Introduction
Sirtuin 1 (Sirt1), a conserved NAD+
dependent deacetylase, has been implicated in modulating
transcriptional silencing and cell survival, and is known to serve
a role in tumorigenesis via deacetylation of important
transcriptional factors, including tumor protein p53 (p53)
(1), E2F transcription factor 1
(E2F1) (2) and nuclear factor-κB
(3).
Despite the paradoxical role of Sirt1 in
tumorigenesis, the expression level of Sirt1 is increased in
numerous types of cancer cell. Upregulation of Sirt1 is frequently
observed in non-melanoma skin cancers, including actinic keratosis,
Bowen's disease, squamous cell carcinoma and basal cell carcinoma
(4). Sirt1 expression level is also
significantly increased in poorly differentiated mouse and human
prostate cancer (5), acute myeloid
leukemia (AML) (6) and lymphoma
(7).
The expression level of Sirt1 is differentially
regulated by oncogenes and tumor suppressor genes. p53 has been
reported to repress the expression of Sirt1 and its removal by
forkhead box O3 activated Sirt1 transcription in PC12 neuronal
cancer cells (8,9). This model is further supported by
studies investigating transformation-associated p53 knockout mice,
which expressed constitutively higher Sirt1 mRNA expression levels
in numerous tissues (10).
Of note, hypermethylated in cancer 1 (HIC1), another
tumor suppressor that acts as a repressor of Sirt1 expression
level, formed a transcriptional repression complex with Sirt1,
which induced inhibition of Sirt1 transcription (11). Sirt1 expression is regulated by the
transcriptional factor E2F1, which binds two sites within the Sirt1
promoter. E2F1 is a crucial activator of Sirt1 expression level in
response to DNA damage (2). Of note,
the majority of the identified transcriptional factors associated
with Sirt1 expression regulation are also subjected to Sirt1
deacetylation and form positive or negative feedback loops to
fine-tune cell fate.
c-Myc is another oncogenic transcriptional factor
that is deacetylated by Sirt1 (12).
It remains unclear whether c-Myc may be involved in feedback loops
of Sirt1 deacetylation. In the present study it was revealed that
c-Myc directly binds to the conserved E-box (−189 to 183 bp) of the
Sirt1 promoter and induces its transcription in p53 deficient
cells. p53 inhibited the c-Myc mediated upregulation of Sirt1
promoter activity by blocking its binding to the Sirt1 promoter.
This novel regulation of Sirt1 identified it as the prototype for a
novel class of c-Myc and p53 target genes and aids understanding of
its role in certain tumor cells.
Materials and methods
Cell culture, transfection and,
reagents
Mouse embryonic fibroblasts MEF/3T3, human embryonic
kidney 293A, human colorectal cancer HCT116, human osteosarcoma
U2OS, human leukemic K562 and human non-small cell lung carcinoma
H1299 cells were purchased from the American Type Culture
Collection (Manassas, VA, USA). All cells were maintained in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with glutamine,
penicillin/streptomycin and 10% fetal bovine serum (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). All transfections were performed
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. 10058-F4 was purchased from Sigma-Aldrich
(Merck KGaA).
Constructs and antibodies
Flag-tagged c-Myc and human influenza
hemagglutinin-tagged p53 expression vectors were provided by Wuhan
Sanyinh Biotechnology (Wuhan, China).
For the luciferase reporter assay, various length
fragments of the Sirt1 promoter, starting from either-2852, −200,
or −102 to the transcriptional start site (+1) were amplified from
human genomic DNA. The primers used in luciferase reporter
generation were as follows: −2852 fragment (E1/2/3),
5′-GACTCGAGCGTCAGCCACCGTGCT-3′ and 5′-TTGCTAGCTCTTCCAACTGCCTCT-3′;
−200 fragment (E2/3), 5′-TTAAGCTTCCTCCGCCCGCCACGT-3′ and
5′-TCGGTACCTCTTCCAACTGCCTCT-3′; −102 fragment,
5′-TTAAGCTTGGGTTTAAATCTCCCG-3′ and 5′-ATGGTACCTCTTCCAACTGCCTCT-3′.
In the −200 fragment, two putative c-Myc binding sites containing
the consensus ‘CACGTG’ were identified using rVISTA2.0 (https://rvista.dcode.org/). Mutant promoter constructs
were obtained by changing each ‘CACGTG’ sequence within the −200
fragment into ‘TTTGGG’ and named as E2/mutant (mut) or E3/mut
respectively. All amplified promoter fragments were cloned into the
pGL3-basic luciferase reporter (Promega Corporation, Madison, WI,
USA) within restriction enzyme sites SmaI and
HindIII. The scrambled siRNA (control) and siRNAs targeting
p53 were purchased from Thermo Fisher Scientific, Inc. Indicated
overexpression constructs were generated by amplifying the coding
region from cDNA obtained from MEF/3T3 and H1299 cells and
sub-cloned into the pEF-HA vector (Addgene, Inc., Cambridge, MA,
USA). MEF/3T3 and H1299 cells were transiently transfected with
siRNAs targeting p53 using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol.
