Introduction
Glutamate, a major excitatory neurotransmitter in
the brain that activates glutamate receptors including
N-methyl-D-aspartate receptors (NMDARs), is primarily associated
with neuronal communication; and it is involved in normal brain
functions including cognition, memory and learning (1). Glutamate-secreting brain tumors have
been reported to exhibit enhanced growth (2). Thus, it was speculated that the blockade
of NMDARs may be a beneficial approach to the treatment of such
brain tumors. Indeed, among NMDAR antagonists, MK801 and memantine
were demonstrated to inhibit the growth of glutamate-secreting
tumors (2). MK801 is used only for
basic research due to its psychotomimetic side effects, including
schizophrenic symptoms (3). In
contrast, memantine is the agent approved for moderately severe to
severe Alzheimer disease in Europe and for moderate-to-severe
Alzheimer disease in the United States (4). However, neither MK801 or memantine can
be used for anticancer treatment in the clinical setting.
In contrast, the NMDAR antagonist ketamine is widely
used in clinical practice as an intravenous or intramuscular
anesthetic in humans as well as animals, and it may be an
anesthetic candidate for use in tumor resection. However, even
basic data on the effects of ketamine in this regard are lacking.
The present study hypothesized that ketamine can also inhibit the
growth of glutamate-secreting tumors in a similar fashion to MK801
and memantine. To test our hypothesis, the present study thus
evaluated the effects of ketamine on the proliferation of gliomas,
known as glutamate-secreting brain tumors, using a cell line
culture model. The objective of this study is to provide basic data
on the anticancer effect of NMDAR antagonist, ketamine.
Materials and methods
Cell culture
The rat brain glioma C6 cell line and the neonatal
rat astrocyte RNB cell line were obtained from The Health Science
Research Resources Bank (Tokyo, Japan). Since gliomas arise from
astrocytes, the RNB cells were selected for use in the present
study as representative non-malignant astrocytes for comparison.
For C6 and RNB cell cultures, cells were seeded in 100-mm culture
plates at 1×105 cells/well and cultured in Hams F10
medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 15% horse serum (ICN Flow 2070033; MTX Lab
Systems, Inc., Vienna VA, USA) and 2.5% fetal bovine serum (ICN
Flow 101083; MTX Lab Systems, Inc.), and grown in a humidified
atmosphere of 95% air and 5% CO2 at 37°C for 72 h.
Following a 72 h-incubation, the cells were harvested from the
dishes using trypsin/EDTA in each culture medium for further
passages.
Cell proliferation assay following
ketamine treatment
The C6 and RNB cell lines were detached using
trypsin/EDTA and seeded in 6-well plates, and cultured at 37°C in a
humidified incubator containing 95% air and 5% CO2 with
medium as aforementioned. Cell cultures were maintained for 24 h to
allow cells to adhere, at which time cultures were incubated for a
further 72 h in medium containing ketamine at a range of
concentrations (0, 3, 10, 30 and 100 µM; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). To assess the dose-response of ketamine at 72
h of culture, cell proliferation was determined from each treatment
group by counting the numbers of cells using a cell counter (Nihon
Kohden, Tokyo, Japan). To determine the time-response of cell
proliferation to ketamine treatment, cells were incubated in medium
with/without ketamine (30 µM; Sigma-Aldrich; Merck KGaA), and cell
proliferation was assessed using the aforementioned method at 0,
24, 48 and 72 h time points.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) detection of apoptotic cells
following ketamine treatment
The C6 and RNB cell lines were detached using
trypsin/EDTA and seeded in 24-well plates, and incubated at 37°C in
a humidified incubator containing 95% air and 5% CO2
with Hams F10 medium (Thermo Fisher Scientific, Inc.). Cell
cultures were maintained for 24 h to allow cells to adhere, at
which time cells were incubated for a further 72 h in medium
containing ketamine at a range of concentrations (0, 3, 10, 30 and
100 µM; Sigma-Aldrich; Merck KGaA), renewed each day. At the
termination point of 72 h of culture, the cells were subjected to
in situ labeling and a quantitative analysis of late-phase
apoptotic cells using the TUNEL method (5). TUNEL staining, which detects fragmented
DNA, was performed using an In Situ Apoptosis Detection kit,
according to the manufacturers protocol (Takara Biotechnology Co.,
Ltd., Dalian, China). Calcium ionophore A23187-induced apoptosis in
each cell line and was used as a positive control for TUNEL
staining. Apoptotic cells were visualized with
3,3′-diaminobenzidine at 15–25°C-incubation for 20 min and detected
by light microscopy (12 fields of view and magnification, ×400).
Cell nuclei were counterstained with methyl green. at
15–25°C-incubation for 15 min The numbers of apoptotic cells were
counted, and the percentages of apoptotic cells relative to the
total cells were calculated.
