Introduction
Kidney cancer accounts for approximately for 3% of
all malignant tumors in humans but its incidence worldwide is
growing. Around 90% of all cases is classified as renal cell
carcinoma (RCC). RCC is characterized as highly aggressive with
20–30% of the patients presenting metastasized disease at the
moment of diagnosis, and another 30% of the patients treated for
localized disease developing metastases during follow-up. RCC
comprises a heterogeneous group, of which clear cell RCC (ccRCC) is
the most common (70–80%) followed by papillary RCC (pRCC) (15%)
(1,2).
Molecular bases of RCC are complicated and not completely
discovered. The most common alteration include inactivation of Von
Hippel-Lindau gene (VHL), mutations in cMET and TP53 genes,
upregulation of vascular endothelial growth factor A (VEGFA), and
plateled-derived growth factor B (PDGFB) (3,4). In the
case of localized disease, RCC is curable with surgery. However,
the prognosis is poor for patients with distant metastases. RCC is
not responsive to conventional radiotherapy and chemotherapy but in
recent years significant improvement was achieved with use of small
targeted molecules, like tyrosine kinase inhibitors (TKIs) and
mammalian target of rapamycin (mTOR) inhibitors. New molecular
targets are tested aiming to improve survival of patients with RCC.
Some authors report the role of the renin-angiotensin system (RAS)
in cancer development (5–7) but studies concerning RCC have not been
comprehensively revised so far. Derosa et al (8) has publish recently review on RAS
inhibitors in RCC concentrating mostly on hypertension and impact
on patients survival. In this paper we present review of current
knowledge about role of RAS in RCC development and progression,
including not only clinical aspects but also molecular mechanisms
and possible future directions in clinical and basic research in
this field.
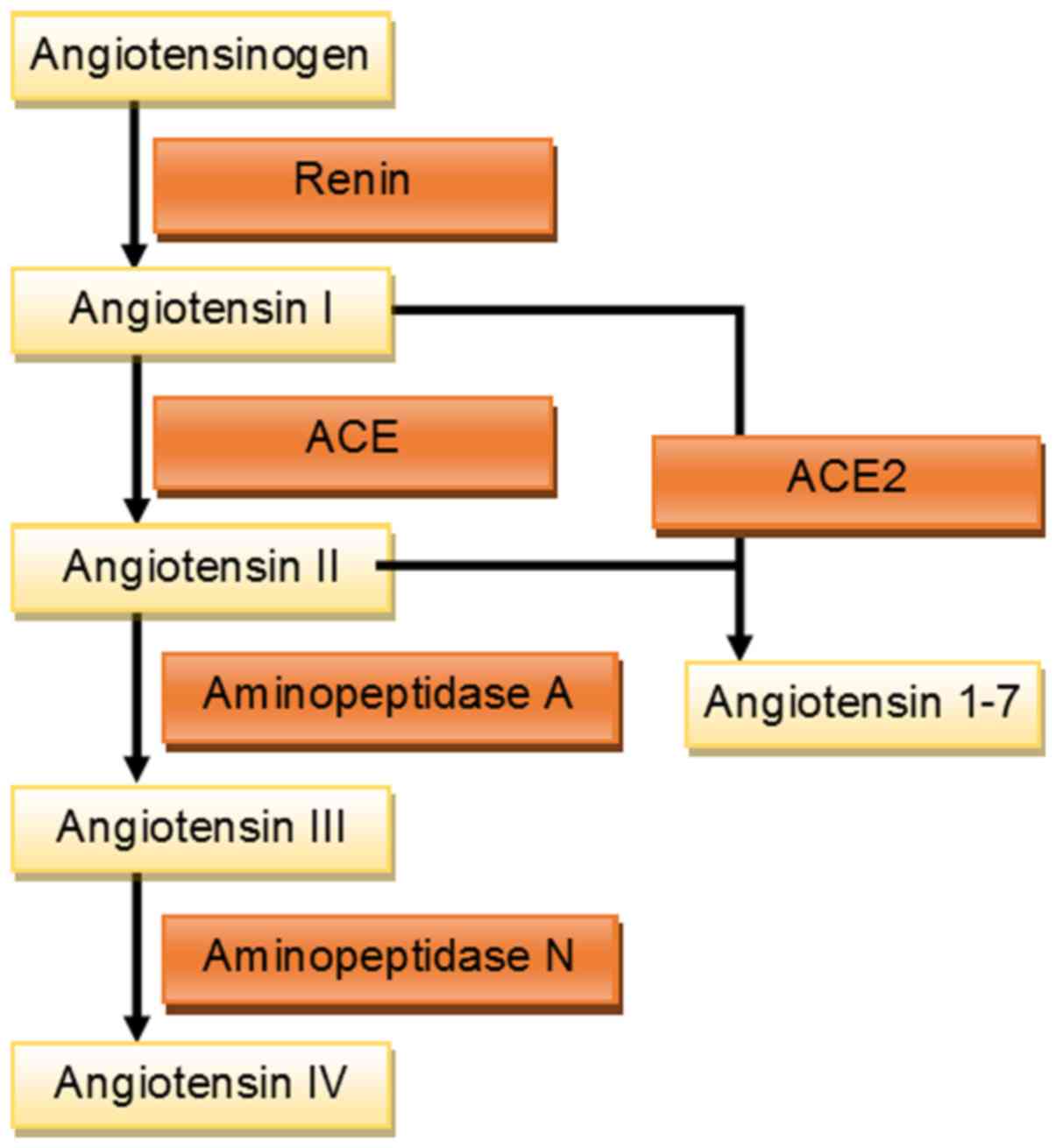
Renin-angiotensin system
The RAS is one of the most comprehensively described
hormonal and polypeptide axes, involved in many physiological and
pathological processes (Fig. 1). The
single and obligate precursor of all angiotensin peptides is
angiotensinogen (AGT), which is synthesized and released from the
liver. In response to such factors as blood pressure (BP) or plasma
sodium level, the kidneys release renin-proteinase, which cleaves
AGT to produce angiotensin I (Ang-I). Next, Ang-I is converted by
angiotensin-converting enzyme (ACE) to angiotensin II (Ang-II), a
key particle of the RAS posing a variety of functions (5). Ang-II acts through two types of G
protein-coupled receptors: Angiotensin type 1 receptor (AT1-R) and
angiotensin type 2 receptor (AT2-R) (9). Ang-II can also undergo further
modifications by aminopeptidases A and N to produce angiotensin III
(Ang-III) and angiotensin IV (Ang-IV), respectively. Ang-III binds
to AT1-R and AT2-R while Ang-IV has its own receptor, AT4-R
(5).
Recently, a new converting enzyme called ACE2 has
been discovered. It is capable of cleaving carboxy-terminal amino
acids from Ang-II to generate angiotensin 1–7 (Ang 1–7), which
signals through the G protein-coupled receptor Mas (MasR) to
antagonize the cardiovascular function of Ang-II (10,11).
Ang-II can be also generated from Ang-I through an alternative
pathway by cathepsin G, CAGE, or chymase (5). Ang-II, through AT-1R, promotes
vasoconstriction, increases plasma aldosterone, retains water and
sodium, and enhances thirst and salt appetite. Altogether, this
results in maintaining fluid and salt homeostasis and increasing
BP. Dysregulation or over-activity of the system is associated with
cardiovascular diseases, predominantly hypertension (5). Besides classical RAS, many organs, such
as the brain, kidneys, heart, and blood vessels, can locally
produce RAS components that work independently or synergistically
with circulating RAS molecules. Locally generated, angiotensins are
likely to contribute to tissue homeostasis and dysfunction
(12).
Angiotensin receptors
Angiotensin receptors in the
kidneys
Angiotensin receptors play an important role in
kidney development. Both AT1-R and AT2-R are present in the early
days of embryogenesis and persist through embryonic life. AT1-R
reaches peaks of its expression at embryonic day 20 and preserve
this level until adulthood, whereas AT-2R is observed until day 28
of the postnatal period (13). In
adult kidneys, its expression is significantly lower (14). Both receptor types co-localize at
differentiated nephrons and blood vessels, while AT2-R also
concentrates in actively differentiating cells of the cortex
(13). AT1-R is the most common
angiotensin receptor in human kidneys with an 8–10-fold higher mRNA
expression than AT2-R. In healthy adult kidneys, AT1-R is
predominantly expressed in the kidney glomeruli, interlobular
arteries, and tubule-interstitial fibrous regions surrounding the
interlobular arteries, while AT2-R is expressed in large
preglomerular vessels of the human cortex and by interlobular
endothelial arterial cells (14–16).