Anti-c-Myc (N262; dilution, 1:100; cat. no.
sc-500771), anti-human Sirt1 (dilution, 1:200; cat. no. sc-15404),
anti-p53 (dilution, 1:500; cat. no. sc-126) and anti-GAPDH
(dilution, 1:500; cat. no. sc-47724) antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Luciferase reporter assay
H1299 cells were harvested at 36 h after
transfection. Cellular promoter activity was determined using the
Dual Luciferase Assay System kit, according to the manufacturer's
protocol (Promega Corporation). pRL-CMV renilla luciferase was used
as an internal control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The expression level of Sirt1 was determined by RT
of total RNA followed by qPCR analysis. Total RNA was extracted
from K562, 293A and MEF/3T3 cells using an RNA extraction kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. A total of 2 µg total RNA was reverse
transcribed by extension of oligo dT primers using M-MLV reverse
transcriptase (New England Biolabs, Inc., Ipswich, MA, USA),
according to the manufacturer's protocol. qPCR was performed on a
Bio-Rad IQ5 cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
using SYBR-Green Master Mix (Takara Biotechnology, Co., Ltd.,
Dalian, China). The primers used were as follows: Human sirt1, CAG
TGG CTG GAA CAG TGA GA (forward) and TCT GGC ATG TCC CAC TAT CA
(reverse); human GAPDH, GAG TCA ACG GAT TTG GTC GT (forward) and
GAC AAG CTT CCC GTT CTC AG (reverse); mouse sirt1, GTA AGC GGC TTG
AGG G (forward) and TTC GGG CCT CTC CGT A (reverse); mouse GAPDH,
CGT CCC GTA GAC AAA ATG GT (forward) and GAA TTT GCC GTG AGT GGA GT
(reverse). The program was as follows: 95°C for 2 min, followed by
30 cycles of denaturation at 95°C for 5 sec, and
annealing/extension temperature (55/68°C) for 1 min. GAPDH was used
as reference gene and the method of 2−ΔΔCq was applied
for quantification (13).
Chromatin immunoprecipitation
(ChIP)
K562 and 293A cells were treated with 1%
formaldehyde for 15 min at room temperature. Cross-linked chromatin
was sheared by sonication for 5 cycles (30 sec on/30 sec off) at
4°C to 500–1,000 bp and then used for IP with anti-cMyc antibodies
as previously described (dilution, 1:100) or non-immune rabbit
immunoglobulin G (dilution, 1:500; cat. no., 2729S; Cell Signaling
Technology, Inc., Danvers, MA, USA) as a negative control.
Immunoprecipitated complexes were collected by protein A Sepharose
beads (Upstate Biotechnology, Inc., Lake Placid, NY, USA). The
immunoprecipitated chromatin was amplified by PCR with DNA
polymerase (Takara Biotechnology, Co., Ltd.) using primers for the
human telomerase reverse transcriptase (hTERT) or Sirt1 promoter.
The primers were as follows: hTERT, 5′-AGGCCGGGCTCCCAGTGGATTC-3′
and 5′-CGTGGCCAGCGGCAGCACCTC-3′; Sirt1 E2/3,
5′-GGAGCGGTAGACGCAACA-3′ and 5′-CTTCCAACTGCCTCTCTGG-3′; Sirt1 E1:
5′-AGGCCAAGTCATTTCCTTCC-3′ and 5′-ACCTTTGACGTGGAGGTTTG-3′. The
program was as follows: 95°C for 5 min, followed by 30 cycles of
denaturation at 95°C for 5 sec, annealing/extension temperature
(55/68°C) for 1 min, and a final extension of 68°C for 10 min. The
resulting PCR products were separated by 2% agarose gel
electrophoresis.
DNA mediated precipitation
The DNA-protein complex was cross-linked and
chromatin DNA was sheared by sonication as previously described. A
total of 4 µg biotinylated double strand oligonucleotide from human
Sirt1 promoter 222
(5′-CCCAGGCGGAGCGGTAGACGCAACAGCCTCCGCCCGCCACGTGACCCGTA-3′) or the
control 276
(5′-CGCCACAAAGAGGAAGGGCCGCCGGCCGCCGGGGCCGAGTGCGCTTCCAG-3′) were
incubated with HeLa cell lysate (Abcam, Cambridge, MA, USA) and 30
µl streptavidin agarose beads (BioVision, Inc., Milpitas, CA, USA)
slurry at 4°C for 2 h with agitation. The streptavidin agarose
beads specifically bind to the DNA probe. The mixture was pelleted
by centrifugation at 700 × g at 4°C for 10 min and washed with PBS
three times. The bound proteins were analyzed by subsequent western
blotting.