Inhibition of apoptosis using
DIDS
Confluent C6 glioma cells were seeded in 24-well
plates, and cultured for 24 h to allow for adherence. C6 cells were
incubated for 1 h in Hams F10 medium supplemented with various
concentrations of 4,4′-diisothiocyanate-2,2′-disulfonic acid
stilbene (DIDS; 0, 10, 30 and 100 µM; Wako Pure Chemicals
Industries, Ltd., Osaka, Japan). Following 1 h incubation, the
cells were incubated for a further 72 h with/without various drugs
(medium alone as a control, or 30 µM ketamine +0, 10, 30 or 100 µM
DIDS). Apoptotic cells were detected using the TUNEL method as
aforementioned. DIDS was dissolved in dimethyl sulfoxide (DMSO).
The final concentration of DMSO in the experimental tubes did not
exceed 0.04% and did not affect the viability of the cells. Cell
proliferation was assessed in this assay by counting cells using a
cell counter (Nihon Kohden).
Cell proliferation and apoptosis assay
following D-AP5 treatment
Confluent C6 glioma cells were seeded in 6-well
plates at 2×104 cells/well, and incubated in Hams F10
medium (Thermo Fisher Scientific, Inc.) as aforementioned.
Following 24 h of culture to allow for adherence, cells were
incubated for a further 72 h in medium containing various
concentrations (0, 3, 10, 30 and 100 µM) of D-AP5 (Sigma-Aldrich,
Merck KGaA) or ketamine (0–100 µM; Sigma-Aldrich, Merck KGaA). To
assess the dose-dependent response of D-AP5 after 72 h of culture,
cell proliferation was determined from each treatment group, by
counting the numbers of cells using a cell counter (Nihon Kohden).
In addition, to assess the apoptotic response of cells following 72
h of culture with various concentrations of D-AP5 (0–100 µM),
renewed every 3 days, the TUNEL assay was performed as
aforementioned.
Large-scale gene expression analysis
of tumor growth from a cDNA microarray
To determine an estimate of the effects of ketamine
on C6 glioma cell growth, a large-scale gene expression survey of
cell growth, using a cDNA microarray was performed, as follows.
RNA isolation
Following incubation of C6 cells for 30 min at 37°C
with Hams F10 medium (Thermo Fisher Scientific, Inc.) containing
100 µM ketamine (Sigma-Aldrich; Merck KGaG), total RNA was
extracted from the cell monolayer using an RNeasy Mini kit (Qiagen,
Inc. Valencia, CA, USA) according to the manufacturer's protocol.
The total RNA was treated with RNase free-DNase (Qiagen, Inc.) for
eliminating genomic DNA contamination from RNA samples prior to the
cDNA microarray.
cDNA labeling and microarray
hybridization
Reverse transcription, labeling and hybridization
were performed with LabelStar (Qiagen, Inc.) according to the
manufacturers protocol. Briefly, total RNA was reverse-transcribed
into target cDNA using LabelStar with Cy3 and Cy5 for control and
ketamine-treated cells, respectively. Labeled cDNAs were purified
using MiniElute spin columns (Qiagen, Inc.). Cy3-labeled and
Cy5-labeled target cDNAs were combined, dried and resuspended in
hybridization buffer. The target cDNA mixture was hybridized with
rat ADME cDNA microarrays (Asahi Techno Glass; Tokyo, Japan) that
included 1,936 cDNA elements. The microarrays were scanned in the
Cy3 and Cy5 channels with an Affymetrix 428 Array Scanner
(Affymetrix; Thermo Fisher Scientific, Inc.), and analyzed using
the ImaGene and GeneSight-Lite software 7.0 packages (BioDiscovery,
El Segundo, CA, USA). Following background subtraction and dye bias
normalization, poor-quality features were excluded from further
analysis. Features with low signal intensity in the reference
channel were filtered if the signal-to-noise ratio was <2.5. The
fold-change in the expression of each gene is expressed as the
logarithm of the ratio (Cy5 fluorescence/Cy3 fluorescence). The log
ratio ≥0.3 or ≤-0.3 (i.e., normalized intensity ≥2 or ≤0.5) was
used to define genes with significant changes in expression.
Statistical analysis
Multiple group comparisons were performed by one-way
analysis of variance, followed by a Tukey-Kramer post hoc test.
Data were presented as mean ± SEM. Student's unpaired t-tests were
performed to compare differences between two groups. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using the software KyPlot 5.0
(KyensLab, Inc., Tokyo, Japan).
Results
Large-scale gene expression survey of
tumor growth using the cDNA microarray
Analysis of the large-scale gene expression survey
obtained from the cDNA microarray, revealed that the expression of
multiple growth factor genes [epidermal growth factor, transforming
growth factor (TGF)β1-induced transcript 1 and TGFβ3) and
transcriptional regulators (wilms tumor 1], tumor protein p53 and
signal transducer and activator of transcription 3) of C6 glioma
cells remained unchanged; although several genes were downregulated
in response to ketamine treatment (Table
I).
 | Table I.Gene expression ratio of cell growth
in C6 cells as demonstrated by cDNA microarray analysis. |
Table I.
