Introduction
Breast cancer is the most common type of malignancy,
and the leading cause of cancer-associated female mortality, in a
number of countries (1,2) Chemotherapy with or without resection of
the tumor is the only known treatment strategy for long-term
survival, and survival is limited with standard chemotherapeutic
options (3). Breast cancer cells
exhibit intrinsic and acquired resistance to numerous anticancer
drugs (4), this is a major problem
for the effective treatment of breast cancer (5). Improved treatment protocols and
alternative chemotherapeutic strategies are therefore required.
Statins competitively inhibit
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the
rate-limiting enzyme in cholesterol biosynthesis (6), and are used to treat hyperlipidemia by
reducing serum lipids such as cholesterol and triglycerides
(7,8).
This activity, combined with their protective effects on the blood
vessels and heart, allows statins to be used in the treatment and
prevention of cardiovascular events (9,10). In
addition, statins perform roles in immune regulation (11), the inhibition of inflammation
(12) and the modulation of
angiogenesis (13). Statins also
exhibit anticancer activities (14,15),
decrease cellular proliferation (15–17) and
induce apoptosis (15,18,19) in
breast, colorectal, lung, prostate and pancreatic cancer (20). Notably, statins inhibit cancer cell
growth in vivo and decrease metastasis at clinically
therapeutic doses (21).
The suppression of HMG-CoA reductase results in the
reduction of several important cholesterol intermediates, including
mevalonate, geranylgeranyl pyrophosphate (GGPP) and farnesyl
pyrophosphate (FPP) (22). These
proteins are necessary for the post-translational modification of
intracellular G-proteins, including Rho, ras-related C3 botulinum
toxin substrate (Rac) and Ras, which regulate cellular mechanisms
including cytoskeletal reorganization and cellular transformation,
migration, invasion and proliferation (23). Statins activate cancer cell death,
including apoptosis, via a reduction of GGPP and FPP levels.
Additionally, Ras-related C3 botulinum toxin substrate 1 (RAC1)
regulates and modulates several signaling pathways that control
cellular proliferation (24), and
direct inhibition of RAC1 activity or gene expression induces cell
cycle arrest and apoptosis in breast cancer cells (25).
In clinical studies, the role of statins in cancer
treatment remains debatable, and appears to be dependent on the
molecular identity of the type of cancer. In order to better
understand the interaction between statins and breast cancer cells,
the ability of simvastatin to potentiate the doxorubicin-induced
inhibition of cellular proliferation and apoptosis using the MCF-7
cancer cell line was investigated. It was hypothesized that
simvastatin sensitizes MCF-7 cells to doxorubicin, and may be a
viable strategy for improving the efficacy of other anticancer
drugs against breast cancer.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum and other cell culture reagents were purchased from
Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Simvastatin (cat. no. s6196), doxorubicin (cat. no. D1515),
protease inhibitor cocktail (cat. no. P8340), dihydroethidium (DHE;
cat. no. D7008), radioimmunoprecipitation assay (RIPA) lysis buffer
(cat. no. R0278), sulforhodamine B (SRB; cat. no. s1402) and a
caspase 3 activity assay kit (cat. no. CASP3F-1KT) were obtained
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The primary
antibodies against cyclin-dependent kinase inhibitor 1 (p21; cat.
no. 2947), caspase 3 (cat. no. 9662), cytochrome c (cat. no. 4272),
cyclin D1 (cat. no. 2922), β-actin (cat. no. 4967) and the
secondary anti-rabbit immunoglobulin G horseradish peroxidase
(HRP)-linked antibody (cat. no. 7074) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). iScript reverse
transcription Supermix for reverse transcription quantitative
polymerase chain reaction (RT-qPCR; cat. no. 170-8841) and SsoFast
EvaGreen Supermix (cat. no. 172-5200) were supplied by Bio-Rad
Laboratories, Inc. (Hercules, CA, USA).
Cell line and cell culture
The human breast cancer MCF-7 cell line was obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and maintained according to ATCC's recommendations at 37°C and 5%
CO2 in DMEM medium supplemented with 10% fetal bovine
serum, 100 U/ml penicillin G and 100 µg/ml streptomycin. The DMEM
media was renewed every 2–3 days, trypsinized with 0.25%
trypsin-EDTA and subcultured in the same media.
Cell viability assay
The SRB assay was used to determine the effect of
simvastatin and doxorubicin, alone and in combination, on the
viability of MCF-7 cells. A 96-well plate was seeded with
1×104 MCF-7 cells/well and incubated for 24 h at 37°C.
Subsequent to exposure to 0–100 µM simvastatin for 24–48 h, 0–10 µM
doxorubicin for 24–48 h and in combination (cells were treated with
0–100 µM simvastatin with or without 1 µM doxorubicin for 24 h) at
37°C, the cultured cells were fixed with ice-cold 10%
trichloroacetic acid and stained with 0.4% SRB for 30 min at room
temperature. Excess dye was removed by rinsing several times with
1% acetic acid, and protein-bound dye was dissolved with 200 µl 10
mM Tris base solution for the determination of absorbance with a
microplate reader with a filter wavelength of 540 nm.
Colony formation assay
Approximately 800 MCF-7 cells were seeded in 6-well
plates and allowed to grow for 24 h at 37°C. The cells were then
treated with 0–50 µM simvastatin and in combination of 0–50 µM
simvastatin with or without 0.5 µM doxorubicin treatment for 24 h
at 37°C. Following this, the cells were washed with PBS and fresh
medium was added. The cells were then grown for another 14 days.