Western blotting
Cells were lysed with radioimmunoprecipitation assay
buffer which containing a mixture of protease inhibitors (Santa
Cruz Biotechnology, Inc.). Bicinchoninic acid protein assay kit
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to determine
the protein concentration of the cells. A total of 40 µg of protein
was electrophoresed and separated by 10% SDS-PAGE. The protein was
transferred to a polyvinylidene fluoride (PVDF) membrane
(Invitrogen; Thermo Fisher Scientific, Inc.). Subsequent to
blocking the PVDF membrane using blocking buffer for 1 h at 37°C,
the blots were incubated with the anti-c-Myc (dilution, 1:100),
anti-Sirt1 (dilution, 1:200), anti-p53 (dilution, 1:500) and
anti-GAPDH (dilution, 1:500) antibodies as previously described, at
4°C for 12 h. The PVDF membrane was then washed with TBS-Tween-20
buffer and incubated with horseradish peroxidase-conjugated
anti-mouse IgG (dilution, 1:100; cat. no. sc-358914; Santa Cruz
Biotechnology, Inc.) for 1 h at 37°C. An enhanced chemiluminescence
kit (Beyotime Institute of Biotechnology, Haimen, China) and gel
imaging system (Shanghai Furi Science and Technology Co., Ltd.,
Shanghai, China) were used for immune complex detection, according
to the manufacturer's protocol. GAPDH was used as the internal
control protein. The bands were analyzed by densitometry using
Image J software version 1.47 (National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
All the statistical analyses were performed using
SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). All analyses were
repeated three times and the data are expressed as the mean ±
standard deviation. The comparison between the two groups was
performed using Student's t-test. The comparison between the three
groups was based on one-way analysis of variance. When the
difference was significant, the SNK-q test method was used to
compare between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
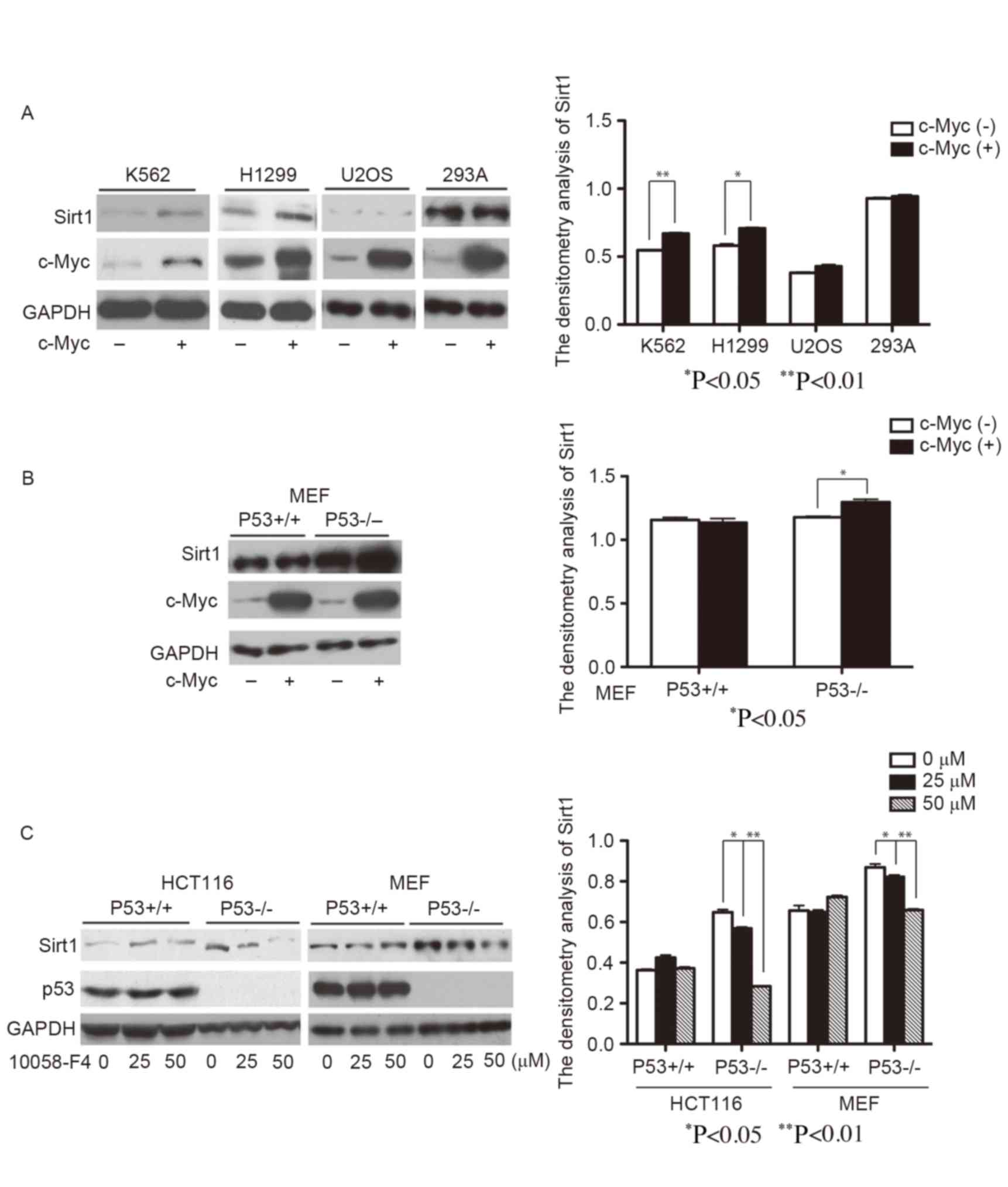
c-Myc upregulates the protein
expression level of Sirt1 in p53 deficient cells
In order to determine the effect of c-Myc on Sirt1
expression level, c-Myc was overexpressed in various cells lines,
including K562, H1299, U2OS and 293A. Overexpression of c-Myc
significantly increased the protein expression level of Sirt1 in
K562 and H1299 cells compared with the control (Fig. 1A; P<0.05); however, an increase of
Sirt1 protein expression level was not observed accompanying c-Myc
overexpression in U2OS and 293A cells. K562 and H1299 cells are p53
deficient, whereas in U2OS and 293A cells, p53 is expressed
(14–16). This implied the involvement of p53 in
the c-Myc regulation of Sirt1 expression level.
