs (25). Thus, the
H2O2-induced death of CPAECs predominantly
occurs via apoptosis. However, MAPK inhibitors that affect the
sub-G1 cell proportion in
H2O2-treated CPAECs did not alter the levels
of Annexin V-FITC positive cells. Therefore, MAPK inhibitors may
promote the death of CPAECs via necrosis rather than apoptosis. In
addition, treatment with the JNK inhibitor alone increased the
number of Annexin V-FITC positive cells in the control CPAECs,
suggesting that the inhibition of JNK signaling increases the
susceptibility of CPAECs to exogenous
H2O2.
ROS can disturb the natural oxidation/reduction
equilibrium in cells by triggering a reduction in MMP (42). Accordingly, H2O2
treatment induced a loss of MMP in CPAECs in the present study.
Similar to the effect on sub-G1 cells, treatment with
the JNK inhibitor increased the loss of MMP in
H2O2-treated CPAECs, whereas treatment with
the p38 inhibitor reduced the MMP loss in the cells. In addition,
treatment with the JNK inhibitor alone increased the loss of MMP in
CPAECs without H2O2 treatment, suggesting
that JNK signaling may be involved in the maintenance of MMP in
CPAECs. Treatment with H2O2 slightly
increased the MMP level of CPAECs; treatment with the MEK and JNK
inhibitors decreased the MMP levels of
H2O2-treated and -untreated CPAECs, whereas
treatment with the p38 inhibitor slightly increased the MMP level
in H2O2-treated and -untreated CPAECs. These
results indicate that each MAPK signaling pathway has distinct and
specific effects on MMP in CPAECs.
The primary ROS associated with cell signaling
pathways are O2·− and
H2O2. ROS toxicity is generally mediated by
·OH (6). As treatment with
30 µM H2O2 significantly induced the death of
CPAECs, it is possible that exogenous H2O2
was converted into the more cytotoxic ·OH through the
Fenton reaction to eliminate CPAECs (43). Notably ROS levels, including the
levels of O2·−, decreased in
H2O2-treated CPAECs after 24 h. It is
possible that the actual ROS level of the
H2O2-treated CPAECs was distorted, as dead
cells have a reduced capacity for the uptake of DCF and DHE. Our
previous study also reported a decrease in
O2·− levels following 24 h of treatment with
5–50 µM H2O2 in CPAECs (25). As ROS have a short half-life in the
cell (44), further study on
H2O2-treated CPAECs is required to assess ROS
levels at an earlier time point, such as 30 min or 1 h. None of the
MAPK inhibitors significantly altered the levels of ROS, including
O2−, in H2O2-treated
CPAECs. However, MEK or p38 inhibitor treatments non-significantly
increased ROS levels, including O2·−, in the
control CPAECs without a corresponding induction of cell death.
Treatment with the JNK inhibitor, as induced cell death and the
loss of MMP in the control CPAECs, also increased the levels of
ROS, including O2·−. The results suggest that
the death of CPAECs subsequent to H2O2 and/or
individual MAPK inhibitor treatment could only weakly be attributed
to an increase in ROS levels, and that treatment with each MAPK
inhibitor changed the ROS levels in CPAECs via dissimilar
mechanisms. The molecular mechanisms underlying these effects
require further study, ideally with small interfering RNA knockdown
of the MAPKs.
The extent of the induction of apoptosis is
inversely proportional to the GSH content of cells (37,45,46). In
the present study, H2O2 treatment increased
the number of GSH-depleted cells in CPAECs. Furthermore, JNK
inhibitor treatment increased the number of GSH-depleted cells in
H2O2-treated CPAECs, whereas treatment with
the p38 inhibitor decreased it. The results appear to reflect the
proportion of sub-G1 cells. In our previous study,
treatment with the JNK inhibitor, as is associated with a
pro-apoptotic effect on pyrogallol-treated CPAECs, enhances GSH
depletion, whereas treatment with the p38 inhibitor had the
opposite effect on pyrogallol-induced GSH depletion (16). Only JNK inhibitor treatment was
associated with cell death in control CPAECs while also inducing
GSH depletion in the present and previous studies. These results
support the hypothesis that intracellular GSH content has a
decisive role in cell death (45–47).
Notably, GSH levels in the viable cells among
H2O2-treated CPAECs increased, which may be a
defense mechanism in response to exogenous
H2O2. While MEK inhibitor treatment did not
alter the GSH level in H2O2-treated CPAECs,
treatment with JNK or p38 inhibitors did increase the GSH levels.