Gene expression ratio of cell growth
in C6 cells as demonstrated by cDNA microarray analysis.
| A, Growth
factors |
|---|
|
|---|
| Gene name | Gene ID | Log expression ratio
(n) | Regulation |
|---|
| Bone morphogenetic
protein 6 | 25,644 | −0.15±0.62 (8) | n.s. |
| Brain derived
neurotrophic factor | 24,225 | −1.23±0.50 (4) | d |
| Connective tissue
growth factor | 64,032 | 0.10±0.09 (4) | n.s. |
| Early B-cell factor
(olfactory neuronal transcription factor 1) | 11,6,543 | −0.35±0.72 (4) | d |
| Early growth response
1 | 24,330 | 0.08±0.15 (2) | n.s. |
| Epidermal growth
factor | 25,313 | −1.29±0.33 (2) | d |
| Growth arrest and
DNA-damage-inducible-45 α | 25,112 | −0.11±0.76 (4) | n.s. |
| Growth factor
receptor bound protein 2 | 81,504 | −0.12±0.11 (4) | n.s. |
| Insulin-like growth
factor 1 | 24,482 | 0.49±0.48 (2) | u |
| Insulin-like growth
factor binding protein 1 | 25,685 | −0.49±0.50 (4) | d |
| Insulin-like growth
factor binding protein 2 | 25,662 | −0.22±0.93 (4) | n.s. |
| Insulin-like growth
factor binding protein 3 | 24,484 | −0.16±0.54 (6) | n.s. |
| Insulin-like growth
factor binding protein, acid labile subunit | 79,438 | −0.43±0.37 (2) | d |
| Insulin-like growth
factor II (somatomedin A) | 24,483 | 0.06±0.24 (2) | n.s. |
| Insulin-like growth
factor-binding protein 5 | 25,285 | −0.29±0.81 (6) | n.s. |
| Midkine | 81,517 | −1.07±0.30 (4) | d |
| Neural cell adhesion
molecule | 24,586 | −0.32±1.00 (4) | d |
| Platelet-derived
growth factor A chain | 25,266 | −0.01±0.13 (2) | n.s. |
| Platelet-derived
growth factor- receptor α | 25,267 | −0.17±0.73 (4) | n.s. |
| Latent TGF
β binding protein 1 | 59,107 | 0.32±0.28 (2) | u |
| TGF β
inducible early growth response | 81,813 | −0.38±0.15 (2) | d |
| TGF β 1
induced transcript 1 | 84,574 | −0.34±0.42 (2) | d |
| TGF β
3 | 25,717 | −0.79±0.47 (4) | d |
| Vascular endothelial
growth factor | 83,785 | 0.16±0.06 (2) | n.s. |
| Vascular endothelial
growth factor C | 11,4,111 | 0.44±0.37 (2) | u |
|
| B, Transcriptional
regulators | Gene ID | Log expression ratio
(n) | Regulation |
|
| Nuclear factor kappa
B p105 subunit | 81,736 | 0.57±0.63 (2) | u |
| V-myc avian
myelocytomatosis viral-oncogene homolog | 24,577 | 0.37±0.75 (4) | u |
| Wilms tumor 1 | 24,883 | −0.72±1.37 (4) | d |
| PRKC, apoptosis, WT1,
regulator | 64,513 | −0.88±0.32 (4) | d |
| Tumor protein p53
(Li-Fraumeni syndrome) | 24,842 | −1.34±1.00 (2) | d |
| Retinoblastoma 1
(including osteosarcoma) | 24,708 | −1.40±0.02
(2) | d |
| Fos like antigen
2 | 25,446 | 0.40±0.53 (4) | d |
|
Retinoblastoma-binding protein 1-related
protein | 84,481 | 0.12±1.19 (4) | n.s. |
|
Retinoblastoma-related gene | 81,758 | 0.24±1.21 (2) | n.s. |
| Signal transducer
and activator of transcription 1 | 25,124 | −0.03±2.06
(4) | n.s. |
| Signal transducer
and activator of transcription 3 | 25,125 | −0.35±0.32
(4) | d |
| Suppression of
tumorigenicity 13 (colon carcinoma) | 81,800 | −0.01±0.38
(4) | n.s. |
| Hsp70-interacting
protein |
|
|
|
The effects of ketamine on the growth
of C6 glioma and RNB cells
The effects of ketamine treatment significantly
inhibited the proliferation of C6 cells at every concentration
examined. However, there were no ketamine-induced effects on the
proliferation of RNB cells at any given concentration (Fig. 1A). The proliferation of C6 cells was
most significantly inhibited in response to ketamine at a dose of
30 µM (n=7, 66.4%, P<0.001). The 30 µM dose of ketamine
significantly inhibited C6 cell proliferation at 24-, 48-, and 72-h
incubations compared with that of the control, however no notable
effect was observed on RNB cell proliferation at any time point
tested (n=4, Fig. 1B).
