Introduction
Helicobacter pylori is a gram-negative,
spiral-shaped bacterium present in the human stomach. It is
estimated that H. pylori infects ~50% of the world's
population, and it is a risk factor not only for the pathogenesis
of chronic gastritis and peptic ulcer disease (1), but has also been classified as a
carcinogen by the World Health Organization (2). Chronic infection with H. pylori
results in prolonged inflammation which is mediated by inflammatory
mediators, including cytokines and prostaglandin E2
(PGE2) (3). In this
regard, upregulation of the inducible type of cyclooxygenase-2
(COX-2) (4) and elevated levels of
pro-inflammatory cytokines (1) have
been observed in the gastric mucosa of patients with H.
pylori infection.
COX-2 is a key rate-limiting and inducible enzyme
responsible for the formation of prostanoids and thromboxanes
(5). PGE2 is the main
prostanoid generated from arachidonic acid, catalyzed by COX-2, and
it exerts physiological as well as pathological effects through
prostaglandin receptors (6,7). Elevation of PGE2 has been
observed in multiple types of cancer and is associated with tumor
growth and angiogenesis (7,8). PGE2 levels are regulated not
only by its synthetic enzyme, COX-2, and microsomal prostaglandin
synthase (mPGES), but also by its degrading enzyme, 15-hydroxy
prostaglandin dehydrogenase (15-PGDH) (9). 15-PGDH is the key enzyme that
inactivates PGE2 by catalyzing the oxidation of its 15
(S)-hydroxy group, which results in the formation of inactive
15-keto metabolites (10). Consistent
with its function of regulating the local concentration of
PGE2, studies have demonstrated that 15-PGDH is
expressed in normal colonic epithelial cells, but transcription of
15-PGDH mRNA is lost in the majority of colon cancers (10–12).
Although the incidence of gastric cancer is
declining, it remains the fourth most common cancer and the second
leading cause of cancer-associated mortality globally (13). Multiple factors may be involved in the
carcinogenesis of gastric cancer, including genetics and
environmental factors. H. pylori is one of the most
important environmental risk factors for gastric malignancies
(13,14). Persistent inflammation in response to
H. pylori infection is hypothesized to contribute to tumor
cell proliferation and metastasis, and to affect survival (15,16). While
the molecular mechanisms of gastric tumors remain poorly
understood, modulation of COX-2 expression and 15-PGDH mRNA, and
the resultant elevated local PGE2 levels, may be
responsible for H. pylori-associated carcinogenesis.
Therefore, prostaglandins, particularly PGE2, may be
involved in maintaining gastric mucosal integrity through
regulating gastric mucosal blood flow, kinetics of epithelial
cells, synthesis of mucus and inhibition of gastric acid secretion
(17). By contrast, PGE2
transactivates epidermal growth factor receptor and initializes
mitogenic signal transduction in gastric epithelial and colon
cancer cells (18), by which
mechanism PGE2 may trigger carcinogenesis and
proliferation of gastrointestinal cancers.
In the present study, the expression of COX-2 and
15-PGDH was investigated in gastric cancer specimens in comparison
with normal tissues, and the levels of these enzymes were analyzed
by grouping the samples with or without H. pylori
infection.
Materials and methods
Study population
The present study enrolled a total of 66 patients
who had received surgical resection of gastric cancer at the Second
Affiliated Hospital of Tianjin Medical University (Tianjin, China)
between January 2011 and August 2012, in order to study the
potential involvement of H. pylori in the development of
gastric cancer. Of these patients, 38 (57.6%) were male; the
average age was 65.8 (range, 35–85) years; 23 patients were tumor
node metastasis (TNM) stage I and II; 43 were TNM stage III and IV
(19); and 44 patients had lymph node
metastasis. In comparison, 70 patients who had no atrophic
gastritis (as evidenced by gastric tissue biopsies) were also
enrolled into the present study. Of these patients 32 patients were
male, with an average age of 39.7 (range, 20–60) years. All the
enrolled patients had no history of using non-steroid
anti-inflammation drugs, antibiotics within 4 weeks of the study,
anti-acid drugs or bismuth. Written consent was obtained from all
patients and the study protocol was approved by the Ethics
Committee of The Tianjin Medical University (Tianjin, China).
Determination of H. pylori
infection
H. pylori infection was determined prior to
surgery by the 14C-urea breath test (3).