Subsequently, the DMEM medium was discarded, the cells were washed
with PBS buffer three times, fixed with 100% methanol at −20°C for
1 h, stained with 0.5% crystal violet in 100% methanol for 1 h at
room temperature, washed with tap water, and the colonies were then
viewed and captured using a digital camera (Nikon D3100, Nikon
Corporation, Tokyo, Japan). Colonies containing >50 individual
cells were counted using Image-Pro Plus software version 2.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
Reactive oxygen species (ROS)
production assay
Intracellular ROS generation was measured using the
cell-permeable fluorescent probe, DHE. Black 96-well plates were
seeded with ~1×104 MCF-7 cells/well and incubated for 24
h at 37°C. The medium was discarded and the cells were washed with
PBS. The cells were then treated with 0–50 µM of simvastatin alone,
or 50 µM simvastatin in combination with 1 µM doxorubicin, for 90
min. The cells were then assessed for ROS production by incubation
with 25 µM DHE in serum-free medium, in a 5% CO2
atmosphere, at 37°C, for 90 min, in the dark. The fluorescence
intensity was measured at a 518 nm excitation and 605 nm emission
wavelength on a fluorescence microplate reader. The data were
expressed as the percentage of ROS relative to the untreated
controls.
Caspase 3 activity assay
Caspase 3 activity was measured using fluorimetric
assay kits (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's protocol. Subsequent to treatment with the test
compounds for 24 h, the medium was removed, the cells were
trypsinized and the cell pellet was lysed with cell lysis buffer on
ice for 10 min. The lysed pellet was then centrifuged (10,000 × g,
4°C, 30 min), and protein concentrations were measured with
Bradford's reagent (Bio-Rad Laboratories, Inc.), using albumin as a
standard. Briefly, 50 µl of cell protein or the albumin standard
was mixed with 200 µl Bradford reagent and incubated for 15 min at
room temperature in the dark. The absorbance at 620 nm was measured
with a spectrophotometer, and the protein concentration was
calculated using a standard. A total of 5 µl cell lysates (0.5
mg/ml) were added to 195 µl of buffer containing an
Ac-DEVD-7-amino-4-methylcoumarin (AMC)-conjugated substrate for
caspase (Sigma-Aldrich; Merck KGaA). This was followed by 90 min
incubation at 37°C in the dark. The concentration of the released
AMC was calculated from the fluorescence intensity, which was read
using a fluorescence plate reader with the excitation and emission
wavelengths of 360 and 460 nm, respectively, and using AMC standard
to calculate caspase 3 activity. Data were adjusted according to
the protein content.
Gene expression assay
The MCF-7 cells were seeded in 6 well-plates and
allowed to grow for 24 h. Cells were treated with the test
compounds, and RNA was isolated using TRIzol® reagent
according to the manufacturer's protocol (Sigma-Aldrich; Merck
KGaA). Recovered RNA was quantified by using a spectrophotometer to
measure the 260/280 nm absorbance ratio. Complementary DNA (cDNA; 1
µg) was prepared by reverse transcription of isolated RNA using the
iScript Reverse Transcription Supermix for RT-qPCR. PCR
amplification was performed using primers specific for RAC1, cdk2,
cdk4 and cdk6, and using β-actin (ACTB) as an internal control. The
PCR primer sequences were as follows: RAC1 (GenBank accession no.
NM_018890) forward, 5′ATG-TCC-GTG-CAA-AGT-GGT-ATC3′ and reverse,
5′CTC-GGA-TCG-CTT-CGT-CAA-ACA3′; Cdk2 (GenBank accession no.
NM_001798) forward, 5′CCA-GGA-GTT-ACT-TCT-ATG-CCT-GA3′ and reverse,
5′TTC-ATC-CAG-GGG-AGG-TAC-AAC3′; Cdk4 (GenBank accession no.
NM_000075) forward, 5′ATG-GCT-ACC-TCT-CGA-TAT-GAG-C3′ and reverse,
5′CAT-TGG-GGA-CTC-TCA-CAC-TCT3′; Cdk6 (GenBank accession no.
NM_001145306) forward, 5′GCT-GAC-CAG-CAG-TAC-GAA-TG3′ and reverse,
5′GCA-CAC-ATC-AAA-CAA-CCT-GAC-C3′; ACTB (GenBank accession no.
NM_001101) forward, 5′CAT-GTA-CGT-TGC-TAT-CCA-GGC3′ and reverse,
5′CTC-CTT-AAT-GTC-ACG-CAC-GAT3′.
qPCR was carried out in a final reaction volume of
20 µl containing SYBR Green PCR Master mix (Bio-Rad Laboratories,
Inc.), 0.5 µM of each target gene and the internal control. The
expression of each gene was monitored using an Applied
Biosystems® StepOne™ real-time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with the following
conditions: Denaturation at 95°C for 3 min, then amplification by
cycling 40 times at 95°C for 15 sec and 60°C for 30 sec. The
differences in gene expression levels were calculated using the
2−ΔΔCq method for relative quantification (26), and expressed as the fold change
relative to the untreated control. Data from 3 independent
experiments were normalized to the expression of ACTB mRNA, which
included on the same PCR array plate as the target genes.
Protein extraction and western blot
analysis
The MCF-7 cells were lyzed with RIPA lysis buffer
for 30 min on ice. The lysates were collected, and the protein
concentrations were determined using Bradford's reagent, as
described in the caspase 3 activity assay. A total of 20 µg protein
was separated by 12% SDS-PAGE, and transferred to an
Immobilon® polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The blots were blocked for 2 h at
room temperature with 5% (w/v) skimmed milk in Tris buffered saline
containing 0.1% Tween-20 (TBST). The membrane was probed with each
primary antibody at 4°C overnight (dilution, 1:1,000). Subsequent
to washing with TBST, the blots were incubated with the
HRP-conjugated secondary antibody for 2 h at room temperature
(dilution, 1:2,500). The immunoactive bands were detected using an
Enhanced Clarity™ Western enhanced chemiluminescence substrate
(Bio-Rad Laboratories, Inc.). Images of the specific protein bands
were captured and analyzed using the ImageQuant™ LAS-4000 and Image
Gauge version 3.1 (GE Healthcare Life Sciences, Chalfont, UK).