Each MAPK inhibitor influenced the GSH levels in
H2O2-treated CPAECs in different ways when
considering the GSH levels of the non-GSH-depleted cells. The
increased GSH levels in the control cells following MAPK inhibitor
treatment may be a direct response to ROS generated by these
inhibitors. GSH levels are high in typical cells (≤10 mM) and GSH
transferase is ubiquitously present (48). Thus, measuring CMF fluorescence, which
is produced upon reacting with thiol groups via a glutathione
S-transferase-mediated reaction, is useful to evaluate GSH levels
(49). However, CMF dye has
limitations in accurately determining whole GSH content and
GSH:glutathione disulfide (GSSG, the oxidized form of GSH) ratios
in cells, as this dye may also detect other thiol groups (48). The determination of exact GSH levels
and GSH:GSSG ratios in H2O2-treated CPAECs
with or without each MAPK inhibitor are further required in order
to understand the precise role of GSH in the regulation of CPAEC
redox status.
In conclusion, treatment with
H2O2 induced cell growth inhibition and death
in CPAECs through GSH depletion. Treatment with the JNK inhibitor
boosted cell growth inhibition and death, whereas the p38 inhibitor
diminished the growth inhibition and death of
H2O2-treated CPAECs.
Acknowledgements
The present study was supported by a grant from the
National Research Foundation of Korea funded by the Korean
government (MSIP; grant nos. 2008-0062279 and
2016R1A2B4007773).
Glossary
Abbreviations
Abbreviations:
|
ECs
|
endothelial cells
|
|
CPAECs
|
calf pulmonary arterial endothelial
cells
|
|
ROS
|
reactive oxygen species
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein kinase
kinase 1
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MMP
|
mitochondrial membrane potential
|
|
FITC
|
fluorescein isothiocyanate
|
|
DCF
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMF
|
5-chloromethylfluorescein
diacetate
|
References
|
1
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: A review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai H: Hydrogen peroxide regulation of
endothelial function: Origins, mechanisms, and consequences.
Cardiovasc Res. 68:26–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ameziane-El-Hassani R and Dupuy C:
Detection of reactive oxygen species in cells undergoing
oncogene-induced senescence. Methods Mol Biol. 1534:139–145. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perez-Vizcaino F, Cogolludo A and Moreno
L: Reactive oxygen species signaling in pulmonary vascular smooth
muscle. Respir Physiol Neurobiol. 174:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bassenge E: Endothelial function in
different organs. Prog Cardiovasc Dis. 39:209–228. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Atay O and Skotheim JM: Spatial and
temporal signal processing and decision making by MAPK pathways. J
Cell Biol. 216:317–330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blenis J: Signal transduction via the MAP
kinases: Proceed at your own RSK. Proc Natl Acad Sci USA. 90:pp.
5889–5892. 1993; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eguchi S, Dempsey PJ, Frank GD, Motley ED
and Inagami T: Activation of MAPKs by angiotensin II in vascular
smooth muscle cells. Metalloprotease-dependent EGF receptor
activation is required for activation of ERK and p38 MAPK but not
for JNK. J Biol Chem. 276:7957–7962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gupta K, Kshirsagar S, Li W, Gui L,
Ramakrishnan S, Gupta P, Law PY and Hebbel RP: VEGF prevents
apoptosis of human microvascular endothelial cells via opposing
effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res.
247:495–504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sundaresan M, Yu ZX, Ferrans VJ, Irani K
and Finkel T: Requirement for generation of H2O2 for
platelet-derived growth factor signal transduction. Science.
270:296–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: JNK and p38 inhibitors increase and decrease
apoptosis, respectively, in pyrogallol-treated calf pulmonary
arterial endothelial cells. Int J Mol Med. 24:717–722.
2009.PubMed/NCBI
|
|
17
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant injury. J Biol. Chem.
271:4138–4142. 1996.
|
|
18
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Latimer HR and Veal EA: Peroxiredoxins in
regulation of MAPK signalling pathways; sensors and barriers to
signal transduction. Mol Cells. 39:40–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee YJ, Kang IJ, Bünger R and Kang YH:
Enhanced survival effect of pyruvate correlates MAPK and NF-kappaB
activation in hydrogen peroxide-treated human endothelial cells. J
Appl Physiol (1985). 96:792–801. 2004. View Article : Google Scholar
|
|
21
|
Zhai L, Zhang P, Sun RY, Liu XY, Liu WG
and Guo XL: Cytoprotective effects of CSTMP, a novel stilbene
derivative, against H2O2-induced oxidative stress in human
endothelial cells. Pharmacol Rep. 63:1469–1480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang B, Oo TN and Rizzo V: Lipid rafts
mediate H2O2 prosurvival effects in cultured endothelial cells.