To further confirm the roles of p53 and c-Myc on
Sirt1 expression regulation, c-Myc was overexpressed in p53 wild
type and knockout MEF/3T3 cells. In agreement with a previous study
(17), the protein expression level
of Sirt1 was markedly increased in p53 knockout MEF/3T3s compared
with wild-type MEF/3T3s (Fig. 1B). As
expected, enforced expression of c-Myc significantly upregulated
the protein expression level of Sirt1 in p53−/− MEF/3T3
cells (P<0.05), but had no effects on Sirt1 expression level in
wild type MEF/3T3 cells (Fig. 1B). In
addition, c-Myc inhibitor 10058-F4 treatment largely reduced Sirt1
protein expression level in p53−/− MEF/3T3 and
p53−/− HCT116 cells (P<0.05 and P<0.01 with 25 and
50 µM 10058-F4, respectively, for the two cell lines) but not in
p53+/+ MEF/3T3 and p53+/+ HCT116 cells
(Fig. 1C). The aforementioned results
suggest that c-Myc may function as a positive regulator of Sirt1
expression level, whereas p53 inhibited regulation.
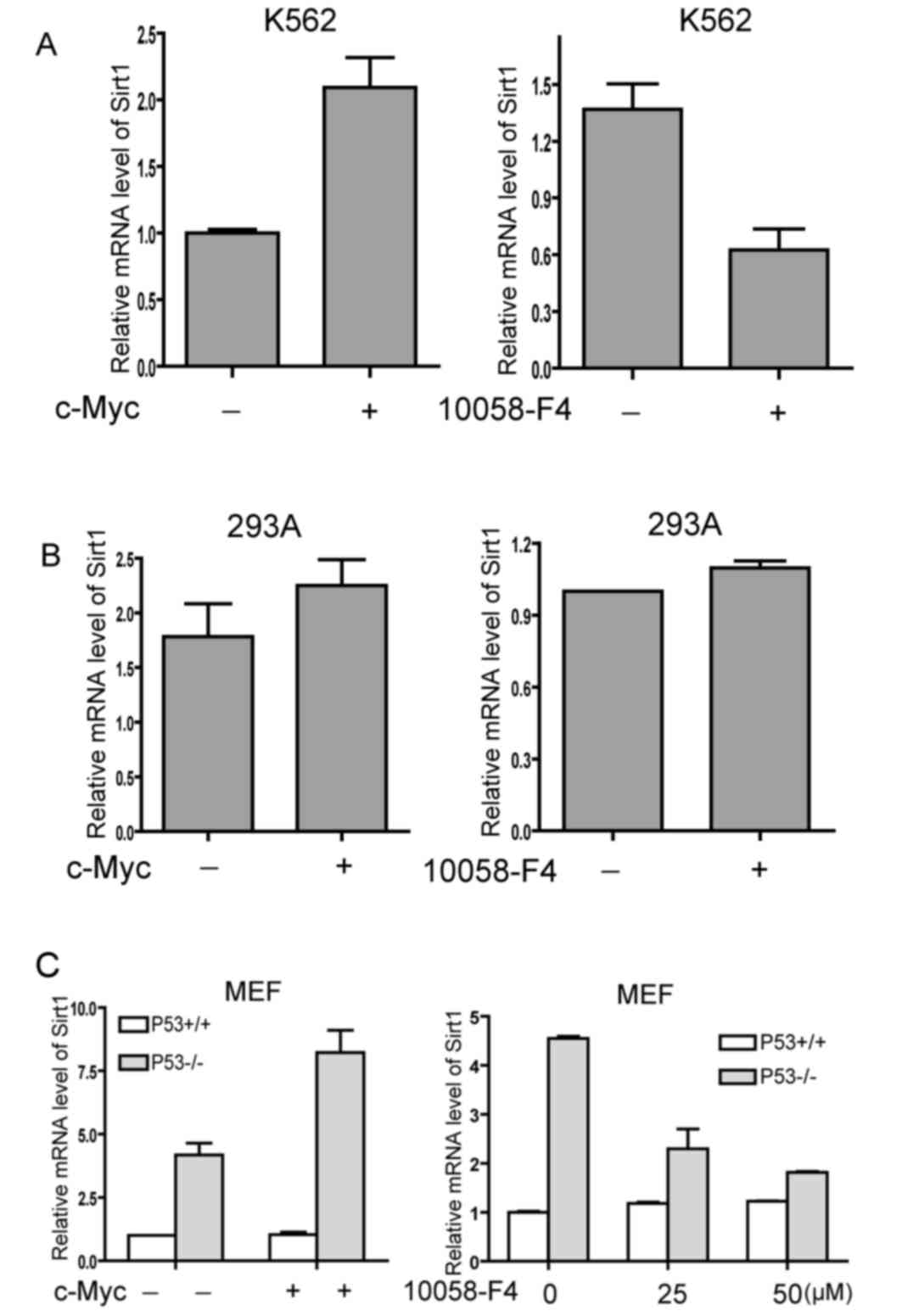
c-Myc upregulates the mRNA expression
level of Sirt1 in p53 deficient cells
To further explore the regulatory effect of c-Myc on
Sirt1 transcription and the underlying mechanism of p53 involvement
in c-Myc upregulation to Sirt1, the change of Sirt1 mRNA expression
level was examined upon experimental modulation of c-Myc activity
in p53 wild type or deficient cells by qPCR. Overexpression of
c-Myc resulted in an increased mRNA expression level of Sirt1 in
p53 deficient K562 cells (Fig. 2A,
left panel). Consistently, 10058-F4 treatment induced
downregulation of Sirt1 mRNA expression level in K562 cells (right
panel). Conversely, neither overexpression of c-Myc or 10058-F4
treatment altered the mRNA expression level of Sirt1 in 293A cells,
which express p53 (Fig. 2B).
Subsequently, the present study detected the mRNA expression level
of Sirt1 in primary MEF/3T3 cells from p53 wild type or knockout
mice. The mRNA expression level of Sirt1 was significantly elevated
in p53−/− MEF/3T3s and overexpression of c-Myc induced a
further increase of Sirt1 mRNA expression level in the absence of
p53 (Fig. 2C, left panel).
Furthermore, 10058-F4 impaired the mRNA expression level of Sirt1