Statistical analysis
Statistical comparison between the control and
treatment groups was analyzed with an unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of simvastatin and doxorubicin
on MCF-7 cellular viability and colony formation efficacy
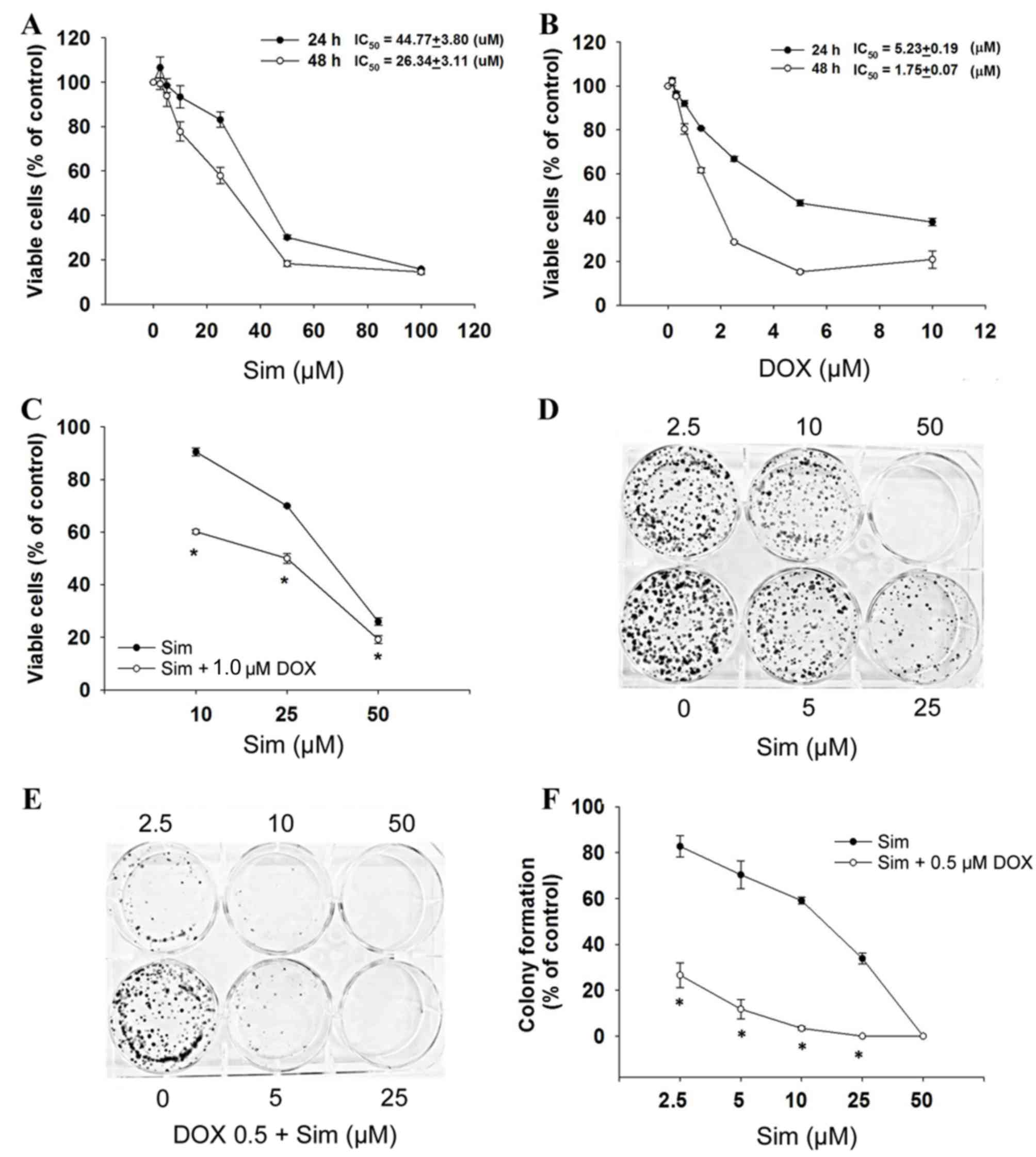
To evaluate the cytotoxicity of simvastatin with or
without doxorubicin, the MCF-7 cells were exposed to simvastatin
and doxorubicin and assessed for viability by the SRB assay. The
results demonstrated that cell growth was inhibited after 24 and 48
h of treatment, with simvastatin IC50 values of 44.8±3.8
µM at 24 h and 26.3±3.1 µM at 48 h (P<0.05, Fig. 1A), and doxorubicin IC50
values of 5.2±0.2 µM at 24 h and 1.8±0.1 µM at 48 h (P<0.05,
Fig. 1B) in a dose- and
time-dependent manner. The combination of simvastatin and
doxorubicin significantly enhanced cytotoxicity (Fig. 1C).
To determine the effect of simvastatin and
doxorubicin on the longer-term viability and replicative potential
of the MCF-7 cells, a colony formation assay was used. Treatment
with simvastatin alone caused a dose-dependent decrease in the
colony forming ability of the MCF-7 cells (Fig. 1D). When simvastatin and doxorubicin
were used in combination, colony formation was significantly
reduced compared with simvastatin treatment alone (Fig. 1E and F; P<0.05). These results
indicated that simvastatin enhances the activity of doxorubicin in
breast cancer cells, and prompted the investigation of the
mechanism(s) by which this increase in activity occurs.