Tissue specimen collection and
immunohistochemistry
The tissues obtained via surgical resection from
cancer patients and via biopsy from control subjects were fixed
with 10% formalin, paraffin-embedded and sectioned (4 µm). The
slides were deparaffinized with xylene and then gradually
rehydrated with 100, 95, 85 and 75% ethanol followed by water.
Following antigen retrieval with 10 mM citrate buffer (pH 6.0) at
96°C for 20 min and non-specific blocking with normal goat serum
(Beijing Zhongshan Jinqiao Biotechnology Co., Ltd., Beijing, China)
at room temperature for 1 h, the tissue slides were allowed to
react with monoclonal anti-COX-2 (cat no. 160112; dilution, 1:100)
and anti-15-PGDH (cat no. 160615; dilution 1:200) (both from Cayman
Chemical, Ann Arbor, MI, USA) overnight at 4°C. Following
incubation with horseradish peroxidase-conjugated second antibody
(1:1,000; cat no. ZB-2301; ZSGB-Bio Co., Ltd., Beijing, China) at
room temperature for 20 min, the slides were developed with
3,3′-diaminobenzidine solution (ZSGB-Bio Co., Ltd.), followed by
counterstaining with hematoxylin. Samples were examined under a
light microscope (BX41; Olympus Corporation, Tokyo, Japan) and ≥5
random fields were captured.
All slides were reviewed and scored by one
gastrointestinal pathologist. For 15-PGDH, the number of cells with
positive 15-PGDH was scored under 5 high power field
(magnification, ×400) as follows: 0, <5% cells positively
stained; 1, 5–25% cells positively stained; 2, 25–50% cells
positively stained; 3, 50–75% cells positively stained; and 4,
>75% cells positively stained. COX-2 staining >10% was
considered to be a positive sample of COX-2 expression. 15-PGDH was
semi-quantitatively scored by counting positive cell number as well
as intensity of positive staining as follows: 1, light brown; 2,
brown; 3, dark brown. Total 15-PGDH positive staining score was
calculated as follows: Score by positive cell number × score
intensity of positive staining.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Expression of 15-PGDH mRNA was determined by RT-PCR.
Briefly, total RNA was extracted using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and quantified
using a spectrometer. Sequences of the primers were as follows:
15-PGDH forward, 5′-GCTGACCAGCAACAACTGAGA-3′ and reverse,
5′-CTGGACAAATGGCATTCAGTC-3′; and β-actin forward,
5′-CTGGGACGACATGGAGAAAA-3′ and reverse, 5′-AAGGAAGGCTGGAAGAGTGC-3′.
Briefly, cDNA was synthesized using 1 µg total RNA with the RT kit
(Aoke Biology Research Co., Ltd., Beijing, China). PCR was then
performed using a commercially available kit (Aoke Biology Research
Co., Ltd.). RT-PCR was performed according to the manufacturer's
protocol (Aoke Biology Research Co., Ltd.). The thermocycling
conditions performed were as follows: 95°C for 5 min, 94°C for 30
sec, 57°C for 30 sec, and 72°C for 30 sec; total 30 cycles followed
by 72°C for 10 min.
Statistical analysis
Data were analyzed by SPSS 17.0 software (SPSS,
Inc., Chicago, IL, USA). Quantitative data was expressed as the
mean ± standard deviation. Paired data was analyzed by Student's
t-test. Event occurrence was examined by the χ2 test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Clinical features of patients with or
without H. pylori infection
Of the 66 patients with gastric cancer, 33 (50%)
were infected with H. pylori, while 38 (54.3%) of the 70
control subjects had H. pylori infection. No significant
difference was observed in age and gender between the patients with
or without H. pylori infection (P>0.05; Table I).
 | Table I.Characteristics of the control
patients and patients with gastric cancer with or without H.
pylori infection. |
Table I.
Characteristics of the control
patients and patients with gastric cancer with or without H.
pylori infection.
|
| H.
pylori-positive, n | H.