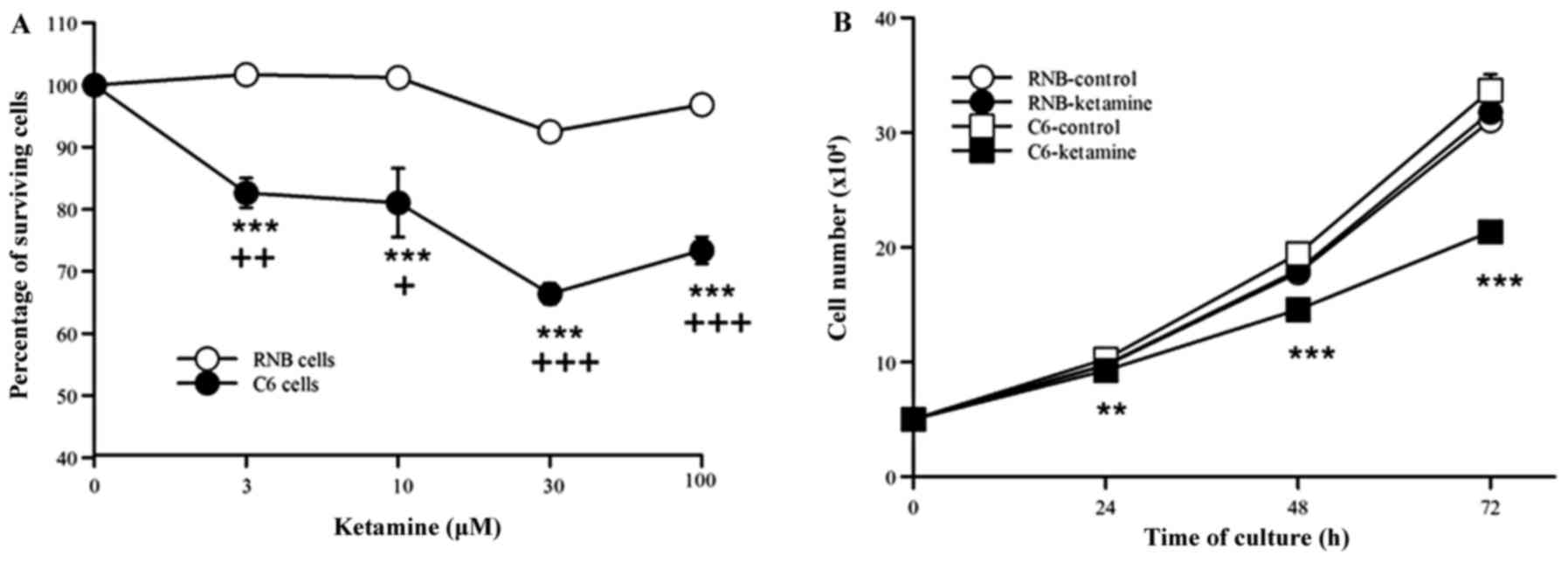 | Figure 1.(A) Ketamine concentration-response
curves and the percentage of survival of C6 and RNB cells. Cell
proliferation was examined by cell counts following culture for 72
h (C6 control, 72.2±0.17×104 cells; RNB control,
68.8±0.13×104 cells). The results (mean ± SEM) are
expressed as percentages compared with control experiments (C6,
n=7; RNB, n=3). (B) Time-response curves of ketamine for the number
of C6 and RNB cells. Cell proliferation was examined in ketamine
treated (30 µM) C6 and RNB cells at 0-, 24-, 48-, and 72-h
incubations. The results are expressed as the mean ± standard error
of the mean. **P<0.01, ***P<0.001 vs. C6 control;