in p53 knockout, but not in p53 wild type MEF/3T3 (Fig. 2C, right panel). The results indicated
that p53 may affect the transactivation ability of c-Myc on the
Sirt1 promoter.
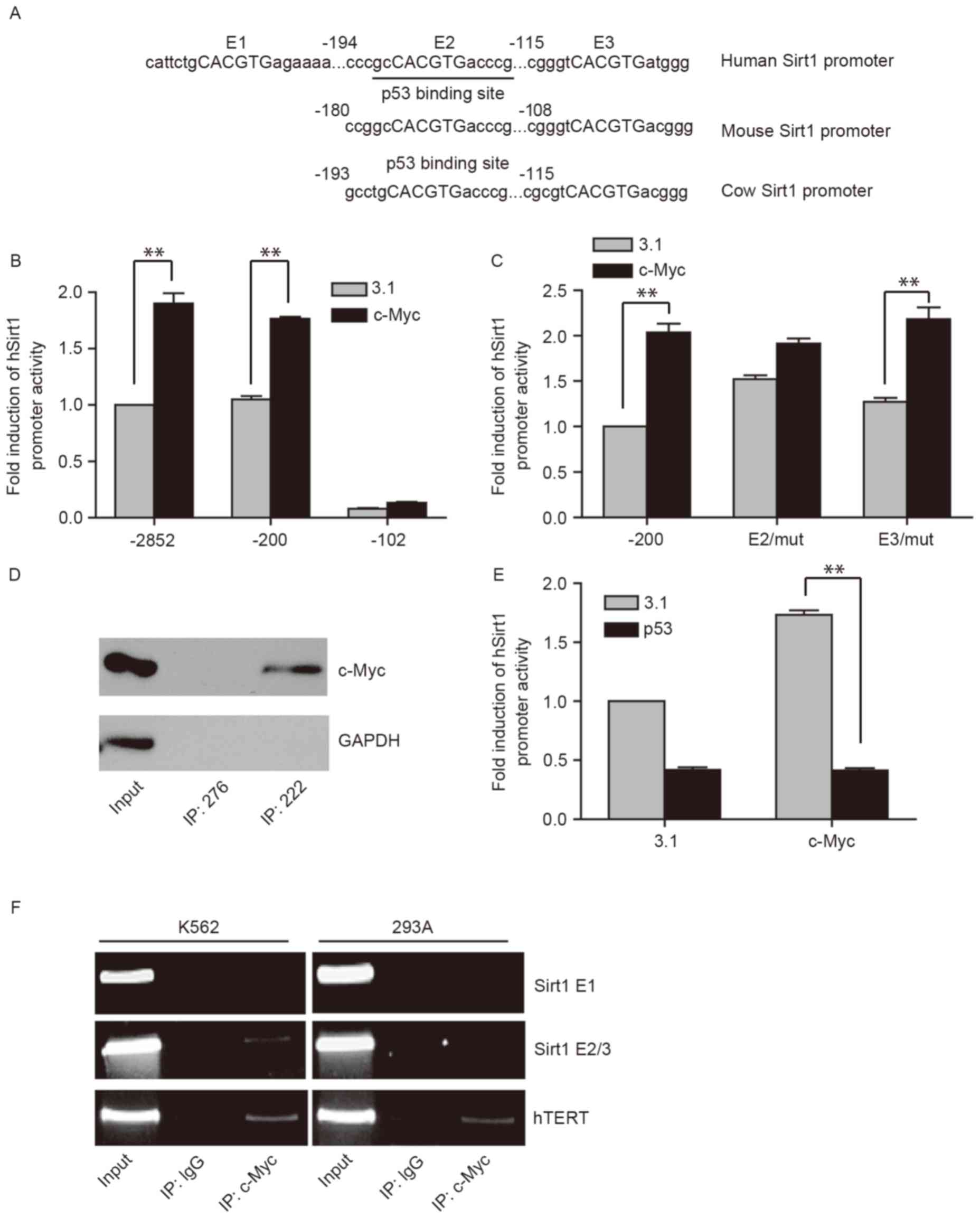
p53 inhibits the binding of c-Myc on
the Sirt1 promoter
To further elucidate the mechanism underlying
p53-mediated inhibition of the transactivation ability of c-Myc on
Sirt1, the present study first predicted putative c-Myc binding
elements in the human Sirt1 promoter using an online program
(rVISTA2.0). A total of three potential c-Myc binding elements,
consensus sequence CACGTG, were identified in the human Sirt1
promoter 5′-flanking region, and were named E1, E2 and E3,
respectively (Fig. 3A). However, E1
was not conserved across species when comparing the human with
mouse and cow counterparts. In contrast, E2 and E3 are highly
conserved among various types of species. The present study
hypothesized that E2 and E3, but not E1, were the response elements
for c-Myc.
To verify this possibility, the dual luciferase
assay was performed to evaluate the activity of truncated deletion
constructs of the Sirt1 promoter, −2852, −200 and −102. Truncation
of the promoter to −102 significantly decreased the baseline
activity of the promoter in the control group compared with the
full length Sirt1 promoter (−2852; Fig.
3B) whereas truncation to −200 did not, indicating that the
proximal region of the promoter (−200 to −102) is important for
promoter activity. Overexpression of c-Myc significantly
upregulated full length and −200 truncated Sirt1 promoter activity
up to 2-fold compared with the control (Fig. 3B; P<0.05); however, deletion of the
proximal region (−200 to −102) abolished c-Myc dependent
transactivation (Fig. 3B). These
results suggested that c-Myc transactivates the Sirt1 promoter via
E2 or E3 but not E1.
To further define the exact response element of
c-Myc on human Sirt1 promoter, E2 or E3 point mutants of the Sirt1
promoter were generated, where the individual c-Myc binding sites
were mutated. As presented in Fig. 3C
(P<0.05), the point mutation of E2 markedly reduced the
induction of Sirt1 promoter activity by c-Myc compared with the
truncated wild-type Sirt1 promoter −200, whereas E3/mut was