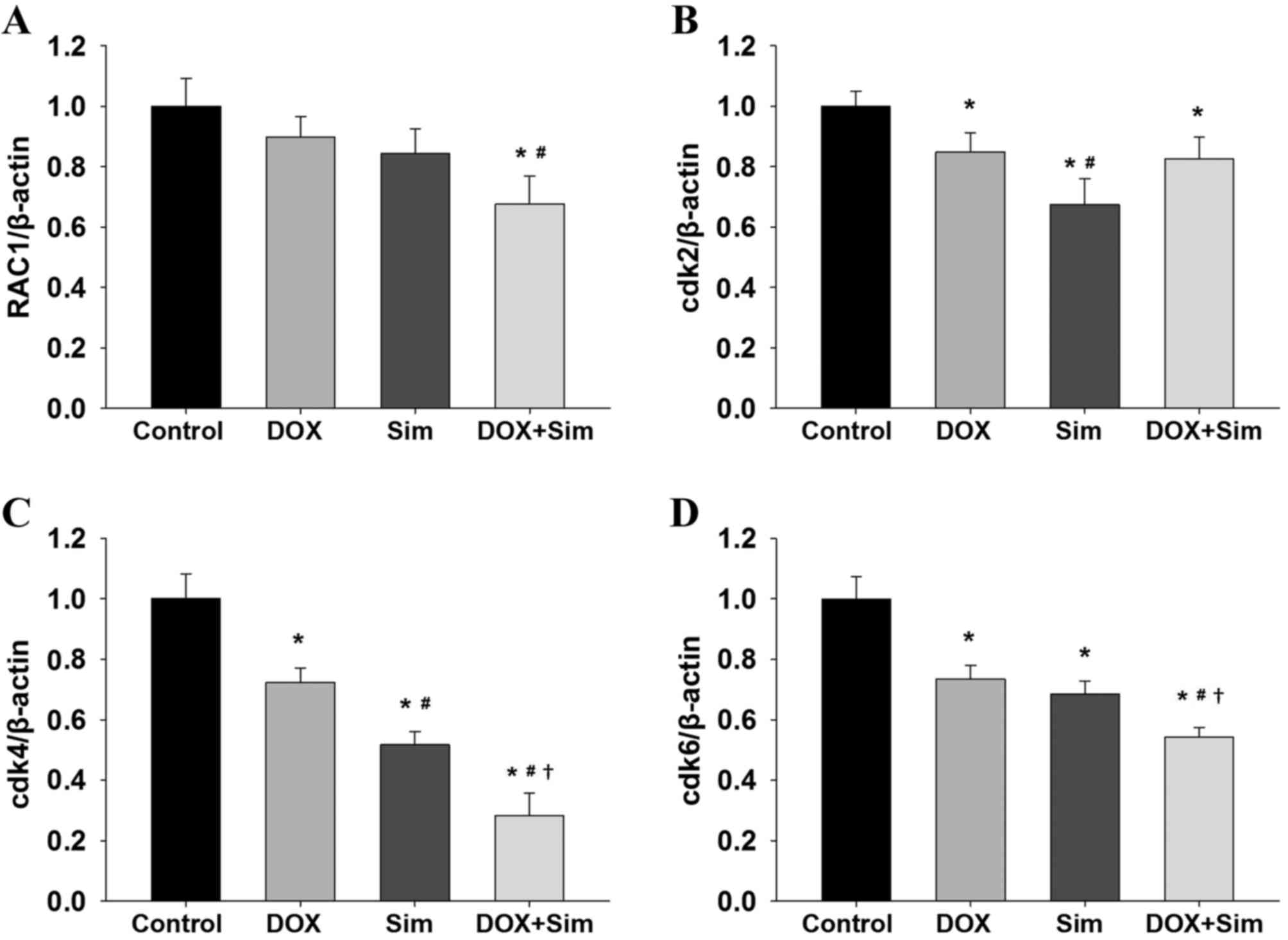
Effects of simvastatin and doxorubicin
on RAC1 and downstream gene expression
To investigate whether simvastatin enhances the
effects of doxorubicin on the cell cycle regulator RAC1, mRNA
expression of RAC1, cdk2, cdk4 and cdk6 were measured. The
treatment of cells with a combination of simvastatin and
doxorubicin decreased RAC1 mRNA expression significantly more than
treatment with simvastatin or doxorubicin alone (Fig. 2A; P<0.05). Cdk2 mRNA expression was
reduced by simvastatin and doxorubicin individually; however, there
was no additive effect when the two were used in combination
(Fig. 2B; P<0.05). Notably, it was
observed that simvastatin or doxorubicin treatment significantly
reduced cdk4 and cdk6, and that the combination treatment exhibited
an additive effect when compared with simvastatin or doxorubicin
treatment alone (Fig. 2C and D;
P<0.05).