+P<0.05, ++P<0.01 and
+++P<0.001 vs. RNB cells. SEM, standard error of the
mean. |
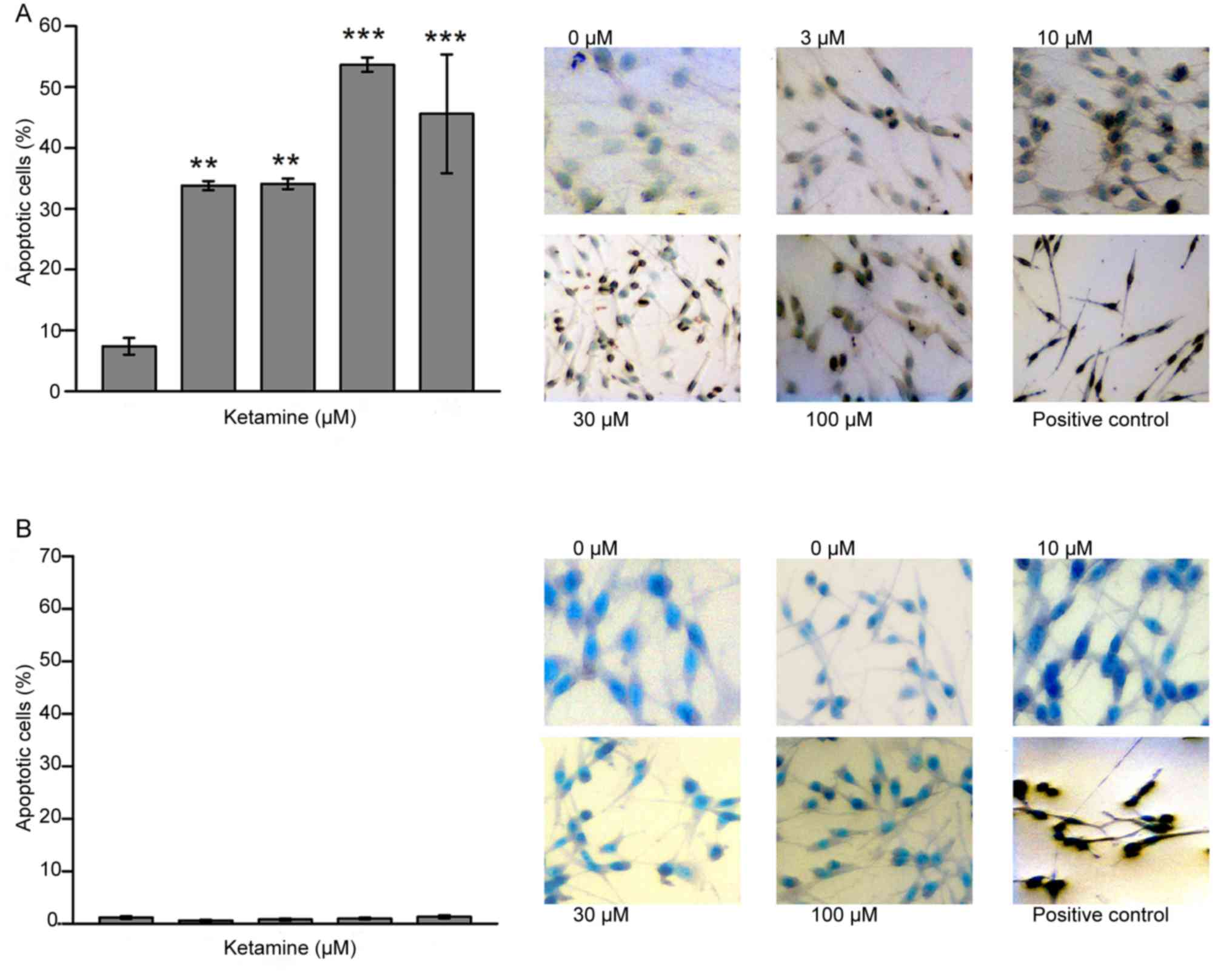
Detection of apoptotic cells following
ketamine treatment
As presented in Fig.
2, the ratio of apoptotic cells following a 72-h incubation was
significantly more pronounced in all ketamine-treated C6 cells
compared with C6-control cells Control, (7.4±2.8%; 3 µM, 33.8±1.5%;
10 µM, 34.1±1.7%; 30 µM, 53.7±2.4%, and 100 µM, 45.6±19.5%; n=4;
Fig. 2A). In contrast, no significant
differences in the proportion of apoptotic cells were observed
between the RNB control cells and any of the ketamine-treated RNB
cells (n=6; Fig. 2B).