activated by c-Myc to a similar level compared with the truncated
wild-type Sirt1 promoter −200. The results suggested that E2 may be
the response element for c-Myc.
Subsequently, the results were confirmed by DNA
mediated precipitation assay. Biotinylated double strand
oligonucleotide 222 (Sirt1 promoter sequence containing E2) or 276
(50 bp upstream of 222, used as the negative control) were
incubated with HeLa nuclear extracts and streptavidin agarose beads
slurry and the precipitates were analyzed by western blotting. As
presented in Fig. 3D, c-Myc bound to
the probe 222 but not 276, whereas the negative control GAPDH did
not bind to the two probes. This further demonstrated that E2 was
the binding site of c-Myc.
Of note, the E2 site forms part of the element
previously identified to respond to p53 (10). The overlapping two elements may serve
a role in the differential regulatory effects of c-Myc on Sirt1 in
the absence or presence of p53. In the present study, c-Myc and p53
expressing plasmids (−200) were co-transfected together with Sirt1
promoter reporter plasmids into p53 deficient H1299 cells. As
presented in Fig. 3E, overexpression
of p53 decreased the Sirt1 promoter activity and abolished the
c-Myc-dependent activation of the promoter, which suggested that
c-Myc-dependent upregulation of Sirt1 was inhibited by p53 at the
transcriptional level.
Based on the aforementioned results, it was
hypothesized that p53 and c-Myc share ≥1 response element on the
Sirt1 promoter. The binding of p53 may block the recruitment of
c-Myc onto its response element. Therefore, a ChIP assay was
performed with K562 and 293A cells. The results revealed that the
Sirt1 promoter fragment containing E2 co-precipitated with
endogenous c-Myc in p53 deficient K562 cells but not in 293A cells
expressing p53 (Fig. 3F). In
agreement with the luciferase assay results, E1 was not present in
the DNA precipitate of c-Myc in K562 or 293A cells. The hTERT
promoter was amplified as a positive control. Taken together, these
results suggested that p53 inhibited the binding of c-Myc on the
Sirt1 promoter, thus blocking the upregulation of Sirt1 expression
level mediated by c-Myc.