Moreover, Ang-II through AT1-R is responsible for the proliferation
of proximal tubule cells (17), and
through AT2-R, it triggers tubular cell proliferation, apoptosis
and neo-angiogenesis (18).
Angiotensin receptors in RCC
Goldfarb et al (16) analyzed with autoradiography the
expressions of angiotensin receptors in RCC specimens. The
receptors were present in all cancer samples in proportions of 60
and 40% for AT1-R and AT2-R, respectively. Dolley-Hitze et
al (19) broadly analyzed the
expressions of AT1-R and AT2-R in clear cell RCC (ccRCC) and
correlated it with tumor aggressiveness and clinical outcome. In
immunohistochemical staining, 82 out of 84 tumor samples expressed
AT1-R and 76 out of 84 AT2-R. When the number of positive tumor
cells/total tumor cells was expressed as a percentage, the AT1-R
and AT2-R median ratios were 12.5 and 10%, respectively. Both types
were significantly overexpressed by Fuhrman's grade 4. The
correlation between receptor expressions and tumor grade was also
confirmed by western blotting. Interestingly, at the mRNA level, a
significant decrease in AT1-R mRNA levels according to Fuhrman's
grade was present. This is one of the arguments proving that the
altered function of the RAS is mainly an effect of
post-transcriptional or post-translational modifications (19). The expressions of Ang-II receptors can
be also used as a predictor of patient survival. The PFS of ccRCC
patients is longer when the AT1-R and AT-2R expressions are below
12.5% (median) and 10% (median), respectively. By univariate
analyses, AT1-R was correlated with PFS, but it was not confirmed
in a multivariate analysis. AT2-R's influence on PFS was confirmed
in both univariate and multivariate analyses with HR 1.021
(P=0.006), when adjusted for stage, grade, nodal invasion, distant
metastasis, tumor size, and ECOG status (19). Even though, correlation between AT2-R
expression and PFS was statistically significant, HR is very close
to 1 meaning no differences between subgroups. Data should be
analyzed in the bigger cohort to prove statistical improvement in
PFS. Additional, Authors did not present confidence intervals for
HR.
Angiotensin-converting enzyme
General information
ACE is a bivalent dipeptidil carboxyl
metallopeptidase present in body fluids in a soluble form and in a
membrane-bound form, attached to endothelial, epithelial, and
neuroepithelial cells. The main function of ACE is the cleavage of
Ang-I into Ang-II. Beyond Ang-II production, ACE cleaves many other
substrates, including bradykinin, substance P, tetrapeptide AcSDKP,
and many others (20). Altogether,
ACE plays an important role in maintaining balance between the
vasodilatatory and natriuretic actions of bradykinin and the
salt-retaining properties of Ang-II. Additionally, experiments on
ACE in knock-out mice showed that a lack of the enzyme results in
male infertility and a variety of hematological abnormalities, such
as anemia and immature myeloid cells production (21,22). ACE2
is a carboxypeptidase that cleaves single amino acids from Ang-I to
produce angiotensin-(1–9) or to degrade Ang-II to Ang 1–7. It is not
expressed as widely as ACE and is mainly localized in the kidneys,
heart, and testis. However, it shares 42% of the genomic structure
of ACE (12).
ACE in healthy kidney
The kidneys are among the organs with the highest
expression of ACE, which is mainly bound to cell membranes of
endothelial, mesangial, and epithelial cells of proximal and distal
structures of nephron. The highest expression is present in the
brush border of the proximal tubule. In kidney development, its
activity is crucial to provide sufficient levels of Ang-II
necessary for organogenesis (20). In
physiological conditions in the kidneys, ACE is responsible for the
production of Ang-II, a main effector of the RAS.
ACE2 is responsible for the production of Ang 1–7,
acting on the kidneys through the MasR, predominantly expressed in
the kidney's blood vessels and proximal tubules (12). Activation of the ACE2/Ang 1–7/Mas
pathway causes a decrease in BP, vasodilatation of the renal
vessels, and increases in renal blood flow, the glomerular
filtration rate, and diuresis (23).
Generally, Ang 1–7 counteracts the effects of Ang-II receptors.
ACE and ACE2 in RCC
Takada et al (24) had already reported on the presence of
ACE in specimens of RCC, while it was not observed in extra-renal
tumors. The enzyme activity of ACE is significantly decreased in
homogenized tissue samples of chromophobe RCC (chRCC), ccRCC, and
renal oncocytoma (RO) with respect to healthy tissue. In ccRCC and
chRCC, the activity is four-fold lower and in RO, it is seven-fold
lower. Similar results were also observed for ACE2 regarding
activity, with a nearly two-fold decrease in ccRCC and four-fold
decrease in chRCC (25). Larrinaga
et al reported as well that ACE activity correlates
positively with tumor grade, but not with stage. ACE activity was
nearly twice as high in the high Fuhrman grade group (G3-G4) than
in the low grade group (G1-G2). ACE activity levels could be used
as predictors for poor prognosis in ccRCC. In the same study, the
authors described the immunohistochemical pattern of the ACE and
ACE2 expressions. ACE-specific staining was negative in cancer
cells, whereas it was present at a high rate in tumor vessels.
Contrarily, the ACE2 expression was observed within ccRCC cells,
with differences between cancers originating from proximal and
distal nephrons (25). Based on the
presented data, distinct patterns of immunostaining for ACE and
ACE2 may be helpful in clinics in differential diagnosis between
distal and proximal nephron tumors and in the selection of
appropriate therapy. At the mRNA level, significantly, an almost
100-times decrease ACE2 mRNA in chRCC was noticed. In ccRCC, the
mRNA expression was not different than in healthy tissue (25). Different trends in the expression of
mRNA and enzymatic activity may rely on post-translation
modifications, leading to decreased ACE activity. Hence, we cannot
solely rely on mRNA levels to assess changes in protein levels in
cancers.
ACE gene polymorphisms in RCC
It is hypothesized that ACEI influences cancer
incidence, and prognosis may be associated with differences in ACE
activity in plasma, which is strongly correlated with the
insertion/deletion (I/D) polymorphisms of 287 base pairs in intron
16 of the ACE gene (rs4646994). Healthy homozygotes for allele D
have been shown to increase ACE plasma levels and are correlated
with high Ang-II levels, while homozygotes for the I allele have
the lowest ACE activity (26,27). In the large cohort analyzed for a
correlation between the rs4646994 polymorphism and cancer incidence
among RAS inhibitors users, (28) Van
der Knaap et al concluded that using RAS inhibitors is
associated with a significant decrease in the risk of four of the
most common non-skin cancers (colorectal, lung, breast and
prostate) in individuals with the DD genotype, especially among
long-term users. Among RCC patients, the DD genotype and D allele
are more frequent than among controls (27). Earlier, Usmani et al found a
greater incidence of the D allele in non-ccRCC patients vs. RCC,
which almost reached statistical significance (26). Genotype distribution did not differ
between controls and ccRCC or papillary RCC, while significant
differences were present for chRCC. The receptor II genotype has
not been observed in chRCC (27).
In a multivariate analysis, the ACE genotype was
found to be an independent risk factor for RCC of any kind and for
chRCC without a correlation with tumor grade, stage, or nodal and
metastatic status (27). The DD
genotype and D allele are associated with susceptibility for chRCC,
and the II genotype might be protective. The rs4646994 polymorphism
is linked with RCC development but not progression. The limitation
of the study is a lack of data about plasma levels and ACE activity
in relation to genotype and RCC type (27). Polymorphisms rs4295 and rs4343 are not
associated with RCC risk, neither in hypertensive nor in
normotensive patients (29).