pylori-negative, n |
|
|
|---|
|
|
|
|
|
|
|---|
|
| GC | Con | GC | Con | χ2
(GC/Con) | P-value |
|---|
| Age (years) |
|
|
|
| 0.32/0.23 | >0.05 |
|
<60 | 11 | 28 | 12 | 24 |
|
|
|
>60 | 22 | 10 | 21 | 8 |
|
|
| Sex |
|
|
|
| 0.29/0.03 | >0.05 |
|
Male | 21 | 17 | 17 | 15 |
|
|
|
Female | 12 | 21 | 16 | 17 |
|
|
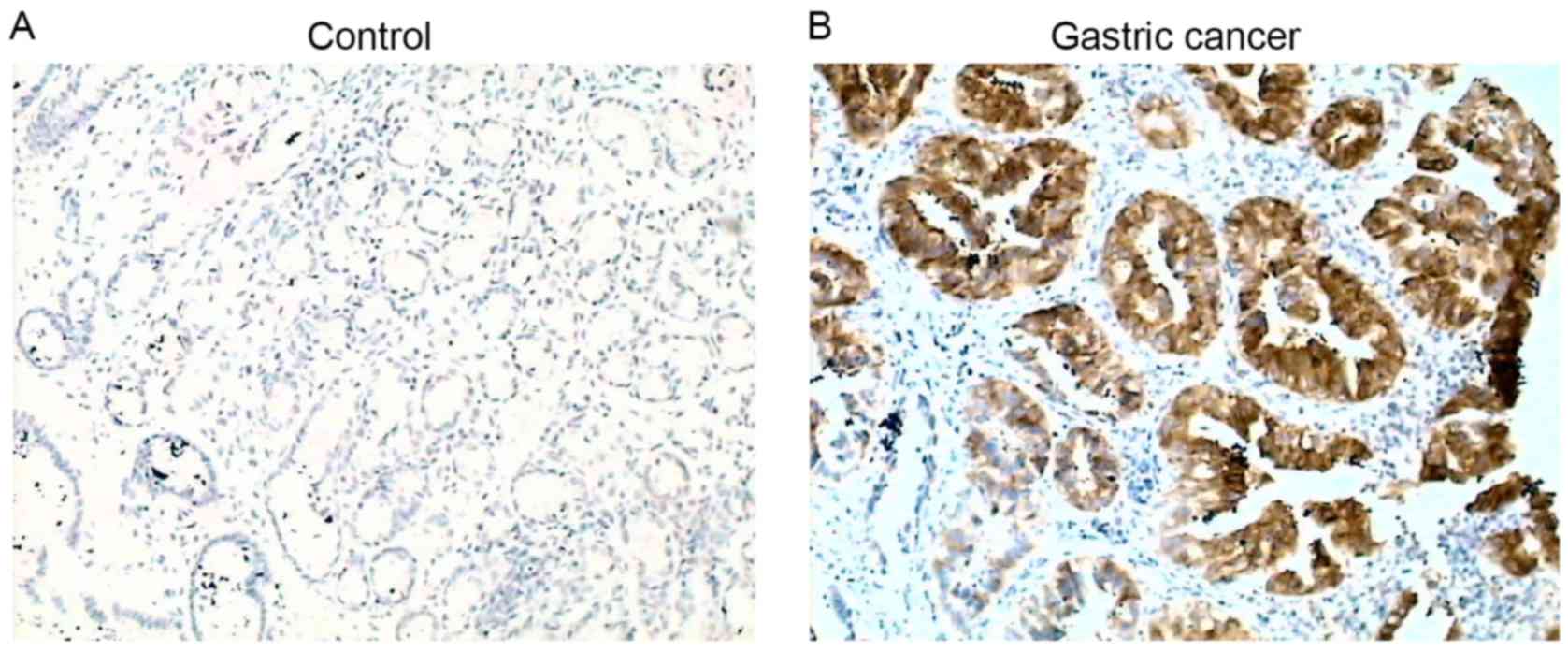
Comparison of COX-2 positivity in
patients with gastric cancer with or without H. pylori
infection
COX-2 protein was detectable in 14.3% (10/70) of
control subjects, while it was detectable in 81.8% (54/66) of
patients with gastric cancer, and was significantly increased
compared with the control (P<0.01; Fig. 1). Of the 66 patients with gastric
cancer, 33 were positive and 33 were negative for H. pylori
infection. COX-2 was positively expressed in 32/33 (97%) patients
with H. pylori infection, while it was positively expressed
in 22/33 (66.7%) patients without H. pylori infection
(P<0.01; Table II).
| Table II.Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in control vs. gastric cancer with
or without H. pylori infection. |
Table II.
Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in control vs. gastric cancer with
or without H. pylori infection.
|
| H. pylori
positive | H. pylori
negative |
|
|---|
|
|
|
|
|
|---|
| Tissue | N | 15-PGDH
Protein | 15-PGDH mRNA | COX-2 +/− | N | 15-PGDH
Protein | 15-PGDH mRNA | COX-2 +/− |
P-valuea |
|---|
| Gastric cancer | 33 | 1.10±0.47 | 0.68±0.31 | 32/1 | 33 | 2.10±0.78 | 1.09±0.51 | 22/11 | <0.01 |
| Control | 38 | 10.02±1.24 | 2.72±1.20 | 6/32 | 32 | 11.54±1.40 | 3.03±1.32 | 4/28 | >0.05 |
|
P-valueb |
| <0.01 | <0.01 | <0.01 |
| <0.01 | <0.01 | <0.01 |
|
In addition, COX-2 protein levels were significantly
increased in patients with gastric cancer with lymphatic metastasis
[100% (23/23) patients with, and 76.2% (16/21) without H.
pylori infection; Table III]
compared with patients without lymphatic metastasis [80% (8/10)
patients with, and 58.3% (7/12) without H. pylori infection;
P<0.01; Table III].
| Table III.Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in gastric cancer with or without
LM and H. pylori infection. |
Table III.
Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in gastric cancer with or without
LM and H. pylori infection.
|
| H.
pylori-positive, n | H.
pylori-negative, n |
|
|---|
|
|
|
|
|
|---|
| Metastasis | Total | 15-PGDH
protein | 15-PGDH mRNA | COX-2 +/− | Total | 15-PGDH
protein | 15-PGDH mRNA | COX-2 +/− |
P-valuea |
|---|
| With LM | 23 | 0.46±0.19 | 0.18±0.07 | 23/0 | 21 | 0.90±0.40 | 0.44±0.20 | 16/5 | <0.01 |
| Without LM | 10 | 1.12±0.50 | 0.56±0.23 | 8/2 | 12 | 1.78±0.80 | 1.02±0.45 | 7/5 | <0.05 |
|
P-valueb |
| <0.01 | <0.01 | <0.01 |
| <0.01 | <0.01 | <0.01 |
|
However, patients had a similar positive rate of
COX-2 expression in stage III–IV gastric cancer [100% (22/22)
patients with H. pylori infection and 66.7% (14/21) without
H. pylori infection; Table
IV] compared with that in stage I–II gastric cancer [91%
(10/11) patients with H. pylori infection and 66.7% (8/12)
without H. pylori infection; Table IV].
| Table IV.Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in gastric cancer at different
stages with or without H. pylori infection. |
Table IV.
Scoring of 15-PGDH protein and mRNA
levels and COX-2 protein levels in gastric cancer at different
stages with or without H. pylori infection.
|
| H.
pylori-positive, n | H.
pylori-negative, n |
|
|---|
|
|
|
|
|
|---|
| Cancer stage | Total | 15-PGDH
protein | 15-PGDH mRNA | COX-2 +/− | Total | 15-PGDH
protein | 15-PGDH mRNA | COX-2 +/− |
P-valuea |
|---|
| I–II | 11 | 1.79±083 | 0.69±0.26 | 10/1 | 12 | 2.47±1.10 | 1.07±0.46 | 8/4 | <0.05 |