FASEB J. 20:1501–1503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moriue T, Igarashi J, Yoneda K, Nakai K,
Kosaka H and Kubota Y: Sphingosine 1-phosphate attenuates
H2O2-induced apoptosis in endothelial cells. Biochem Biophys Res
Commun. 368:852–857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang N, Verna L, Hardy S, Zhu Y, Ma KS,
Birrer MJ and Stemerman MB: c-Jun triggers apoptosis in human
vascular endothelial cells. Circ Res. 85:387–393. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park WH: The effects of exogenous H2O2 on
cell death, reactive oxygen species and glutathione levels in calf
pulmonary artery and human umbilical vein endothelial cells. Int J
Mol Med. 31:471–476. 2013.PubMed/NCBI
|
|
26
|
Hermann C, Zeiher AM and Dimmeler S: Shear
stress inhibits H2O2-induced apoptosis of human endothelial cells
by modulation of the glutathione redox cycle and nitric oxide
synthase. Arterioscler Thromb Vasc Biol. 17:3588–3592. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han YH and Park WH: Pyrogallol-induced
calf pulmonary arterial endothelial cell death via
caspase-dependent apoptosis and GSH depletion. Food Chem Toxicol.
48:558–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Moon HJ, You BR and Park WH:
Propyl gallate inhibits the growth of calf pulmonary arterial
endothelial cells via glutathione depletion. Toxicol In Vitro.
24:1183–1189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.PubMed/NCBI
|
|
30
|
Han YH, Kim SZ, Kim SH and Park WH:
Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the
changes of intracellular GSH levels rather than ROS levels. Lung
Cancer. 59:301–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park WH: MAPK inhibitors and siRNAs
differentially affect cell death and ROS levels in arsenic
trioxide-treated human pulmonary fibroblast cells. Oncol Rep.
27:1611–1618. 2012.PubMed/NCBI
|
|
34
|
Han YH and Park WH: The effects of MAPK
inhibitors on a proteasome inhibitor, MG132-induced HeLa cell death
in relation to reactive oxygen species and glutathione. Toxicol
Lett. 192:134–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han YH and Park WH: Pyrogallol-induced
As4.1 juxtaglomerular cell death is attenuated by MAPK inhibitors
via preventing GSH depletion. Arch Toxicol. 84:631–640. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han YH, Moon HJ, You BR and Park WH: The
effects of MAPK inhibitors on pyrogallol-treated Calu-6 lung cancer
cells in relation to cell growth, reactive oxygen species and
glutathione. Food Chem Toxicol. 48:271–276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park WH: Pyrogallol induces the death of
human pulmonary fibroblast cells through ROS increase and GSH
depletion. Int J Oncol. 49:785–792. 2016.PubMed/NCBI
|
|
38
|
Park WH and You BR: Antimycin A induces
death of the human pulmonary fibroblast cells via ROS increase and
GSH depletion. Int J Oncol. 48:813–820. 2016.PubMed/NCBI
|
|
39
|
Han BR, You BR and Park WH: Valproic acid
inhibits the growth of HeLa cervical cancer cells via
caspase-dependent apoptosis. Oncol Rep. 30:2999–3005.
2013.PubMed/NCBI
|
|
40
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Machino T, Hashimoto S, Maruoka S, Gon Y,
Hayashi S, Mizumura K, Nishitoh H, Ichijo H and Horie T: Apoptosis
signal-regulating kinase 1-mediated signaling pathway regulates
hydrogen peroxide-induced apoptosis in human pulmonary vascular
endothelial cells. Crit Care Med. 31:2776–2781. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Campo ML, Kinnally KW and Tedeschi H: The
effect of antimycin A on mouse liver inner mitochondrial membrane
channel activity. J Biol Chem. 267:8123–8127. 1992.PubMed/NCBI
|
|
43
|
Winterbourn CC: Toxicity of iron and
hydrogen peroxide: The Fenton reaction. Toxicol Lett. 82-83:1–974.
1995. View Article : Google Scholar
|
|
44
|
Fan LM and Li JM: Evaluation of methods of
detecting cell reactive oxygen species production for drug
screening and cell cycle studies. J Pharmacol Toxicol Methods.
70:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Puchalski RB, Manoharan TH, Lathrop AL and
Fahl WE: Recombinant glutathione S-transferase (GST) expressing
cells purified by flow cytometry on the basis of a GST-catalyzed
intracellular conjugation of glutathione to monochlorobimane.
Cytometry. 12:651–665. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tauskela JS, Hewitt K, Kang LP, Comas T,
Gendron T, Hakim A, Hogan M, Durkin J and Morley P: Evaluation of
glutathione-sensitive fluorescent dyes in cortical culture. Glia.
30:329–341. 2000. View Article : Google Scholar : PubMed/NCBI
|