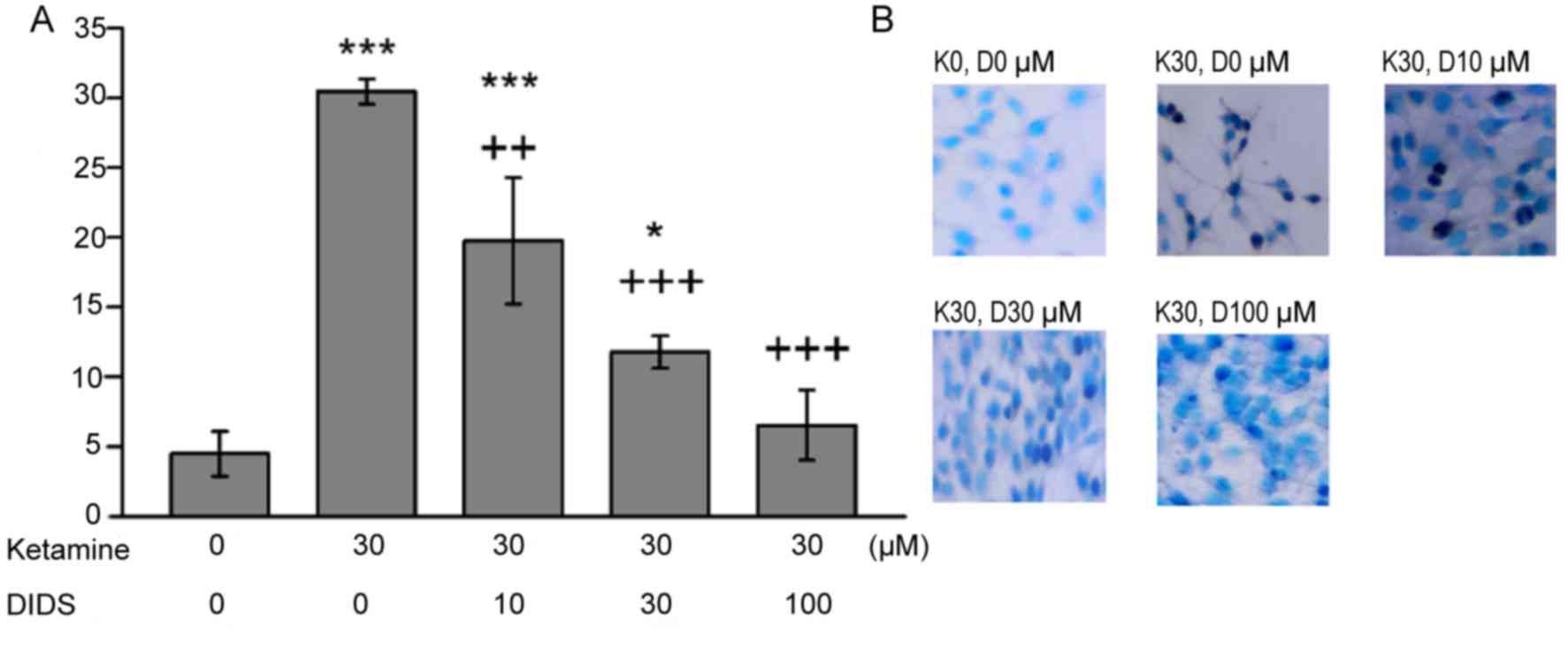
Inhibition of apoptosis using
DIDS
The increased proportion of apoptotic C6 cells
following ketamine treatment were significantly reversed in the
presence of DIDS, in a dose-dependent manner (Fig. 3). The rate of apoptotic C6 cells
observed in response to ketamine and 100 µM DIDS treatment was
comparable to that observed with no treatment. DMSO alone had no
effect on the percentage of apoptotic cells (data not shown).
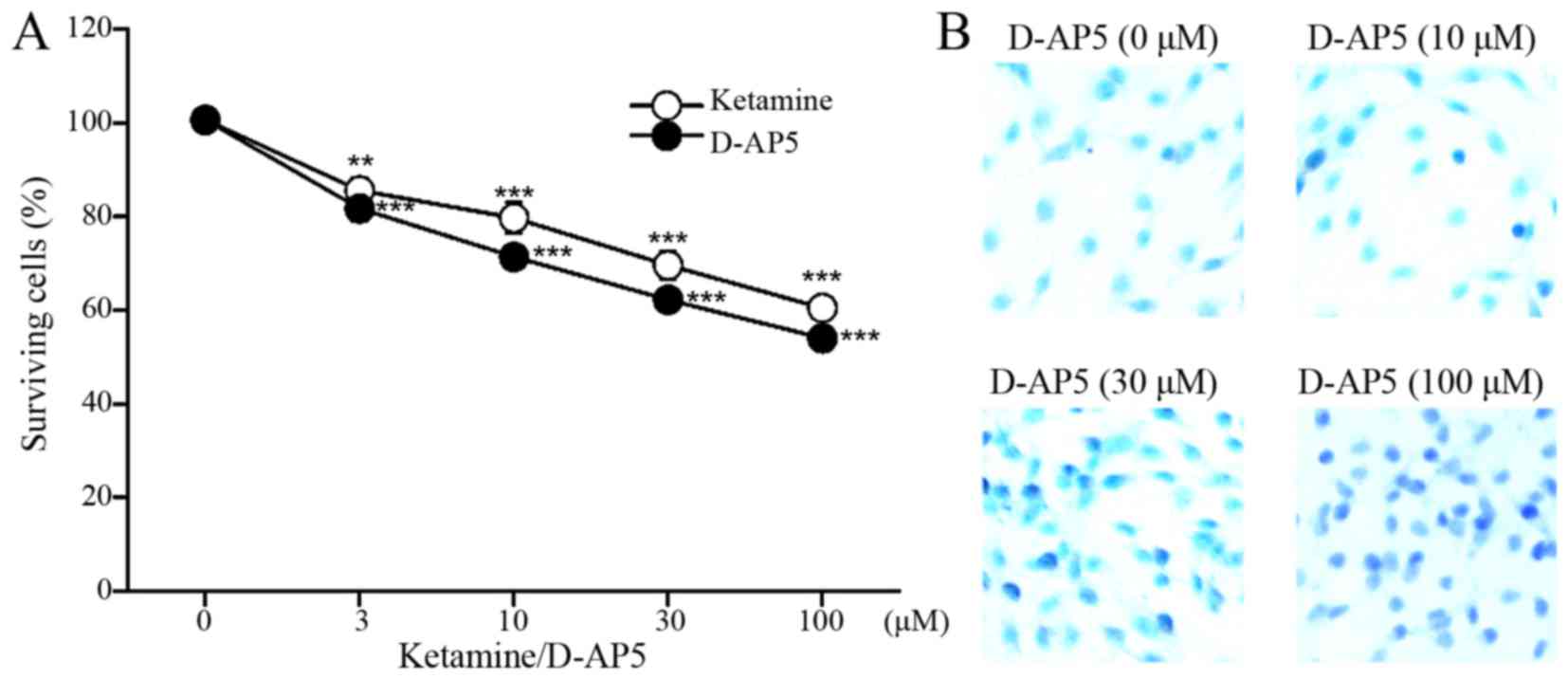
Proliferation and apoptosis assay
results following treatment with selective NMDAR antagonist
D-AP5
Treatment with the selective NMDAR antagonist D-AP5
significantly inhibited the proliferation of C6 cells in the same
manner as ketamine treatment (n=4, Fig.
4A) and induced apoptosis (Fig.
4B).
Discussion
In the present study, the gene expression profile
screening of ketamine-treated C6 glioma cells using a cDNA
microarray indicated that ketamine may suppress the growth of C6
cells via the regulation of growth factors and transcriptional
regulators. The subsequent proliferation assays in ketamine-treated
C6 cells confirmed these results. In addition, the results of the
apoptotic assay demonstrated that ketamine-induced apoptotic cell
death due to apoptosis in malignant astrocytes (C6 glioma cells),
resulted in the suppression of C6 cell proliferation. In contrast,
ketamine treatment had no effect on the growth of the non-malignant
astrocytes (RNB cells). In the present study, it was observed that
the effects of a selective blockade of NMDARs, by D-AP5, mimicked
the ketamine-induced cell death apoptotic effect in C6 cells,
indicating that a blockade of NMDARs is involved in the decreased
proliferation of C6 glioma cells due to ketamine.
The results from the present study suggested that
activation of NMDARs is associated with C6 glioma cell
proliferation. Previous studies, which investigated the