| III–IV | 22 | 0.81±0.36 | 0.27±0.11 | 22/0 | 21 | 1.32±0.53 | 0.42±0.17 | 14/7 | <0.05 |
|
P-valueb |
| <0.01 | <0.01 |
|
| <0.01 | <0.01 |
|
|
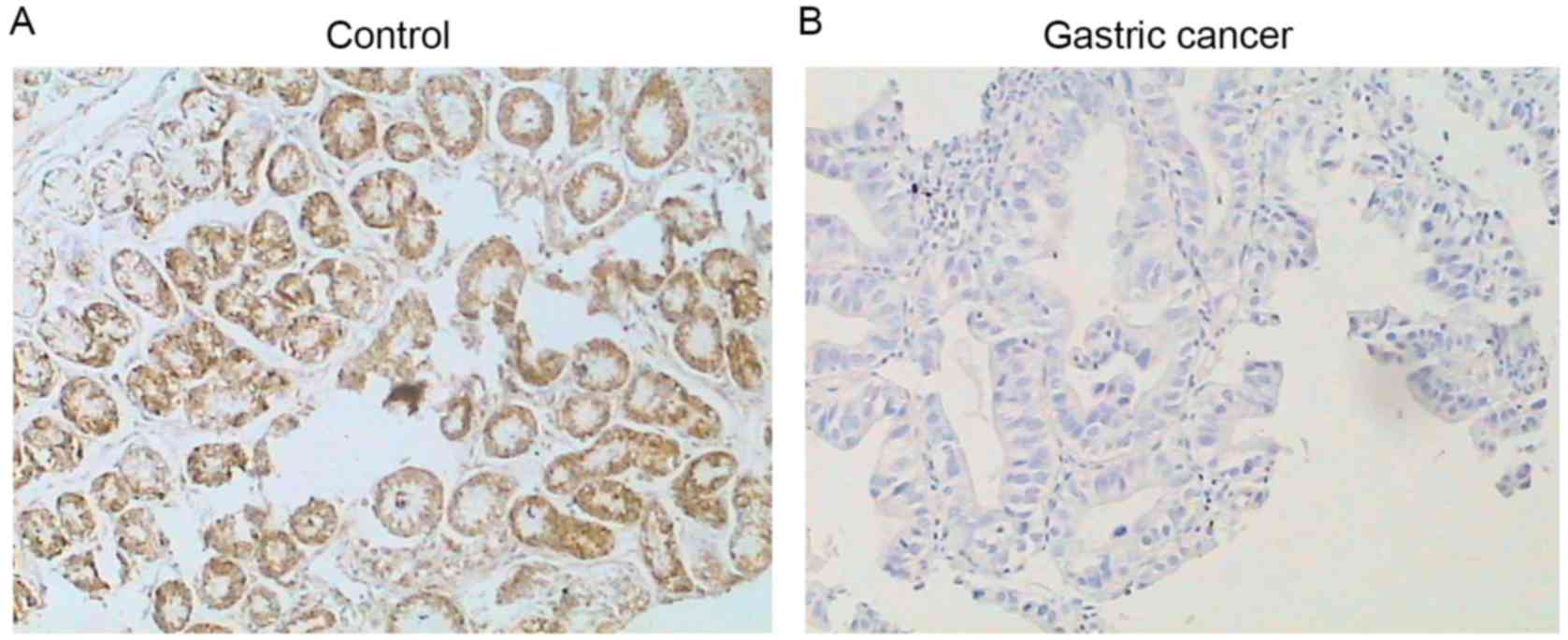
Comparison of 15-PGDH mRNA and protein
expression in patients with gastric cancer with or without H.
pylori infection
15-PGDH protein was detectable by immunostaining in
the control subjects (Fig. 2), and no
difference was observed in the protein level of 15-PGDH in the
control subjects with (10.02±1.24) or without (11.54±1.40) H.
pylori infection (P>0.05; Table
II). Similarly, no difference was observed in the expression of
15-PGDH mRNA between control subjects with (2.72±1.20) or without
(3.03±1.32) H. pylori infection (P>0.05; Table II). However, expression levels of
mRNA and protein of 15-PGDH in the patients with gastric cancer
were significantly reduced compared with normal subjects
(P<0.01; Figs. 2 and 3). In addition, patients with H.
pylori infection had a more significant decrease in 15-PGDH
mRNA (0.68±0.31) and protein (1.10±0.47) compared with patients
without the bacterial infection (1.09±0.51, mRNA; 2.10±0.78,
protein; P<0.01; Table II).
Expression levels of 15-PGDH mRNA and protein were
lowest in the patients with lymphatic metastasis and bacterial
infection (0.18±0.07 for mRNA and 0.46±0.19 for protein; Table III), and were significantly lower
compared with patients without lymphatic metastasis but with
bacterial infection (0.56±0.23, mRNA; 1.12±0.50, protein;
P<0.01; Table III) or patients
with lymphatic metastasis but without bacterial infection
(0.44±0.20, mRNA; 0.90±0.40, protein; P<0.01; Table III). Expression of 15-PGDH mRNA and
protein was highest in the patients without lymphatic metastasis
and negative infection of H. pylori (1.02±0.45, mRNA;
1.78±0.80, protein; P<0.01; Table
III), which were significantly different from the other three
groups (P<0.01; Table III).
Similarly, late stage patients with H. pylori
infection had the lowest mRNA and protein levels of 15-PGDH
(0.27±0.11, mRNA; 0.81±0.36, protein), which were significantly
lower compared with stage I–II patients with bacterial infection
(0.69±0.26, mRNA; 1.79±0.83, protein; P<0.01; Table IV) or stage III–IV patients without
bacterial infection (0.42±0.17, mRNA; 1.32±0.53, protein;
P<0.01; Table IV). Expression of
15-PGDH mRNA and protein was highest in the stage I–II patients
without bacterial infection (1.07±0.46, mRNA; 2.47±1.10, protein)
and were significantly increased compared with the other three
groups (P<0.05; Table IV).