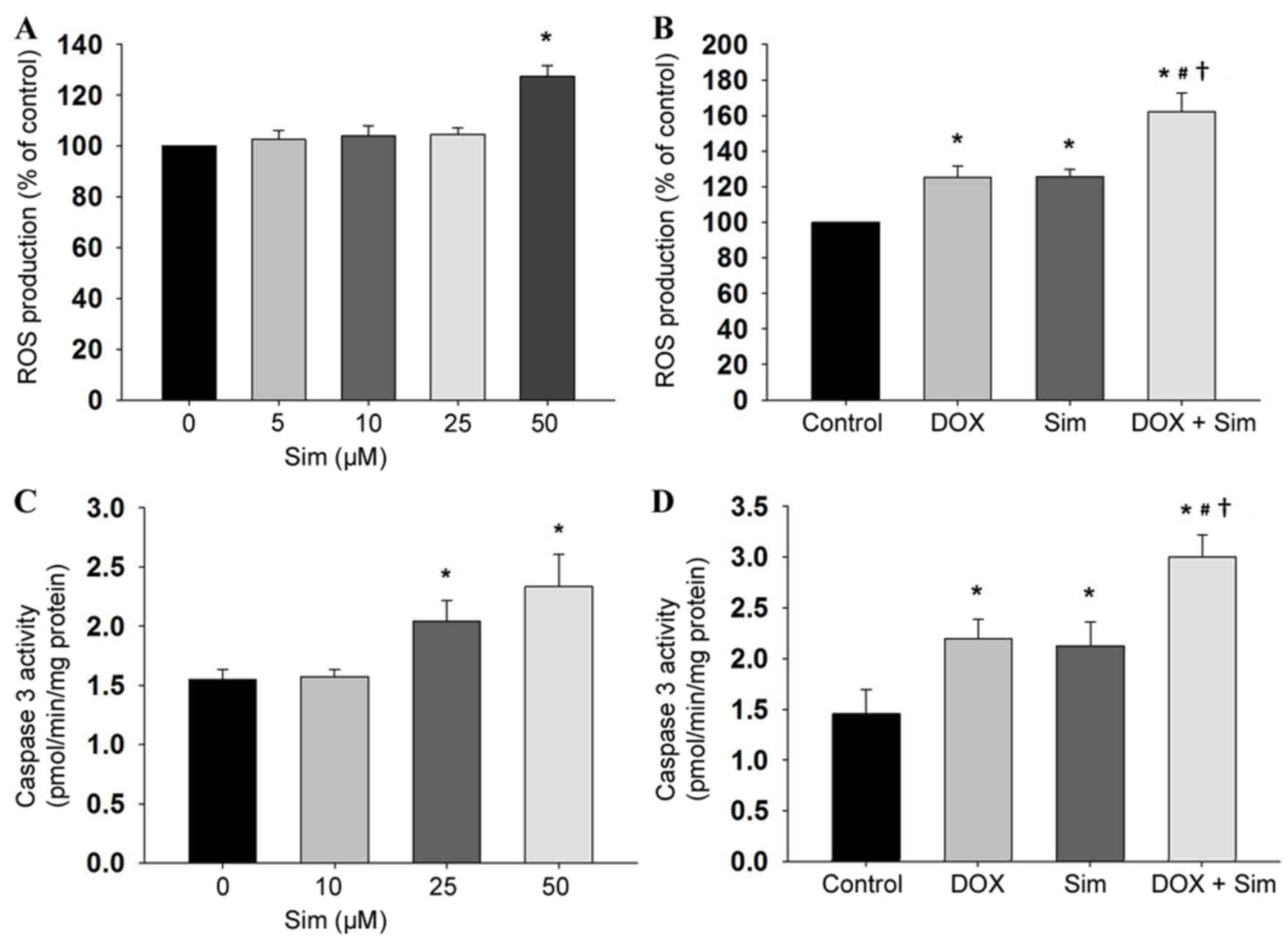
Effects of simvastatin and doxorubicin
on ROS and caspase 3 activity
To establish the mechanism of action by which
simvastatin sensitizes MCF-7 cells to doxorubicin, the
intracellular accumulation of ROS was also monitored by the
DHE-enhanced chemiluminescence method. Simvastatin induced
intracellular ROS production in the MCF-7 cells (Fig. 3A, P<0.05), and cells treated with
simvastatin in combination with doxorubicin demonstrated higher ROS
production compared with those treated with simvastatin or
doxorubicin alone (Fig. 3B,
P<0.05). The involvement of mitochondria in simvastatin- and
doxorubicin-induced cytotoxicity was investigated by measuring the
extent of caspase 3 activity. The results revealed that treatment
with simvastatin increased caspase 3 activity (Fig. 3C, P<0.05), and that the combination
of simvastatin and doxorubicin increased caspase 3 activity, to a
greater extent than treatment with simvastatin alone (Fig. 3D, P<0.05).
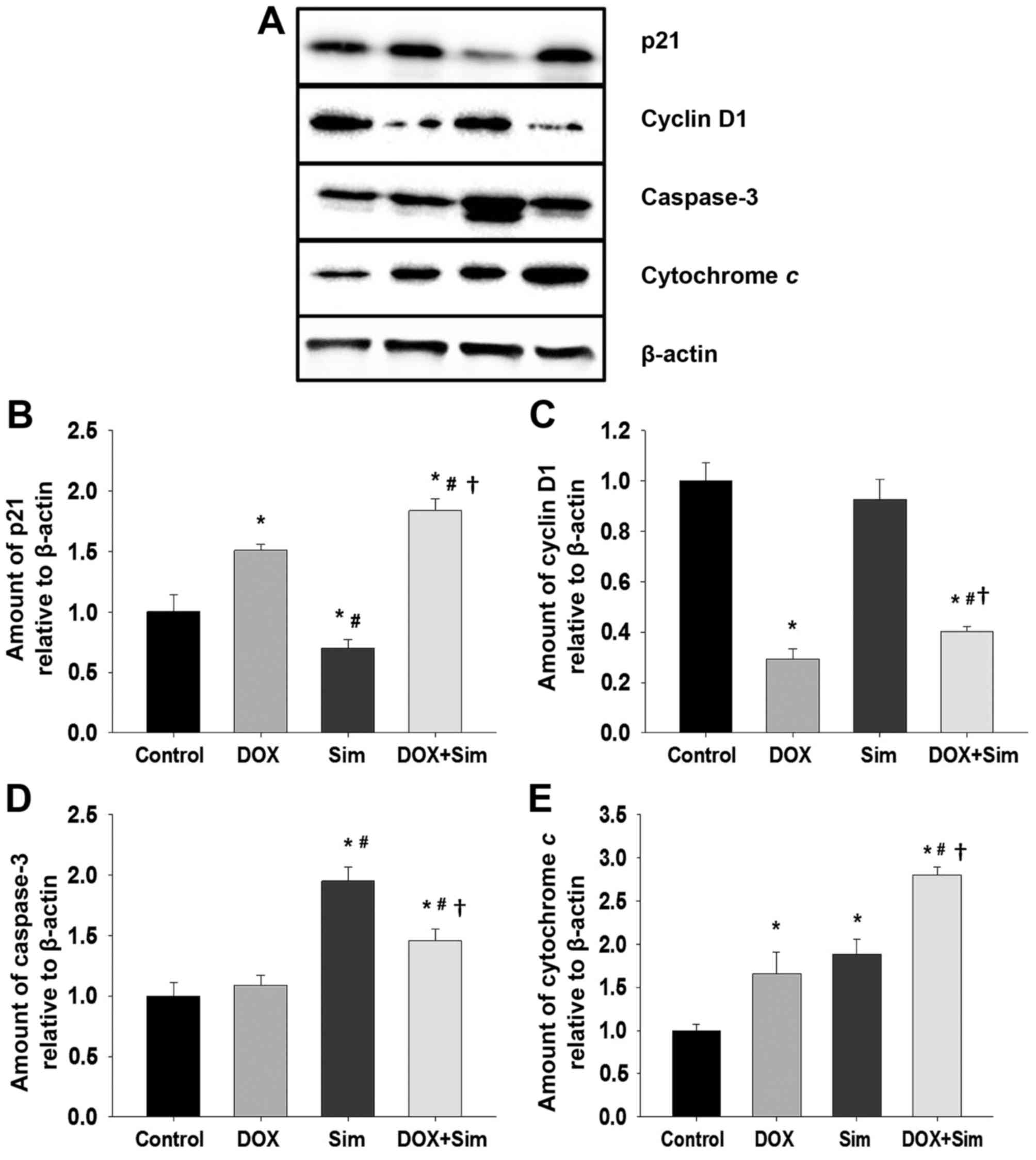
Effects of simvastatin and doxorubicin
on protein-related apoptosis
To understand how simvastatin may enhance
doxorubicin cytotoxicity in MCF-7 cells, the level of proteins
associated with cell survival and apoptosis, including p21, cyclin
D1, caspase 3 and cytochrome c, were assessed with western blotting
(Fig. 4A-E).
The results revealed that the level of p21 was
significantly altered in all treatment groups; p21 was increased
following treatment with doxorubicin, decreased following treatment
with simvastatin and increased by the combination of doxorubicin
with simvastatin, to a greater extent than treatment with
doxorubicin or simvastatin alone (Fig.
4B; P<0.05). Cyclin D1 levels did not change significantly
following treatment with simvastatin; however, treatment with
doxorubicin or the combination significantly suppressed cyclin D1
levels (Fig. 4C; P<0.05). Caspase
3 levels were not significantly altered by treatment with
doxorubicin alone; however, they were significantly increased
following treatment with simvastatin or the combination treatment
(Fig. 4D; P<0.05) Cytochrome c
protein levels were significantly increased in all treatment
groups. The doxorubicin and simvastatin combination had a
significantly greater effect on cytochrome c levels than
doxorubicin or simvastatin alone (Fig.
4E; P<0.05).
The combination of doxorubicin with simvastatin