proliferation of C6 cells, are in accordance with the results from
the present study, as it was reported that C6 cells release
glutamate during proliferation, and glutamate promoted C6
proliferation via the activation of NMDARs (2,6). Normal
glial cells uptake excitatory amino acids, ensuring the appropriate
control of synaptic communication and preventing glutamate
neurotoxicity (6). However, due to
their lack of an efficient glutamate uptake system, C6 cells
release excessive glutamate that facilitates their proliferation
(2).
It was suggested that ketamine may increase
neurodegeneration in neonatal animal brains (7–10). The
continuous blockade of NMDARs due to ketamine upregulates NMDARs,
and the upregulation of NMDARs results in an excessive excitatory
response to glutamate, causing neuronal cell death (8,9). Despite
these reports, in the present study ketamine did not affect the
proliferation of RNB cells, even though RNB cells are neonatal.
This result is probably due to the characteristics of the
immortalized RNB cell line. RNB cells are non-malignant, and do not
represent normal cells. To date, it is still unknown if RNB cells
express NMDARs, and the results from the present study do not
disprove a toxic effect of ketamine on astrocytes in the developing
brain.
The limitations of the present study are as follows;
The cDNA microarray is a screening assay, and the results obtained
from this assay may only be a rough estimate of gene expression
changes. To confirm the results of the cDNA microarray, further
assays including reverse transcription quantitative polymerase
chain reaction (RT-qPCR) may be required. However, for the purposes
of the study, RT-qPCR was not conducted as the proliferation and
apoptosis assays were the primary focus.
Furthermore, a single TUNEL assay was conducted to
verify that the cell death observed was due to apoptosis.