Discussion
Previous studies have revealed that Sirt1 expression
is tightly regulated by certain transcriptional factors, including
p53, HIC1 and E2F1 (2,10,11). The
present study demonstrated that Sirt1 transcription was induced by
c-Myc when p53 was deficient. The response element for c-Myc
induction was co-located with the p53 binding site of the Sirt1
promoter. Therefore, p53 inhibited the upregulation of Sirt1
expression mediated by c-Myc by blocking the binding of c-Myc on
the Sirt1 promoter.
Tumor suppressor p53 serves a critical role in
suppression of cell growth and proliferation, whereas the
oncoprotein c-Myc conversely regulates p53-associated physiological
and pathological processes. It is known that there is a negative
association between p53 and c-Myc (18). However, the precise mechanism
underlying the repression of p53 and c-Myc has not been fully
understood. Early reports suggested that p53 directly suppressed
the transcription of c-Myc (19),
whereas c-Myc inhibited p53 expression level via p19ARF, which
forms a feedback loop (20). Another
post-transcriptional mechanism underlying p53 repression of c-Myc
involves microRNA (miR) −145 (21).
p53 induced miR-145 expression and subsequently repressed c-Myc at
the protein level (21). The results
of the present study suggest a new mechanism underlying p53
repression of c-Myc function; p53 and c-Myc compete for binding to
the same response element, and inversely regulate the expression
level of Sirt1. p53 appears to have a stronger affinity to the
binding site since the present study didn't detect the binding of
c-Myc on the Sirt1 promoter in p53 expressing cells. It's
noteworthy that Sirt1 is not the first target identified to be
regulated by p53 and c-Myc in a similar manner. It is reported that
optimal induction of cyclinB1 promoter by c-Myc only occurs when
p53 is concurrently inactivated (22).
Deregulation of c-Myc oncogene or loss of p53
activity occurs frequently in certain types of human cancer
(23). The results of the present
study suggested that c-Myc overexpression affects the
transactivation ability of c-Myc on the Sirt1 promoter in p53
deficient cells, elevating the expression level of Sirt1. The
combination of c-Myc overexpression and p53 loss of function may
contribute to the elevated expression level of Sirt1 in tumor
cells, as cells with high expression levels of Sirt1, including
AML, squamous cell carcinoma and diffuse large B-cell lymphoma,
demonstrated c-Myc overexpression (24–26) and
p53 loss of function (27). The
regulation of Sirt1 expression level by c-Myc and p53 prompted the
present study to consider Sirt1 as a target to mediate the
oncogenic function of c-Myc.
Taken together, the results of the present study
demonstrated that Sirt1 expression level was directly but
conversely regulated by c-Myc and p53. p53 inhibited the activation
of the Sirt1 promoter by blocking c-Myc recruitment on the Sirt1
promoter. The present study revealed a novel network that regulates
the expression of Sirt1 and further elucidated the counterbalance
of a tumor suppressor and promoter.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81402319) and the
Beijing Nova Program (grant no. Z161100004916133).
References
|
1
|
Liu TF and McCall CE: Deacetylation by
SIRT1 reprograms inflammation and cancer. Genes Cancer. 4:135–147.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang C, Chen L, Hou X, Li Z, Kabra N, Ma
Y, Nemoto S, Finkel T, Gu W, Cress WD and Chen J: Interactions
between E2F1 and SirT1 regulate apoptotic response to DNA damage.
Nat Cell Biol. 8:1025–1031. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
McCall CE, El Gazzar M, Liu T,
Vachharajani V and Yoza B: Epigenetics, bioenergetics, andmicroRNA
coordinate gene-specific reprogramming during acute systemic
inflammation. J Leukoc Biol. 90:439–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hida Y, Kubo Y, Murao K and Arase S:
Strong expression of a longevity-related protein, SIRT1, in Bowen's
disease. Arch Dermatol Res. 299:103–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huffman DM, Grizzle WE, Bamman MM, Kim JS,
Eltoum IA, Elgavish A and Nagy TR: SIRT1 is significantly elevated
in mouse and human prostate cancer. Cancer Res. 67:6612–6618. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hennig D, Müller S, Wichmann C, Drube S,
Pietschmann K, Pelzl L, Grez M, Bug G, Heinzel T and Krämer OH:
Antagonism between granulocytic maturation and deacetylase
inhibitor-induced apoptosis in acute promyelocytic leukaemia cells.
Br J Cancer. 112:329–337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jang KY, Hwang SH, Kwon KS, Kim KR, Choi
HN, Lee NR, Kwak JY, Park BH, Park HS, Chung MJ, et al: SIRT1
expression is associated with poor prognosis of diffuse large
B-cell lymphoma. Am J Surg Pathol. 32:1523–1531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shah ZH, Ahmed SU, Ford JR, Allison SJ,
Knight JR and Milner J: A deacetylase-deficient SIRT1 variant
opposes full-length SIRT1 in regulating tumor suppressor p53 and
governs expression of cancer-related genes. Mol Cell Biol.