stimulated a significant increase in p21, cytochrome c and caspase
3 protein levels, and a significant reduction in cyclin D1 protein
level. It was also associated with a marked increase in MCF-7 cell
death, confirming the potentiating effect of simvastatin upon
doxorubicin treatment of breast cancer cells.
Discussion
Simvastatin is one of the most frequently prescribed
drugs due to the efficacy and low toxicity when used to treat
hyperlipidemia (27). Previously,
statins have been identified to reduce proliferation and induce
apoptosis in several cancer cells (28). In the present study, the mechanisms by
which simvastatin reduces the rates of cell proliferation and
increases the rate of cell death in the doxorubicin-treated breast
cancer line MCF-7 cell line were investigated. The results suggest
that simvastatin potentiation of doxorubicin-induced cell death is
accompanied by a suppression of the cell cycle regulator protein
RAC1 signaling pathway. The enhanced cytotoxicity of the
combination treatment is possibly due to an induction in
intracellular ROS formation, leading to increased levels of p21,
cytochrome c, and caspase 3 and decreased cyclin D1 levels. This
may be associated with the mechanism for MCF-7 breast cancer cell
sensitization to anticancer drugs and the induction of cell death.
Therefore, simvastatin may potentially be used in the prevention
and treatment of breast cancer.
Statins or HMG-CoA reductase inhibitors are drugs
commonly used for the treatment of hypercholesterolemia.
Additionally, statins exhibit a number of effects on cancer cells
(29) including inhibition of cancer
cell growth, metastasis and invasion, angiogenesis and the
induction of apoptosis. By inhibiting the mevalonic acid pathway,
statins may reduce the levels of the isoprenoid intermediates FPP
and GGPP (22). These intermediates
are critical for post-translational modification of the
intracellular G-proteins, including Rho, Rac, and Ras, which
regulate the signal transduction of several proteins. These
proteins, in turn, are essential for the gene transcription
involved in cellular proliferation, differentiation and apoptosis
(29). RAC1 is overexpressed in a
number of tumors and serves a critical role in cytoskeleton
reorganization, cell migration and cell survival (24). An overexpression of RAC1 is associated
with the progression, including the metastasis and staging, of
human breast cancer (30).