Andreotti et al (30) has
examined another 11 polymorphisms in the ACE gene but did not
discover any associations with RCC risk. Polymorphisms in two other
genes encoding RAS proteins-AGT (preangiotensinogen) and AGTR1,
were examined by Andreotti et al, and five AGT single
nucleotide polymorphisms (SNPs) [rs1326889, rs2493137, rs7539020,
rs3889728, rs3789662] are significantly associated with RCC when
adjusted for age, gender, country, smoking status, BMI,
hypertension, and lead exposure. Rs1326889 has the highest impact
on kidney cancer with OR 1.26 (30).
RAS-targeting drugs
Due to the importance of the RAS in maintaining
homeostasis and its involvement in the development of many
cardiovascular diseases, considerable research efforts have focused
on developing drugs that antagonize the RAS. Several antagonists of
enzymes and receptors are now available to inhibit the action of
the system. The first class of such drugs includes
angiotensin-converting enzyme inhibitors (ACEIs), which are
commonly used in the treatment of hypertension, heart failure,
myocardial infarction, proteinuria, and diabetic nephropathy. ACEIs
inhibit ACE activity and thus decrease the conversion of Ang-I into
Ang-II. Examples of this group of drugs are captopril, lisinopril,
ramipril, and perindopril. The second-most common group of RAS
inhibitors are angiotensin receptor blockers (ARBs), generally
known as ‘sartans.’ Examples of this group are candesartan,
irbesartan, losartan, telmisartan, and valsartan. They act as
specific inhibitors of AT1 receptors and block the AT1-R-mediated
effects of Ang-II. Specific inhibitors of AT2-R (PD123319,
PD123117, CGP42114, saralasin), AT4-R (Divalnal-Ang-IV), and MasR
(A-779, D-Pro7-Ang 1–7) receptors have been discovered, but so far
they are used only in research and clinical studies (5).
Effect of ACEI use
Blocking ACE activity results in a decrease in
Ang-II levels and an increase in the concentration of Ang-I.
Reduced levels of Ang-II cause a decrease in the binding and
activation of angiotensin receptors, mainly AT1-R, leading to an
increase in renin release (31). It
was proved in many studies that renin, independently on Ang-II, can
promote fibrosis and cell growth (32,33).
Increased levels of Ang-I, resulting from the blockade of ACE, lead
to the activation of alternative pathways of its cleavage. ACE-2,
which in physiological conditions converts mainly into Ang-II, can
produce high levels of Ang 1–7 by converting Ang-I. Ang 1–7 binds
to the MasR and promotes a variety of processes depending on the
tissue type (34). ACE inhibition
does not block the production of Ang-II completely, which can be
present in the bloodstream as a result of the activation of
alternative enzymes, such as cathepsin G, CAGE, and chymase, which
in normal situations play a minor role in the conversion of Ang-I
(5).
Additionally, ACEIs decrease the degradation of
bradykinin, which stimulates the production of vasodilatatory
factors, such as NO, cGMP, prostaglandin E2, and prostacyclin
(35). Increased levels of bradykinin
may promote carcinogenesis by stimulating growth, survival, and
cancer cells migration. Its B2 receptors are overexpressed in many
tumors, including prostate, renal, and breast cancer tumors.
Bradykinin can also increase the permeability of the blood-brain
barrier and together with the stimulation of cell migration can
result in a higher metastatic potential (36). ACEIs have a few lesser-known
activities that may also play a role in their anti-cancer
activities. Some ACEIs have intrinsic metal-chelating properties,
which are thought to be responsible for the inhibition of matrix
metalloproteinase (37). Plasminogen
activator inhibitor (PAI) levels can be directly reduced by ACEIs.
Drugs containing the free sulfhydryl group lead to the generation
of angiostatin, a protein that inhibits angiogenesis (38). The same group also acts as a free
radical scavenger (39). A reduction
in reactive oxygen species prevents the subsequent activation of
metalloproteinase and VEGF, thus reducing tumor invasion. The
pleiotropic effects of ACEIs include anti-inflammatory,
antioxidant, anti-thrombotic, and pro-fibrinolitic activities. It
also improves arterial compliance through the cytoprotection of the
vascular endothelium (35). As
presented above, ACEIs, through the blocking of ACE, cause a
variety of actions with the activation or inhibition of numerous
processes and pathways. Thus, it is difficult to access and
evaluate the precise mechanism involved in the effect of this group
of drugs on cancer cells.
Role of Ang 1–7 in RCC
The use of ACEI increases the plasma levels of
Ang-I, Ang 1–7, as well as renin activity (31). Thus, it is crucial to underline the
role of Ang 1–7. Zheng et al examined the influence of Ang
1–7 on cell migration and invasion in two RCC cell lines: Caki-1
and 786-O. The migration rates of both types of cells were strongly
promoted by Ang 1–7. The invasion ability of the Caki-1 and 786-O
cells was increased in the presence of Ang 1–7 by approximately
1.5-fold. Both activities are driven by the Mas receptor (40). Moreover, Ang 1–7 through the Mas
receptor causes a pro-inflammatory effect in the kidney through the
local activation of the NFκB pathway, as well as the upregulation
of proinflammatory genes (41). This
altogether suggests Ang 1–7 may have a significant impact on RCC
development and progression. The role of Ang 1–7 in renal cancers
is contrary to that of other tumors, where it was suggested that
Ang 1–7 inhibits cell migration and invasiveness (42). Interestingly, the signaling of Ang 1–7
through MasR inhibits tumorgenesis, probably through its effect on
angiogenesis. It has been proven that Ang 1–7 significantly reduces
tumor growth and microvascular density and significantly reduces
the VEGFA expression (43,44). The effect of this discrepancy in Ang
1–7 on tumors can be explained by different experimental conditions
or cell-specific signaling. The presented data suggests that in
RCC, Ang 1–7 tends to have a pro-cancerous rather than
anti-cancerous effect, but this correlation should be examined in
more detail at the molecular level and in xenograft models.
Role of Bradykinin in RCC
Physiologically, bradykinin works as a modulator of
renal function, is responsible for electrolyte and water balance,
and possesses vasodilatatory properties. Via two G-protein-coupled
receptors, B1 and B2, bradykinin can promote renal cell growth and
proliferation (45–47). Both, the B1 and B2, receptors are
expressed in the membranes of ccRCC cells (48). Kramarenko discovered that bradykinin
promotes the proliferation of RCC cells (A498 cell line) through
the B2 receptor. This activation is dependent on the PLC, PKC, and
ERK pathways, as well as on Ca2+/Cam activity (49). Thus, the increased concentration of
bradykinin observed in ACEI users may promote carcinogenesis and
tumor cell proliferation.
Risk of RCC among ACEI inhibitor
users
Lever, Hole and Gillis (50) reported a 28% reduction in relative
risk of cancer incidence among ACEI users compared with general
subjects (HR 0.72, 95% CI, 0.55–0.92), and more attention was paid
to the role of angiotensin system inhibitors in carcinogenesis.
Many analyses of the impact of ACEI on cancer risk were published,
but only a few contained data about the specific risk of RCC. Yoon
et al performed a meta-analysis of cancer risk among ACEI
and ARB users. When stratified by cancer origin site, ACE use was
associated with an increased risk of kidney cancer (RR 1.50, 95%
CI, 1.01–2.23), as well as melanoma, and a reduced risk of
esophageal tumors. The correlation between RCC and ACEI usage was
no longer observed when conventional case-control studies were
excluded (51). Although, Fryzek
et al reported a 1.6 times increased risk of RCC among users
of any antihypertensive drug, the increased risk was not associated
with any specific class of drugs. The number of prescriptions and
length of follow-up did not influence RCC risk (52). In a large analysis of over 3,000
patients with glomerulonephritis, 1% developed any kind of cancer,
but no differences in estimated cancer incidence were reported
regarding the use of ACEIs and ARBs (53). The use of RAS inhibitors significantly
decreased cancer mortality by 0.2% in ACEI/ARB users vs. 2.1% in
non-users. ARB usage was an independent risk factor for cancer
mortality, which was 0.124-fold compared to non-users of ARBs,
while usage of ACEIs did not affect cancer mortality (53). ARBs and ACEIs are safe and do not
increase cancer incidence, while ARBs are related with a decrease
in all-cause and cancer-related mortality.