32:704–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vahtola E, Louhelainen M, Forstén H,
Merasto S, Raivio J, Kaheinen P, Kytö V, Tikkanen I, Levijoki J and
Mervaala E: Sirtuin1-p53, forkhead box O3a, p38 and post-infarct
cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat.
Cardiovasc Diabetol. 9:52010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nemoto S, Fergusson MM and Finkel T:
Nutrient availability regulates SIRT1 through a forkhead-dependent
pathway. Science. 306:2105–2108. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen WY, Wang DH, Yen RC, Luo J, Gu W and
Baylin SB: Tumor suppressor HIC1 directly regulates SIRT1 to
modulate p53-dependent DNA-damage responses. Cell. 123:437–448.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao B, Zhao G, Lv X, Chen HZ, Xue Z, Yang
B, Liu DP and Liang CC: Sirt1 deacetylates c-Myc and promotes
c-Myc/Max association. Int J Biochem Cell Biol. 43:1573–1581. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamaguchi H, Woods NT, Piluso LG, Lee HH,
Chen J, Bhalla KN, Monteiro A, Liu X, Hung MC and Wang HG: P53
acetylation is crucial for its transcription-independent
proapoptotic functions. J Biol Chem. 284:11171–11183. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rothmann T, Hengstermann A, Whitaker NJ,
Scheffner M and zur Hausen H: Replication of ONYX-015, a potential
anticancer adenovirus, is independent of p53 status in tumor cells.
J Virol. 72:9470–9478. 1998.PubMed/NCBI
|
|
16
|
D'Orazi G, Cecchinelli B, Bruno T, Manni
I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal
G, et al: Homeodomain-interacting protein kinase-2 phosphorylates
p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 4:11–19. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang S, Song P and Zou MH: Inhibition of
AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates
genotoxic stress and cell death in mouse embryonic fibroblasts and
cardiomyocytes: Role of p53 and SIRT1. J Biol Chem. 287:8001–8012.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie Y, Bulbul MA, Ji L, Inouye CM, Groshen
SG, Tulpule A, O'Malley DP, Wang E and Siddiqi IN: P53 expression
is a strong marker of inferior survival in de novo diffuse large
B-cell lymphoma and may have enhanced negative effect with MYC
coexpression: A single institutional clinicopathologic study. Am J
Clin Pathol. 141:593–604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ho JS, Ma W, Mao DY and Benchimol S:
P53-dependent transcriptional repression of c-myc is required for
G1 cell cycle arrest. Mol Cell Biol. 25:7423–7431. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ta VB, de Bruijn MJ, ter Brugge PJ, van
Hamburg JP, Diepstraten HJ, van Loo PF, Kersseboom R and Hendriks
RW: Malignant transformation of Slp65-deficient pre-B cells
involves disruption of the Arf-Mdm2-p53 tumor suppressor pathway.
Blood. 115:1385–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sachdeva M, Zhu S, Wu F, Wu H, Walia V,
Kumar S, Elble R, Watabe K and Mo YY: P53 represses c-Myc through
induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA.
106:3207–3212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Molinuevo R, Freije A, de Pedro I, Stoll
SW, Elder JT and Gandarillas A: FOXM1 allows human keratinocytes to
bypass the oncogene-induced differentiation checkpoint in response
to gain of MYC or loss of p53. Oncogene. 36:956–965. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tanaka H, Tamura A, Sekai M, Hamazaki Y
and Minato N: Increased c-Myc activity and DNA damage in
hematopoietic progenitors precede myeloproliferative disease in
Spa-1-deficiency. Cancer Sci. 102:784–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Osdal T, Ho Y, Chun S, McDonald T,
Agarwal P, Lin A, Chu S, Qi J, Li L, et al: SIRT1 activation by a
c-MYC oncogenic network promotes the maintenance and drug
resistance of human FLT3-ITD acute myeloid leukemia stem cells.
Cell Stem Cell. 15:431–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Savage KJ, Johnson NA, Ben-Neriah S,
Connors JM, Sehn LH, Farinha P, Horsman DE and Gascoyne RD: MYC
gene rearrangements are associated with a poor prognosis in diffuse
large B-cell lymphoma patients treated with R-CHOP chemotherapy.
Blood. 114:3533–3537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Y, Gong LP, Dong XL and Liu HG:
Detection of C-MYC oncogene translocation and copy number change in
the normal-dysplasia-carcinoma sequence of the larynx by
fluorescence in situ hybridization. Diagn Cytopathol. 41:515–519.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Balz V, Scheckenbach K, Götte K, Bockmühl
U, Petersen I and Bier H: Is the p53 inactivation frequency in
squamous cell carcinomas of the head and neck underestimated?
Analysis of p53 exons 2–11 and human papillomavirus 16/18 E6
transcripts in 123 unselected tumor specimens. Cancer Res.
63:1188–1191. 2003.PubMed/NCBI
|