A role for RAC1 in the activation of extracellular
signal-regulated kinases (ERK) 1/2 and phosphoinositide
3-kinase/protein kinase B pro-survival signaling was also
identified and demonstrated to promote cell survival (30,31). The
direct suppression of RAC1 activity induces apoptosis and cell
cycle arrest in breast cancer cells (25). In the present study, simvastatin
significantly inhibited breast cancer cell proliferation with
IC50 values in the low micromolar range. This result is
concomitant with the data of previous studies (16,18).
Simvastatin stimulates cell cycle arrest and apoptosis in a number
of cancer cell types (32) via the
intracellular signaling mechanisms of RAC1 and the associated
downstream pathway. Cell cycle progression is controlled by Cdk
activity (33). The cyclin D1-Cdk4/6
complex also promotes G1 phase cell-cycle progression by modulating
the Cdk inhibitor p21 (34,35). RAC1 suppression may inhibit the
cyclin-Cdk complex, leading to the activation of p21 and inhibition
of cellular proliferation.
The results of the present study indicate that
treatment with simvastatin or doxorubicin alone significantly
inhibits RAC1, Cdk4 and Cdk6 mRNA expression after 24 h, whilst
combination of the two drugs results in increased activity. Cdk2 is
also inhibited by treatment with simvastatin or doxorubicin alone,
but in combination this inhibitory effect is not greater compared
with that observed with doxorubicin alone. Similar to these
observations, a previous study revealed that a blockade of RAC1
activity induces cell cycle arrest or apoptosis in breast cancer
cells (25).
In addition to inhibiting the proliferation of MCF-7
cells, the ability of simvastatin to induce apoptosis has been
demonstrated. Simvastatin-induced apoptosis was characterized by
increased levels of caspase 3, cytochrome c and intracellular ROS.
Several studies have revealed that ROS are key signaling molecules
in mammalian cells. An accumulation of ROS is directly correlated
with mitochondrial dysfunction and promotion of cell apoptosis
(36). The results of the present
study suggest that simvastatin induced ROS production in a
concentration-dependent manner. In combination simvastatin and
doxorubicin generated even larger quantities of ROS, a result
indicative of an additive effect. This is potentially a key reason
why MCF-7 cell apoptosis is induced by simvastatin and doxorubicin.
The observation of the present study that a combination of
simvastatin and doxorubicin increased cytochrome c protein
expression and caspase 3 activity more compared with each drug
individually is consistent with the hypothesis that simvastatin
sensitizes MCF-7 cells.
The present study identified that simvastatin
inhibited MCF-7 cell proliferation and colony formation in a
dose-dependent manner and, notably, that it enhanced the activity
of doxorubicin. The effect on cell cycle progression was also
investigated in the present study by measuring p21 and cyclin D1
protein expression. Simvastatin and doxorubicin treatment resulted
in an increase in p21, and a decrease in cyclin D1 expression
level. These data suggest that simvastatin enhances
doxorubicin-induced cancer cell death by inhibiting cell cycle
progression (37).
In conclusion, the present study has demonstrated
that simvastatin enhances doxorubicin cytotoxicity towards MCF-7
cells through an inhibition of the RAC1 pathway and induction of
caspase- and cytochrome c-dependent apoptosis in a process
involving oxidative stress. These data also reveal that the Cdk
inhibitor p21 is activated in the process of simvastatin-induced
cell death, leading to an inhibition of cell cycle progression.
These results contribute to the current understanding of the
molecular mechanisms of simvastatin, and provide a basis for future
studies seeking to validate the mevalonate pathway as a novel
therapeutic target. The inclusion of statins in anticancer
treatment regimens may potentially reduce the quantity of
anticancer drugs required to achieve therapeutic effects and
thereby reduce the side effects associated with cancer
treatment.
Acknowledgements
The present study was supported by the Office of the
Higher Education Commission (grant no., 2558A10962234), a grant
from the Mahasarakham University (MSU) Faculty of Medicine, a
Mahasarakham University 2016 Thailand Research Fund (Grant no.
TRG5780254) and the National Research Council of Thailand (Grant
no. 2559A10902073). The authors would like to thank Dr. Tim Cushnie
(Faculty of Medicine, Mahasarakham University, Thailand) for
language-editing the manuscript.
References
|
1
|
Berry DA, Cronin KA, Plevritis SK, Fryback
DG, Clarke L, Zelen M, Mandelblatt JS, Yakovlev AY, Habbema JD and
Feuer EJ: Cancer Intervention and Surveillance Modeling Network
(CISNET) Collaborators: Effect of screening and adjuvant therapy on
mortality from breast cancer. N Engl J Med. 353:1784–1792. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D and
Bray F: Cancer incidence and mortality patterns in Europe:
Estimates for 40 countries in 2012. Eur J Cancer. 49:1374–1403.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pagani O, Senkus E, Wood W, Colleoni M,
Cufer T, Kyriakides S, Costa A, Winer EP and Cardoso F: ESO-MBC
Task Force: International guidelines for management of metastatic
breast cancer: Can metastatic breast cancer be cured? J Natl Cancer
Inst. 102:456–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Driscoll L and Clynes M: Biomarkers and
multiple drug resistance in breast cancer. Curr Cancer Drug
Targets. 6:365–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coley HM: Mechanisms and strategies to
overcome chemotherapy resistance in metastatic breast cancer.
Cancer Treat Rev. 34:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Istvan ES and Deisenhofer J: Structural
mechanism for statin inhibition of HMG-CoA reductase. Science.
292:1160–1164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoeg JM and Brewer HB Jr:
3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors in the
treatment of hypercholesterolemia. JAMA. 258:3532–3536. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watanabe Y, Ito T, Shiomi M, Tsujita Y,
Kuroda M, Arai M, Fukami M and Tamura A: Preventive effect of
pravastatin sodium, a potent inhibitor of
3-hydroxy-3-methylglutaryl coenzyme A reductase, on coronary
atherosclerosis and xanthoma in WHHL rabbits. Biochim Biophys Acta.