Influence of ACEIs on cancerogenesis
in in vitro and xenograft models
There are contrary reports on the effect of ACEIs on
renal cancer, suggesting they might have an inhibitory effect on
RCC cells and xenograft tumors or could promote carcinogenesis.
Wysocki et al showed that captopril promotes the growth of
immunogenic tumors in immunocompetent mice through the regulation
of the host's immune system. When mice with a RenCa cell-mediated
tumor were treated with different concentrations of captopril,
decreased survival was observed among the group treated with the
highest concentration of the drug. Similar results were noticed
among animals treated with captopril after nephrectomy in
comparison to the control group (54). Alternatively, Hii et al first
described the inhibitory effect of captopril on tumor growth in a
xenograft model of RCC. Using SN12K-1 cells, they proved that
captopril reduces the size of the tumor in a
concentration-dependent manner with no side effects. A reduction in
tumor growth is not a direct effect of ACEI action on RCC cells,
but rather an indirect effect of decreasing Ang-II levels and
inhibiting of angiogenesis (55).
Araujo et al performed experiments in a
xenograft model using RenCa and a murine cell line of spontaneous
origin. Mice treated with ACEI (captopril 10 mg/kg/day), ARB
(losartan 1,000 mg/kg/day), or both together exhibited
significantly reduced tumor growth, but ACEI and ARB alone had a
better impact when compared with double blockage (ACEI + ARB).
Treatment with ACEI reduced the frequency of lung metastases at
14.3% when used alone and at 28.6% when combined with ARB (56). ACEI and ARB use results in the
decreased expressions of VEGF and CD34, a hemopoietic marker of
stem cells, in xenograft models of renal cancer (56). Captopril at a clinically achievable
concentration of 0.1–10 µM has no effect on proliferation in
SN12K-1 cells, but a higher concentration can reduce it by 14–31%
(55).
Effects of ACEIs on RCC cell lines and
xenograft tumors when combined with TKIs
To evaluate the impact of ACEIs on RCC, a few in
vitro and in silico studies were conducted with a
combination of ACEIs and TKIs, commonly used in the treatment of
metastatic renal cancer. When the 769-P and A-498 cell lines were
incubated with sunitinib and captopril or lisinopril in increasing
concentrations (from 0 to 1,000 µmol/l), a decrease in cell
viability was observed. None of the agents was able to cause a
significant decrease alone. While under physiological conditions,
sunitinib acts primarily on endothelial rather than cancer cell
growth, in the same concentration, but combined with captopril and
lisinopril, it can significantly reduce cell viability. A similar
study was conducted with the use of temsirolimus, showing that the
addition of captopril and lisinopril caused a decrease in cell
viability, but only in high concentrations (1,000 µmol/l) (57).
The combination of interferon alfa, cimetidine,
COX-2 inhibitor, and ASI (I-CCA therapy) was evaluated in a Phase
II trial by Tatokoro et al (2011). Patients were receiving
combined therapy with ACEI perindopril, which during the trial, due
to side effects, was replaced by the AT1-R antagonist candesartan.
In the study, objective response rate (ORR) was 22%, complete
response rate (CR) 8%, median progression free survival (PFS) 12
months and overall survival (OS) was 30 months, and all of those
measurements were significantly better than were those presented in
studies that used IFN-α in monotherapy. The efficacy of I-CCA
therapy was comparable to that of sunitinib or IFN-a plus
bevacizumab (58). The monthly cost
of such treatment is approximately three times lower than
sunitinib.
Impact of ACEIs on RCC survival
Impact of ACEIs on RCC survival in patients after
nephrectomy
The use of drugs targeting the RAS (ACEIs or ARBs)
at the moment of diagnosis or nephrectomy can improve prognosis and
survival. Miyaima et al reported that five-year
metastasis-free survival rates were at 93.7% for ARB/ACEIs users
and 83.9% for their counterparts (P=0.035), while five-year
disease-specific survival rates were 96.8 and 89.8%, respectively.
Proportions are also significantly higher in ARB/ACEI users when
compared to patients receiving other antihypertensive drugs
(59). Neither ACEI nor ARB
administration was an independent risk factor for a decrease in
metastasis-free survival (HR 2.36) and disease-specific survival
(HR 2.69) (59). The use of RAS
inhibitors tends to be an independent predictive factor of
metastasis-free survival and disease-specific survival.
Impact of ACEIs on RCC survival in patients with
systemic treatment
Keizman et al first described the correlation
between ASI use and the clinical outcome of sunitinib treatment. In
the analysis of a small cohort of 127 patients, subjects receiving
ACEIs or ARBs (n=44) showed higher ORRs at the first imaging
follow-up after three months in this group vs. non-users (86% vs.
72%). In addition, a decrease in progression rates from 28 to 14%
was observed. Recipients of ASIs had doubled the median PFS (13 vs.
6 months, HR 0.537, P=0.0055) and demonstrated an insignificant
increase in the median OS (30 vs. 23 months, HR=0.688, P=0.21)
(60). In a multivariate analysis,
ASI use was independently associated with improved PFS (HR=0.54)
(60). McKay performed an analysis of
almost 5,000 RCC patients treated with targeted agents, almost
1,500 of whom used ACEIs, ARBs, or both before and during
treatment. OS was significantly longer in ASI users than in
non-users (adjusted HR 0.848, 95% CI 0.731–0.960) and individuals
receiving no antihypertensive treatment (adjusted HR 0.81, 95% CI
0.707–0.929). Similarly, PFS was significantly longer in ASI users
compared with users of other drugs (HR 0.786, 95% CI 0.707–0.876,
median 8.4 vs. 6.70 months). In a multivariate analysis, a lack of
ASI use was an independent predictor of a worsened OS. When
stratified by therapy type, improvements in OS and PFS among ASI
users were observed only in patients receiving VEGF-targeted
therapy. ORRs in patients receiving ASIs were slightly higher, at
28.31% vs. 22.74%, but the correlation did not reach statistical
significance and should be evaluated in clinical studies (57). Results similar to those of Keizman
et al (60) and McKay et
al (57) were presented by
Izzedine et al, who in a multivariable Cox regression model
also showed the significant association between ASI use and PFS and
OS, with HR=0.4 (0.24–0.66; P<0.001) and HR=0.55 (0.35–0.86;
P=0.009), respectively. The protector effect of an ASI user
decreases over time (61). Another
study of 1,120 patients (361 RCC patients) treated with VEGF
signaling pathway inhibitors (sunitinib, sorafenib, pazopanib and
others) showed no significant improvements in survival rates
between ASI and other drug users (62).
Recently, a secondary pooled analysis of two Phase
III randomized controlled trials of patients with mRCC (NCT00334282
and NCT00720941) was published. After adjustment for baseline
systolic blood pressure (SBP) and the use of non-ASI
antihypertensive drugs and standard prognostic factors in a
multivariable model, no significant association was observed
between ASI use and OS. When individual VEGF-targeted therapies
were analyzed separately, there was a marginally significant
association between ASI use and improved OS in patients receiving
sunitinib (HR 0.73, P=0.03), but not for those receiving pazopanib
or a placebo. A multivariable analysis did not produce a
significant improvement in PFS among ASI users (63). In addition, Penttila et al
(64) did not prove any association
between ASI use and PFS or OS among patients treated with sunitinib
or pazopanib. Because most studies combine in one study group
patients receiving ACEIs and ARBs, it is hard to evaluate the
influence of each drug class separately. An analysis by Sorich
et al (63) did not show a
significant association with OS, PFS, ACEIs, or ARBs using patients
grouped separately. There is no clear evidence of the impact of
other antihypertensive agents, such as beta blockers or calcium
channel blockers. Some authors report no influence on ORRs, PFS, or
OS among RCC patients (60), while
others show a significant association between the baseline use of
calcium channel blockers and improved OS (63). Different results concerning ASI
correlation with patient survival do not help to specify the direct
mechanisms of their activity in RCC patients. Many authors suggest
that a blockage of the RAS leads to decreased cell proliferation
and changes in the tumor microenvironment. Another theory
underlines the role of sarcopenia, which predicts sunitinib-induced
early dose-limiting toxicities in mRCC. The use of ACEIs helps to
preserve muscle mass and therefore may improve the therapeutic
index of VEGF-targeted therapies, subsequently resulting in a
longer duration, higher dose-intensity, and improved outcomes
(65).