Additional assays may prove helpful to examine apoptosis. However,
the TUNEL method used herein is recognized as one of the main
methods for detecting apoptotic programmed cell death, as it
identifies cells in the last phase of apoptosis (11,12). In
addition, the DIDS assay further confirmed the existence of
apoptosis, since it is widely accepted that DIDS prevents apoptotic
cell death. The increase in the level of ketamine-induced cell
death demonstrated by TUNEL-positive signals was significantly
inhibited by DIDS, and it was therefore concluded that the present
protocol was sufficient to detect apoptosis. Finally, a cell
viability assay may have been required. Nevertheless, it is worth
noting that the cell counting assay performed in our study is
effective and sufficient to assess cell proliferation, since the
cell viability was confirmed using an apoptosis assay.
In conclusion, the present study confirmed that
ketamine induced glioma cell death and suppressed C6 cell
proliferation. Gliomas are the most common malignant tumors of the
central nervous system. The majority of brain tumors are gliomas
(~80%), and gliomas represent a leading cause of mortality among
young children and adults with brain tumors, due to intensive
growth and invasion ability (13).
Although the glioma cell lines investigated in the present study
were rodent rather than human cell lines; and it would be premature
to apply the results of this basic study to human cases, these
results contribute to the knowledge regarding ketamine-induced
effects. In current clinical practice, ketamine is the only viable
anesthetic option among NMDAR antagonists, and it is widely used in
neurosurgery. The results from the present study suggest that
ketamine is an anesthetic candidate providing potential benefit for
glioma resection. We thus hope that these results will be useful to
oncologists, including anesthesiologists.
Acknowledgements
The authors would like to thank Dr. Miki Izumi
(Hirosaki University Graduate School of Medicine, Aomori, Japan)
for her technical assistance. The present study was supported in
part by the Ministry of Education, Culture, Sports, Science and
Technology, Tokyo, Japan (grant no. 25861353).
Glossary
Abbreviations
Abbreviations:
|
NMDAR
|
N-methyl-D-aspartate receptor
|
|
DIDS
|
4,4′-diisothiocyanate-2,2′-disulfonic
acid stilbene
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
References
|
1
|
Luján R, Shigemoto R and López-Bendito G:
Glutamate and GABA receptor signalling in the developing brain.
Neuroscience. 130:567–580. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takano T, Lin JH, Arcuino G, Gao Q, Yang J
and Nedergaard M: Glutamate release promotes growth of malignant
gliomas. Nat Med. 7:1010–1015. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kovacic P and Somanathan R: Clinical
physiology and mechanism of dizocilpine (MK-801): Electron
transfer, radicals, redox metabolites and bioactivity. Oxid Med
Cell Longev. 3:13–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bullock R: Efficacy and safety of
memantine in moderate-to-severe Alzheimer disease: The evidence to
date. Alzheimer Dis Assoc Disord. 20:23–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gavrieli Y, Sherman Y and Ben-Sasson SA:
Identification of programmed cell death in situ via specific
labeling of nuclear DNA fragmentation. J Cell Biol. 119:493–501.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vanhoutte N and Hermans E:
Glutamate-induced glioma cell proliferation is prevented by
functional expression of the glutamate transporter GLT-1. FEBS
Lett. 582:1847–1852. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ikonomidou C, Bosch F, Miksa M, Bittigau
P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L and
Olney JW: Blockade of NMDA receptors and apoptotic
neurodegeneration in the developing brain. Science. 283:70–74.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang C, Sadovova N, Hotchkiss C, Fu X,
Scallet AC, Patterson TA, Hanig J, Paule MG and Slikker W Jr:
Blockade of N-methyl-D-aspartate receptors by ketamine produces
loss of postnatal day 3 monkey frontal cortical neurons in culture.
Toxicol Sci. 91:192–201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Slikker W Jr, Zou X, Hotchkiss CE, Divine
RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA,
Hanig JP, et al: Ketamine-induced neuronal cell death in the
perinatal rhesus monkey. Toxicol Sci. 98:145–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paule MG, Li M, Allen RR, Liu F, Zou X,
Hotchkiss C, Hanig JP, Patterson TA, Slikker W Jr and Wang C:
Ketamine anesthesia during the first week of life can cause
long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol
Teratol. 33:220–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Negoescu A, Guillermet C, Lorimier P,
Brambilla E and Labat-Moleur F: Importance of DNA fragmentation in
apoptosis with regard to TUNEL specificity. Biomed Pharmacother.
52:252–258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Negoescu A, Lorimier P, Labat-Moleur F,
Drouet C, Robert C, Guillermet C, Brambilla C and Brambilla E: In
situ apoptotic cell labeling by the TUNEL method: Improvement and
evaluation on cell preparations. J Histochem Cytochem. 44:959–968.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sibenaller ZA, Etame AB, Ali MM, Barua M,
Braun TA, Casavant TL and Ryken TC: Genetic characterization of
commonly used glioma cell lines in the rat animal model system.
Neurosurg Focus. 19:E12005. View Article : Google Scholar : PubMed/NCBI
|