960:294–302. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Downs JR, Clearfield M, Weis S, Whitney E,
Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W and Gotto
AM Jr: Primary prevention of acute coronary events with lovastatin
in men and women with average cholesterol levels: Results of
AFCAPS/TexCAPS. Air Force/Texas coronary atherosclerosis prevention
study. JAMA. 279:1615–1622. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khush KK and Waters DD: Effects of statin
therapy on the development and progression of heart failure:
Mechanisms and clinical trials. J Card Fail. 12:664–674. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi XF, Kim DH, Yoon YS, Li JH, Jin D, Teng
YC, Kim SK and Lee KJ: Fluvastatin inhibits expression of the
chemokine MDC/CCL22 induced by interferon-gamma in HaCaT cells, a
human keratinocyte cell line. Br J Pharmacol. 157:1441–1450. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blum A and Shamburek R: The pleiotropic
effects of statins on endothelial function, vascular inflammation,
immunomodulation and thrombogenesis. Atherosclerosis. 203:325–330.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Elewa HF, El-Remessy AB, Somanath PR and
Fagan SC: Diverse effects of statins on angiogenesis: New
therapeutic avenues. Pharmacotherapy. 30:169–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cai JP, Chen W, Hou X, Liang LJ, Hao XY
and Yin XY: Simvastatin enhances the chemotherapeutic efficacy of
S-1 against bile duct cancer: E2F-1/TS downregulation might be the
mechanism. Anticancer Drugs. 24:1020–1029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamigaki M, Sasaki T, Serikawa M, Inoue M,
Kobayashi K, Itsuki H, Minami T, Yukutake M, Okazaki A, Ishigaki T,
et al: Statins induce apoptosis and inhibit proliferation in
cholangiocarcinoma cells. Int J Oncol. 39:561–568. 2011.PubMed/NCBI
|
|
16
|
Shen Y, Du Y, Zhang Y and Pan Y:
Synergistic effects of combined treatment with simvastatin and
exemestane on MCF-7 human breast cancer cells. Mol Med Rep.
12:456–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibits cancer cell growth by inducing apoptosis
correlated to activation of Bax and down-regulation of BCL-2 gene
expression. Int J Oncol. 40:935–941. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller T, Yang F, Wise CE, Meng F,
Priester S, Munshi MK, Guerrier M, Dostal DE and Glaser SS:
Simvastatin stimulates apoptosis in cholangiocarcinoma by
inhibition of Rac1 activity. Dig Liver Dis. 43:395–403. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Werner M, Sacher J and Hohenegger M:
Mutual amplification of apoptosis by statin-induced mitochondrial
stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br
J Pharmacol. 143:715–724. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sassano A and Platanias LC: Statins in
tumor suppression. Cancer Lett. 260:11–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kusama T, Mukai M, Iwasaki T, Tatsuta M,
Matsumoto Y, Akedo H, Inoue M and Nakamura H:
3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors reduce
human pancreatic cancer cell invasion and metastasis.
Gastroenterology. 122:308–317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wong WW, Dimitroulakos J, Minden MD and
Penn LZ: HMG-CoA reductase inhibitors and the malignant cell: The
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jaffe AB and Hall A: Rho GTPases:
Biochemistry and biology. Annu Rev Cell Dev Biol. 21:247–269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bosco EE, Mulloy JC and Zheng Y: Rac1
GTPase: A “Rac” of all trades. Cell Mol Life Sci. 66:370–374. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoshida T, Zhang Y, Rosado Rivera LA, Chen
J, Khan T, Moon SY and Zhang B: Blockade of Rac1 activity induces
G1 cell cycle arrest or apoptosis in breast cancer cells through
downregulation of cyclin D1, survivin, and X-linked inhibitor of
apoptosis protein. Mol Cancer Ther. 9:1657–1668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martirosyan A, Clendening JW, Goard CA and
Penn LZ: Lovastatin induces apoptosis of ovarian cancer cells and
synergizes with doxorubicin: Potential therapeutic relevance. BMC
Cancer. 10:1032010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hindler K, Cleeland CS, Rivera E and
Collard CD: The role of statins in cancer therapy. Oncologist.
11:306–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schnelzer A, Prechtel D, Knaus U, Dehne K,
Gerhard M, Graeff H, Harbeck N, Schmitt M and Lengyel E: Rac1 in
human breast cancer: Overexpression, mutation analysis, and
characterization of a new isoform, Rac1b. Oncogene. 19:3013–3020.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eblen ST, Slack JK, Weber MJ and Catling
AD: Rac-PAK signaling stimulates extracellular signal-regulated
kinase (ERK) activation by regulating formation of MEK1-ERK
complexes. Mol Cell Biol. 22:6023–6033. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saito A, Saito N, Mol W, Furukawa H,
Tsutsumida A, Oyama A, Sekido M, Sasaki S and Yamamoto Y:
Simvastatin inhibits growth via apoptosis and the induction of cell
cycle arrest in human melanoma cells. Melanoma Res. 18:85–94. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fournier AK, Campbell LE, Castagnino P,
Liu WF, Chung BM, Weaver VM, Chen CS and Assoian RK: Rac-dependent
cyclin D1 gene expression regulated by cadherin- and
integrin-mediated adhesion. J Cell Sci. 121:226–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
36
|
D'Autréaux B and Toledano MB: ROS as
signalling molecules: Mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sadeghi-Aliabadi H, Minaiyan M and
Dabestan A: Cytotoxic evaluation of doxorubicin in combination with
simvastatin against human cancer cells. Res Pharm Sci. 5:127–133.
2010.PubMed/NCBI
|