Hypertension in TKI users
Incidence of TKI-induced
hypertension
Currently, tyrosine kinase inhibitors (TKIs)
targeting the VEGF pathway are commonly used for the treatment of
metastatic RCC. One of the on-side effects of this class of drugs
is hypertension (HTN). Clinical trials with sunitinib malate have
shown a 34% incidence of any-grade hypertension among RCC patients.
Sunitinib-induced hypertension is defined as an SBP over 140 mmHg
or DBP over 90 mmHg during treatment (66). In a study by Rini et al by the
end of cycle two, up to 80% of patients treated with sunitinib had
systolic-defined HTN and 68% had diastolic-defined HTN (67). The development of HTN is associated
with better clinical outcomes. ORR was almost seven times better
among patients who developed HTN (54.8 vs. 8.7%). The median PFS
and OS are significantly longer among patients with HTN than among
those without, with no difference in whether HTN was defined as SBP
or DBP over the normal limit. The use of antihypertensive drugs
does not reduce the antitumor efficacy of sunitinib and ORR,
results that are similar in both groups (67).
The most likely mechanism of the rapid increase in
SBP and DBP at the beginning of treatment is a decrease in nitric
oxide synthase (NOS) activity caused by the inhibition of VEGF.
This leads to reduced nitric oxide production, which in
physiological conditions possess vasodilatatory activity (68). In the long term, anti-angiogenic drugs
can lead to the chronic remodeling of the capillaries, resulting in
a reduction of their density, called capillary rarefraction
(69). The RAS was considered one of
the additional pathways involved in the development of VEGF
inhibitor-mediated hypertension, but one clinical study showed a
decrease in plasma renin concentration and plasma renin activity
among patients treated with sunitinib (70) and no significant changes in renin and
aldosterone levels in the plasma of patients receiving sorafenib
(71). Clinical data were confirmed
in a mouse model where plasma renin activity was decreased in rats
exposed to TKI cediranib (72). Those
observations provide evidence that the activation of the RAS is not
involved in the development of hypertension during antiangiogenic
treatment, which suppresses the system.
Treatment of TKI-induced HTN
According to TKIs' manufacturers recommendations, BP
should be monitored during treatment, and when necessary,
antihypertensive treatment should be started or intensified. No
treatment algorithm suggesting the class of antihypertensive drugs
has been proposed yet, but in most cases, hypertension can be
controlled with standard medications. ACEI and ARBs are the most
commonly prescribed drugs for patients who developed TKI-associated
hypertension. However, some drugs can be more effective in treating
anti-VEGF-associated hypertension and have less toxicity in
combination with targeted agents. It is suggested that the ACEI
enalapril and the ARB candesartan may inhibit angiogenesis and
trigger the effects of anti-VEGF drugs. Those data were confirmed
in the study on myocardial angiogenesis, but not tested in RCC
models (73). In addition, the ASI
has an additional benefit of improving endothelial function and
microvascular density, key factors involved in anti-VEGF-induced
hypertension (74).
Hamnvik et al presented data that 30.4% of
patients receive ACEI/ARBs as a first class drug for TKI-induced
HTN, while calcium channel blockers are started/intensified in
23.7% and beta blockers in 17.8% (62). Szmit et al (75) suggest that treatment with at least
three antihypertensive drugs significantly improves PFS on
sunitinib and OS compared with patients who received one, two, or
no medications.
Another important issue in the treatment of
hypertension is drug-drug interactions. The metabolism of
sunitinib, which undergoes oxidative metabolism mediated by
cytochrome p450, mainly CYP3A4, may be potentially altered by
antihypertensive drugs that inhibit CYP3A4. Such properties have
non-dihydropyridine calcium channel blockers, such as verapamil
(76). In rats with cediranib-induced
hypertension, captopril showed activity of lowering a 10-mmHg
increase in BP, but was ineffective with a 35–50-mmHg increase
(72). The concomitant use of ACEI
captopril with sorafenib can lead to a 30-mmHg decrease in a rise
in BP in comparison with sorafenib alone (155 vs. 182 mmHg,
P<0.05) and can reduce albuminuria by 50%. Those findings
demonstrate that ACEI due to its protective effect on the
glomerular filtration barrier can reduce therapy-related glomerular
and renal tubular injury, which often occurs during TKI treatment.
Captopril improves the renal autoregulatory capacity (77). In another experimental model with rats
exposed to sunitinib, captopril had no BP-lowering effect, an
effect observed for amlodipine. On the contrary, captopril
significantly attenuated a rise in proteinuria. The serum
concentration of sunitinib was increased threefold while combined
with captopril, confirming the renoprotective effect of ACEI is not
associated with the reduced bioavailability of sunitinib.
Sunitinib-induced hypertension was associated with a rise in
endotelin-1 excretion, which was attenuated by captopril and
sildenafil but not amlodipine (78).
Recently, Penttila et al published a first analysis of the
treatment of TKI-induced hypertension with ASIs in patients with
metastatic RCC. The study showed that patients receiving ASI as a
novel anti-HTN treatment had a significantly longer PFS and OS. It
also demonstrated a better OS and PFS among patients with
treatment-related HTN for ASI users vs. non-ASI users (64). The study did not asses the differences
between the use of ACEI and ARB. High doses of captopril are
effective in protecting against renal injury associated with TKIs,
but its effect on increased BP is visible only with a small
increase in BP. Based on recent observations stating hypertension
induced by TKIs is associated with changes in microvasculature and
renin suppression, dihydropyridine calcium channel blockers rather
than ASIs are more effective in lowering BP. With the beneficial
effect on renal protection, ASIs could be combined with calcium
channel blockers.
Summary
Molecular studies have revealed specific alterations
in the RAS components in RCC. Much evidence concerns the
overexpression of angiotensin receptors and the downregulation of
ACE. These changes correlate with tumor aggressiveness and grading
in Fuhrman's scale; thus, AT1-R, AT2-R, ACE, and ACE 2 can be used
as additional biomarkers in histopathological examinations. A
different staining pattern may help to determine the type and
origin of RCC.
Data that are more inconsistent come from the
evaluation of cancer incidence among ACEI users. Despite a general
decrease in cancer incidence, one study showed an increase in RCC
among ACEI recipients. Further studies did not correlate ACEI with
any kind of RCC. Based on this knowledge, this group of drugs can
be used without fear of developing kidney cancer.
Influence on survival was evaluated in both, a
xenograft model and clinical practice. Some authors present data
suggesting ACEIs decrease survival in a mouse model, while others
prove decreases in tumor growth, cell viability, and proliferation
when used alone or combined with sunitinib. Significant
improvements in PFS, OS, and ORR are observed in clinical practice.
Similar effects are described for ARBs, but not for any other class
of antihypertensive drugs.
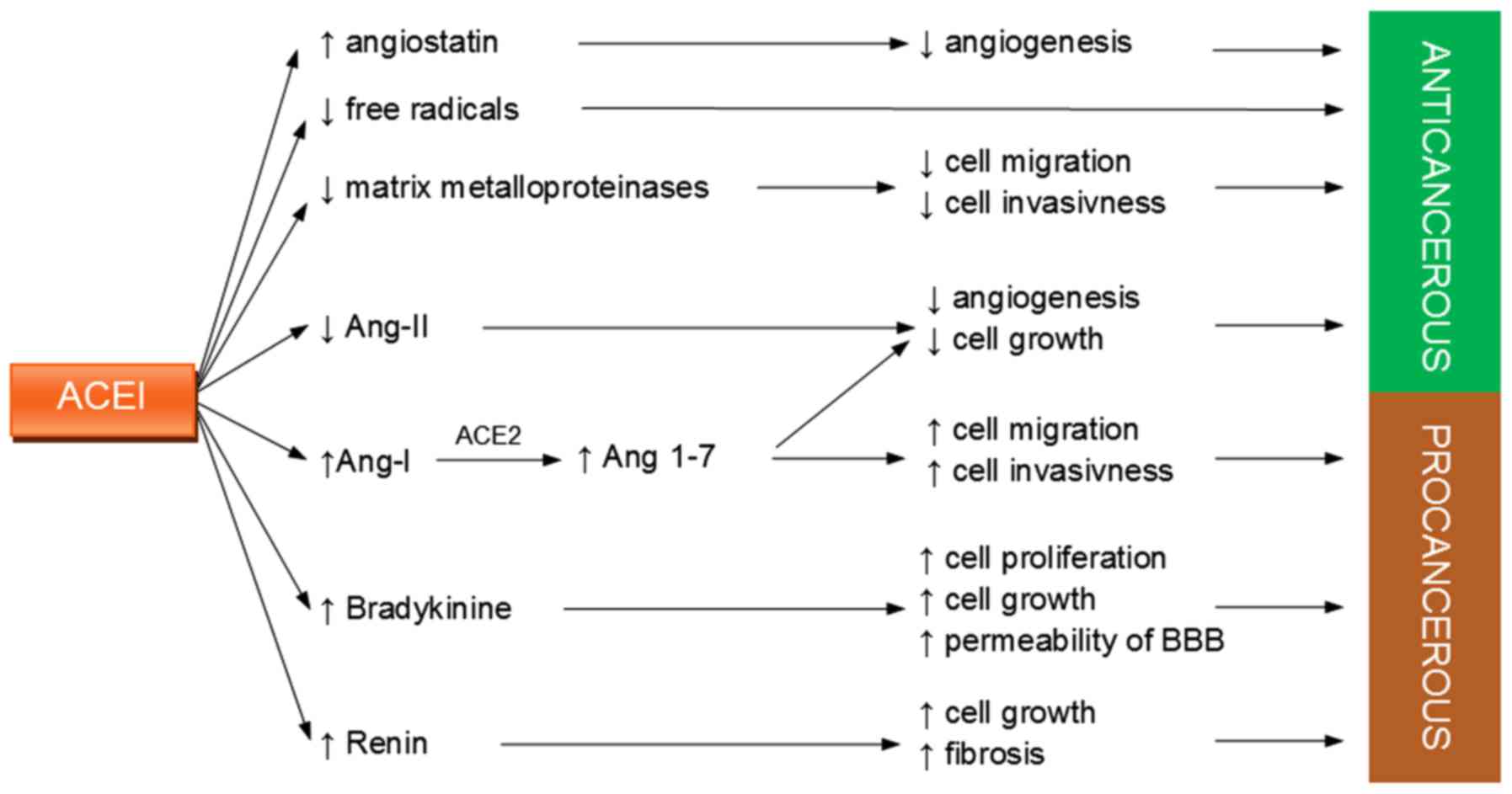
The molecular background of the described effects of
ACEI on RCC is complex and not completely discovered. ACEI affects
the levels of many RAS components: Ang-I, Ang-II, ACE, Ang 1–7, and
ACE2, as well as bradykinin, whose cleavage is mediated by ACE
(Fig. 2). A decrease in Ang-II
activity, NO, cGMP, prostaglandin E2, and prostacyclin causes
anti-cancerous effects intensified by an ACEI-mediated decreased in
the PAI level and the inhibition of metalloproteinase activity.
Less clear is the role of Ang 1–7, whose levels are elevated due to
the inhibition of ACE and the over-reactivity of ACE2. In
experiments on RCC cell lines, Ang 1–7 promoted the migration and
invasion of cells; thus, it is suggested that Ang 1–7 promotes
cancerogenesis. This activity is in opposition to other activities
that increase tumor volume and inhibit angiogenesis and cell
proliferation. Further studies in cell cultures and xenograft
models should be carried out to define the role of Ang 1–7 in
RCC.
Molecular and clinical data leave room for the
possible use of ACEIs in RCC treatment combined with currently used
drugs, such as TKIs. However, translational studies that are more
detailed are necessary to describe molecular bases and prepare
treatment algorithms to use in everyday practice with patients.
Acknowledgements
This study was supported by WIM intramural grant no.
1/8863 (355) founded by Ministry of Science and Higher Education of
Poland.
References
|
1
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Beltran A, Scarpelli M, Montironi R
and Kirkali Z: 2004 WHO classification of the renal tumors of the
adults. Eur Urol. 49:798–805. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brodaczewska KK, Szczylik C, Fiedorowicz
M, Porta C and Czarnecka AM: Choosing the right cell line for renal
cell cancer research. Mol Cancer. 15:832016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rydzanicz M, Wrzesinski T, Bluyssen HA and
Wesoly J: Genomics and epigenomics of clear cell renal cell
carcinoma: Recent developments and potential applications. Cancer
Lett. 341:111–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
George AJ, Thomas WG and Hannan RD: The
renin-angiotensin system and cancer: Old dog, new tricks. Nat Rev
Cancer. 10:745–759. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyajima A, Kosaka T, Kikuchi E and Oya M:
Renin-angiotensin system blockade: Its contribution and
controversy. Int J Urol. 22:721–730. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wegman-Ostrosky T, Soto-Reyes E,
Vidal-Millán S and Sánchez-Corona J: The renin-angiotensin system
meets the hallmarks of cancer. J Renin Angiotensin Aldosterone
Syst. 16:227–233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Derosa L, Izzedine H, Albiges L and
Escudier B: Hypertension and angiotensin system inhibitors in
patients with metastatic renal cell carcinoma. Oncol Rev.
10:2982016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin G, Yan C and Berk BC: Angiotensin II
signaling pathways mediated by tyrosine kinases. Int J Biochem Cell
Biol. 35:780–783. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donoghue M, Hsieh F, Baronas E, Godbout K,
Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan
R, et al: A novel angiotensin-converting enzyme-related
carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9.
Circ Res. 87:E1–E9. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tipnis SR, Hooper NM, Hyde R, Karran E,
Christie G and Turner AJ: A human homolog of angiotensin-converting
enzyme. Cloning and functional expression as a
captopril-insensitive carboxypeptidase. J Biol Chem.
275:33238–33243. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhuo JL and Li XC: New insights and
perspectives on intrarenal renin-angiotensin system: Focus on
intracrine/intracellular angiotensin II. Peptides. 32:1551–1565.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Norwood VF, Craig MR, Harris JM and Gomez
RA: Differential expression of angiotensin II receptors during
early renal morphogenesis. Am J Physiol. 272:R662–R668.
1997.PubMed/NCBI
|
|
14
|
Arendshorst WJ, Brännström K and Ruan X:
Actions of angiotensin II on the renal microvasculature. J Am Soc
Nephrol. 10 Suppl 11:S149–S161. 1999.PubMed/NCBI
|
|
15
|
Matsubara H, Sugaya T, Murasawa S, Nozawa
Y, Mori Y, Masaki H, Maruyama K, Tsutumi Y, Shibasaki Y, Moriguchi
Y, et al: Tissue-specific expression of human angiotensin II AT1
and AT2 receptors and cellular localization of subtype mRNAs in
adult human renal cortex using in situ hybridization. Nephron.
80:25–34. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goldfarb DA, Diz DI, Tubbs RR, Ferrario CM
and Novick AC: Angiotensin II receptor subtypes in the human renal
cortex and renal cell carcinoma. J Urol. 151:208–213. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chatterjee PK, Weerackody RP, Mistry SK,
Hawksworth GM and McLay JS: Selective antagonism of the AT1
receptor inhibits angiotensin II stimulated DNA and protein
synthesis in primary cultures of human proximal tubular cells.
Kidney Int. 52:699–705. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao Z, Kelly DJ, Cox A, Casley D, Forbes
JM, Martinello P, Dean R, Gilbert RE and Cooper ME: Angiotensin
type 2 receptor is expressed in the adult rat kidney and promotes
cellular proliferation and apoptosis. Kidney Int. 58:2437–2351.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dolley-Hitze T, Jouan F, Martin B, Mottier
S, Edeline J, Moranne O, Le Pogamp P, Belaud-Rotureau MA, Patard
JJ, Rioux-Leclercq N and Vigneau C: Angiotensin-2 receptors (AT1-R
and AT2-R), new prognostic factors for renal clear-cell carcinoma?
Br J Cancer. 103:1698–1705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gonzalez-Villalobos RA, Shen XZ, Bernstein
EA, Janjulia T, Taylor B, Giani JF, Blackwell WL, Shah KH, Shi PD,
Fuchs S and Bernstein KE: Rediscovering ACE: Novel insights into
the many roles of the angiotensin-converting enzyme. J Mol Med
(Berl). 91:1143–1154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krege JH, John SW, Langenbach LL, Hodgin
JB, Hagaman JR, Bachman ES, Jennette JC, O'Brien DA and Smithies O:
Male-female differences in fertility and blood pressure in
ACE-deficient mice. Nature. 375:146–148. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin C, Datta V, Okwan-Duodu D, Chen X,
Fuchs S, Alsabeh R, Billet S, Bernstein KE and Shen XZ:
Angiotensin-converting enzyme is required for normal myelopoiesis.
FASEB J. 25:1145–1155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chappell MC: Emerging evidence for a
functional angiotensin-converting enzyme 2-angiotensin-(1–7)-MAS
receptor axis: More than regulation of blood pressure?
Hypertension. 50:596–599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takada Y, Hiwada K, Yokoyama M, Ochi K,
Takeuchi M and Kokubu T: Angiotensin converting enzyme. A possible
histologic indicator for human renal cell carcinoma. Cancer.
56:130–133. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Larrinaga G, Pérez I, Sanz B, Blanco L,
López JI, Cándenas ML, Pinto FM, Gil J, Irazusta J and Varona A:
Angiotensin-converting enzymes (ACE and ACE2) are downregulated in
renal tumors. Regul Pept. 165:218–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Usmani BA, Janeczko M, Shen R, Mazumdar M,
Papandreou CN and Nanus DM: Analysis of the insertion/deletion
polymorphism of the human angiotensin converting enzyme (ACE) gene
in patients with renal cancer. Br J Cancer. 82:550–552.
2000.PubMed/NCBI
|
|
27
|
de Martino M, Klatte T, Schatzl G, Waldert
M, Remzi M, Haitel A, Kramer G and Marberger M: Insertion/deletion
polymorphism of angiotensin I-converting enzyme gene is linked with
chromophobe renal cell carcinoma. Urology. 77:1005.e9–1005.e13.
2011. View Article : Google Scholar
|
|
28
|
van der Knaap R, Siemes C, Coebergh JW,
van Duijn CM, Hofman A and Stricker BH: Renin-angiotensin system
inhibitors, angiotensin I-converting enzyme gene insertion/deletion
polymorphism and cancer: The Rotterdam Study. Cancer. 112:748–757.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deckers IA, van den Brandt PA, van
Engeland M, van Schooten FJ, Godschalk RW, Keszei AP and Schouten
LJ: Polymorphisms in genes of the renin-angiotensin-aldosterone
system and renal cell cancer risk: Interplay with hypertension and
intakes of sodium, potassium and fluid. Int J Cancer.
136:1104–1116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andreotti G, Boffetta P, Rosenberg PS,
Berndt SI, Karami S, Menashe I, Yeager M, Chanock SJ, Zaridze D,
Matteev V, et al: Variants in blood pressure genes and the risk of
renal cell carcinoma. Carcinogenesis. 31:614–620. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kohara K, Brosnihan KB and Ferrario CM:
Angiotensin(1–7) in the spontaneously hypertensive rat. Peptides.
14:883–891. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu T, Miller AG, Deliyanti D, Berka DR,
Agrotis A, Campbell DJ and Wilkinson-Berka JL: Prorenin stimulates
a pro-angiogenic and pro-inflammatory response in retinal
endothelial cells and an M1 phenotype in retinal microglia. Clin
Exp Pharmacol Physiol. 42:537–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shibayama Y, Fujimori T, Nguyen G, Hirose
T, Totsune K, Ichihara A, Kitada K, Nakano D, Kobori H, Kohno M, et
al: (Pro)renin receptor is crucial for Wnt/β-catenin-dependent
genesis of pancreatic ductal adenocarcinoma. Sci Rep. 5:88542015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trask AJ and Ferrario CM:
Angiotensin-(1–7): pharmacology and new perspectives in
cardiovascular treatments. Cardiovasc Drug Rev. 25:162–174. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yacoub R and Campbell KN: Inhibition of
RAS in diabetic nephropathy. Int J Nephrol Renovasc Dis. 8:29–40.
2015.PubMed/NCBI
|
|
36
|
Montana V and Sontheimer H: Bradykinin
promotes the chemotactic invasion of primary brain tumors. J
Neurosci. 31:4858–4867. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Williams RN, Parsons SL, Morris TM,
Rowlands BJ and Watson SA: Inhibition of matrix metalloproteinase
activity and growth of gastric adenocarcinoma cells by an
angiotensin converting enzyme inhibitor in in vitro and murine
models. Eur J Surg Oncol. 31:1042–1050. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
de Groot-Besseling RR, Ruers TJ, van
Kraats AA, Poelen GJ, Ruiter DJ, de Waal RM and Westphal JR:
Anti-tumor activity of a combination of plasminogen activator and
captopril in a human melanoma xenograft model. Int J Cancer.
112:329–334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McMurray J and Chopra M: Influence of ACE
inhibitors on free radicals and reperfusion injury: Pharmacological
curiosity or therapeutic hope? Br J Clin Pharmacol. 31:373–379.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zheng S, Yang Y, Song R, Yang X, Liu H, Ma
Q, Yang L, Meng R, Tao T, Wang S and He J: Ang-(1–7) promotes the
migration and invasion of human renal cell carcinoma cells via
Mas-mediated AKT signaling pathway. Biochem Biophys Res Commun.
460:333–340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Esteban V, Heringer-Walther S,
Sterner-Kock A, de Bruin R, van den Engel S, Wang Y, Mezzano S,
Egido J, Schultheiss HP, Ruiz-Ortega M and Walther T:
Angiotensin-(1–7) and the g protein-coupled receptor MAS are key
players in renal inflammation. PLoS One. 4:e54062009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ni L, Feng Y, Wan H, Ma Q, Fan L, Qian Y,
Li Q, Xiang Y and Gao B: Angiotensin-(1–7) inhibits the migration
and invasion of A549 human lung adenocarcinoma cells through
inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol
Rep. 27:783–790. 2012.PubMed/NCBI
|
|
43
|
Soto-Pantoja DR, Menon J, Gallagher PE and
Tallant EA: Angiotensin-(1–7) inhibits tumor angiogenesis in human
lung cancer xenografts with a reduction in vascular endothelial
growth factor. Mol Cancer Ther. 8:1676–1683. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gallagher PE and Tallant EA: Inhibition of
human lung cancer cell growth by angiotensin-(1–7). Carcinogenesis.
25:2045–2052. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mukai H, Fitzgibbon WR, Bozeman G,
Margolius HS and Ploth DW: Bradykinin B2 receptor antagonist
increases chloride and water absorption in rat medullary collecting
duct. Am J Physiol. 271:R352–R360. 1996.PubMed/NCBI
|
|
46
|
Bagaté K, Grima M, Imbs JL, Jong WD,
Helwig JJ and Barthelmebs M: Signal transduction pathways involved
in kinin B(2) receptor-mediated vasodilation in the rat isolated
perfused kidney. Br J Pharmacol. 132:1735–1742. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
El-Dahr SS, Dipp S and Baricos WH:
Bradykinin stimulates the ERK->Elk-1->Fos/AP-1 pathway in
mesangial cells. Am J Physiol. 275:F343–F352. 1998.PubMed/NCBI
|
|
48
|
Moodley R, Snyman C, Odhav B and Bhoola
KD: Visualisation of transforming growth factor-beta 1, tissue
kallikrein, and kinin and transforming growth factor-beta receptors
on human clear-cell renal carcinoma cells. Biol Chem. 386:375–382.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kramarenko II, Morinelli TA, Bunni MA,
Raymond JR Sr and Garnovskaya MN: The bradykinin B(2) receptor
induces multiple cellular responses leading to the proliferation of
human renal carcinoma cell lines. Cancer Manag Res. 4:195–205.
2012.PubMed/NCBI
|
|
50
|
Lever AF, Hole DJ, Gillis CR, McCallum IR,
McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL and
Robertson JW: Do inhibitors of angiotensin-I-converting enzyme
protect agains risk of cancer? Lancet. 352:179–184. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yoon C, Yang HS, Jeon I, Chang Y and Park
SM: Use of angiotensin-converting-enzyme inhibitors or
angiotensin-receptor blockers and cancer risk: A meta-analysis of
observational studies. CMAJ. 183:E1073–E1084. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fryzek JP, Poulsen AH, Johnsen SP,
McLaughlin JK, Sørensen HT and Friis S: A cohort study of
antihypertensive treatments and risk of renal cell cancer. Br J
Cancer. 92:1302–1306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chin HJ, Oh SW, Goo HS, Oh J, Noh JW, Cho
JT, Na KY, Kim S and Chae DW: Members of The PREMIER Group: Effect
of RAAS inhibition on the incidence of cancer and cancer mortality
in patients with glomerulonephritis. J Korean Med Sci. 26:59–66.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wysocki PJ, Kwiatkowska EP, Kazimierczak
U, Suchorska W, Kowalczyk DW and Mackiewicz A: Captopril, an
angiotensin-converting enzyme inhibitor, promotes growth of
immunogenic tumors in mice. Clin Cancer Res. 12:4095–4102. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hii SI, Nicol DL, Gotley DC, Thompson LC,
Green MK and Jonsson JR: Captopril inhibits tumour growth in a
xenograft model of human renal cell carcinoma. Br J Cancer.
77:880–883. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Araujo WF, Naves MA, Ravanini JN, Schor N
and Teixeira VP: Renin-angiotensin system (RAS) blockade attenuates
growth and metastatic potential of renal cell carcinoma in mice.
Urol Oncol. 33:389.e1–e7. 2015. View Article : Google Scholar
|
|
57
|
McKay RR, Rodriguez GE, Lin X, Kaymakcalan
MD, Hamnvik OP, Sabbisetti VS, Bhatt RS, Simantov R and Choueiri
TK: Angiotensin system inhibitors and survival outcomes in patients
with metastatic renal cell carcinoma. Clin Cancer Res.
21:2471–2479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tatokoro M, Fujii Y, Kawakami S, Saito K,
Koga F, Matsuoka Y, Iimura Y, Masuda H and Kihara K: Phase-II trial
of combination treatment of interferon-α, cimetidine,
cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor
(I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci.
102:137–143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Miyajima A, Yazawa S, Kosaka T, Tanaka N,
Shirotake S, Mizuno R, Kikuchi E and Oya M: Prognostic impact of
renin-angiotensin system blockade on renal cell carcinoma after
surgery. Ann Surg Oncol. 22:3751–3759. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Keizman D, Huang P, Eisenberger MA, Pili
R, Kim JJ, Antonarakis ES, Hammers H and Carducci MA: Angiotensin
system inhibitors and outcome of sunitinib treatment in patients
with metastatic renal cell carcinoma: A retrospective examination.
Eur J Cancer. 47:1955–1961. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Izzedine H, Derosa L, Le Teuff G, Albiges
L and Escudier B: Hypertension and angiotensin system inhibitors:
Impact on outcome in sunitinib-treated patients for metastatic
renal cell carcinoma. Ann Oncol. 26:1128–1133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hamnvik OP, Choueiri TK, Turchin A, McKay
RR, Goyal L, Davis M, Kaymakcalan MD and Williams JS: Clinical risk
factors for the development of hypertension in patients treated
with inhibitors of the VEGF signaling pathway. Cancer. 121:311–319.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sorich MJ, Kichenadasse G, Rowland A,
Woodman RJ and Mangoni AA: Angiotensin system inhibitors and
survival in patients with metastatic renal cell carcinoma treated
with VEGF-targeted therapy: A pooled secondary analysis of clinical
trials. Int J Cancer. 138:2293–2299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Penttila P, Rautiola J, Poussa T, Peltola
K and Bono P: Angiotensin inhibitors as treatment of
Sunitinib/Pazopanib-induced hypertension in metastatic renal cell
carcinoma. Clin Genitourin Cancer. 15:384–390.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Huillard O, Xylinas E, Peyromaure M,
Alexandre J and Goldwasser F: Angiotensin system inhibitors in
renal cell carcinoma-letter. Clin Cancer Res. 22:5242016.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chobanian AV, Bakris GL, Black HR, Cushman
WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright
JT Jr, et al: Seventh report of the Joint National Committee on
prevention, detection, evaluation, and treatment of high blood
pressure. Hypertension. 42:1206–1252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rini BI, Cohen DP, Lu DR, Chen I,
Hariharan S, Gore ME, Figlin RA, Baum MS and Motzer RJ:
Hypertension as a biomarker of efficacy in patients with metastatic
renal cell carcinoma treated with sunitinib. J Natl Cancer Inst.
103:763–773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Robinson ES, Khankin EV, Choueiri TK,
Dhawan MS, Rogers MJ, Karumanchi SA and Humphreys BD: Supression of
the nitric oxide pathway in metastatic renal cell carcinoma
patients receiving vascular endothelial growth factor-signaling
inhibitors. Hypertension. 56:1131–1136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gupta R and Maitland ML: Sunitinib,
hypertension, and heart failure: A model for kinase
inhibitor-mediated cardiotoxicity. Curr Hypertens Rep. 13:430–435.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kappers MH, van Esch JH, Sluiter W,
Sleijfer S, Danser AH and van den Meiracker AH: Hypertension
induced by the tyrosine kinase inhibitor sunitinib is associated
with increased circulating endothelin-1 levels. Hypertension.
56:675–681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Veronese ML, Mosenkis A, Flaherty KT,
Gallagher M, Stevenson JP, Townsend RR and O'Dwyer PJ: Mechanisms
of hypertension associated with BAY 43–9006. J Clin Oncol.
24:1363–1369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Curwen JO, Musgrove HL, Kendrew J,
Richmond GH, Ogilvie DJ and Wedge SR: Inhibition of vascular
endothelial growth factor-a signaling induces hypertension:
Examining the effect of cediranib (recentin; AZD2171) treatment on
blood pressure in rat and the use of concomitant antihypertensive
therapy. Clin Cancer Res. 14:3124–3131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Siddiqui AJ, Mansson-Broberg A, Gustafsson
T, Grinnemo KH, Dellgren G, Hao X, Fischer H and Sylvén C:
Antagonism of the renin-angiotensin system can counteracts cardiac
angiogenic vascular endothelial growth factor gene therapy and
myocardial angiogenesis in normal heart. Am J Hyprtens.
18:1347–1352. 2005. View Article : Google Scholar
|
|
74
|
Agabiti-Rosei E: Structural and functional
changes of the microcirculation in hypertension: Influence of
pharmacological therapy. Drugs. 63:19–29. 2003.(In French).
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Szmit S, Langiewicz P, Zlnierek J,
Nurzyński P, Zaborowska M, Filipiak KJ, Opolski G and Szczylik C:
Hypertension as a predictive factor for survival outcomes in
patients with metastatic renal cell carcinoma treated with
sunitinib after progression on cytokines. Kidney Blood Press Res.
35:18–25. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhu X, Stergiopoulos K and Wu S: Risk of
hypertension and renal dysfunction with an angiogenesis inhibitor
sunitinib: Systematic review and meta-analysis. Acta Oncol.
48:9–17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nagasawa T, Khan Hye MA and Imig JD:
Captopril attenuates hypertension and renal injury induced by the
vascular endothelial growth factor inhibitor sorafenib. Clin Exp
Pharmacol Physiol. 39:454–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lankhorst S, Kappers MH, van Esch JH,
Smedts FM, Sleijfer S, Mathijssen RH, Baelde HJ, Danser AH and van
den Meiracker AH: Treatment of hypertension and renal injury
induced by the angiogenesis inhibitor sunitinib: Preclinical study.
Hypertension. 64:1282–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|