Discussion
Chronic inflammation is involved in the development
of cancer. Bacterial infection of the gastrointestinal tract may
trigger inflammation through elevating PGE2. COX-2 and
15-PGDH are two crucial enzymes that synthesize and degrade
PGE2, respectively. In order to investigate the
expression of COX-2 and 15-PGDH and its potential association with
H. pylori infection in gastric cancer, 33 patients with
gastric cancer with H. pylori infection and 33 patients with
gastric cancer without the bacterial infection were enrolled in the
present study. COX-2 protein was detectable by immunohistochemistry
in almost all the gastric cancer samples infected with H.
pylori, and by contrast it was detectable in <2/3 of the
patients without H. pylori infection. In contrast to gastric
cancer samples, COX-2 was positively expressed in <1/6 of
control subjects regardless of H. pylori infection. However,
the PGE2 degrading enzyme, 15-PGDH, was expressed in
control samples, but its expression was markedly suppressed in
gastric cancer samples. H. pylori infection resulted in
slight inhibition of 15-PGDH in control subjects, but significant
inhibition of 15-PGDH mRNA levels and protein synthesis in patients
with gastric cancer. These findings indicated that enzymes that
regulate PGE2 levels were significantly altered in
gastric cancer, and that H. pylori may be involved in
modulating the synthesis of COX-2 and 15-PGDH.
Previous studies have indicated that 15-PGDH acts as
a tumor suppressor in multiple types of tumors, including breast,
lung, colon, thyroid and pancreatic cancers (12,20–23). Song
et al (24) reported that
15-PGDH was not expressed in 70.1% of gastric cancer specimens. In
addition, Ryu et al (11)
reported that 15-PGDH was suppressed in patients with mild
gastritis who were positive for H. pylori infection.
Consistently, the present study demonstrated that expression of
15-PGDH mRNA and protein was significantly suppressed in gastric
cancer samples compared with control samples, and infection with
H. pylori resulted in additional downregulation of 15-PGDH
mRNA and protein levels. These findings indicated that H.
pylori infection may trigger carcinogenesis of stomach cancer
by elevating PGE2 levels through modulating expression
of 15-PGDH.
Previous studies have also demonstrated that gastric
tumors may be induced by activated macrophages in transgenic mice
overexpressing COX-2 and the PGE2-converting enzyme
microsomal prostaglandin E synthesis (25), and that COX-2 was induced by H.
pylori infection (26,27). A significant upregulation of COX-2 was
also previously identified in >70% of gastric cancer specimens,
and this was reversed when H. pylori was eliminated
(28). The present study revealed
that COX-2 was expressed in >80% (54/66) gastric cancer
specimens, and the patients with H. pylori infection had a
significantly increased positive rate of COX-2 expression compared
with patients without H. pylori infection, indicating that
the activation of prostaglandin synthesis may mediate H.
pylori-induced gastric cancer.
PGE2 levels are controlled not only by
PGE2 synthesis through inducible enzymes, including
COX-2 and mPGES-1/2, but also by the PGE2 inactivating
enzyme 15-PGDH. Notably, the present analysis of gastric cancer
specimens and control subjects revealed that H. pylori
infection was associated with not only significant downregulation
of 15-PGDH, but also upregulation of COX-2. In the present study,
upregulation of COX-2 and suppression of 15-PGDH was demonstrated,
indicating that elevated PGE2 levels in the pathogenesis
of gastric cancer are a consequence of not only increased synthesis
through upregulating COX-2 but also reduced degradation by
suppressing 15-PGDH. Therefore, in addition to antibiotics for
eradication of H. pylori infection, medicines targeting
PGE2 levels through inhibiting COX-2 or stimulating
15-PGDH may be an effective strategy. In this regard, drugs that
decrease the risk of colon carcinogenesis by inhibiting COX-2 have
been tested in clinical trials, however, these drugs are
problematic due to their unfavorable cardiovascular side effects
(29). Thus, a strategy of combining
H. pylori eradication and expression of 15-PGDH in the
gastrointestinal tract may be an effective way to prevent gastric
carcinogenesis.
Taken together, the present study demonstrated that
COX-2 was expressed in almost all gastric cancers infected with
H. pylori, but it was also expressed in <2/3 of gastric
cancers without H. pylori infection. In addition, COX-2 was
positively expressed in <1/6 of control subjects regardless of
H. pylori infection. By contrast, 15-PGDH was expressed in
control samples and it was markedly downregulated in gastric cancer
samples. H. pylori infection resulted in slight inhibition
of 15-PGDH in control subjects, but significant inhibition of
15-PGDH mRNA expression and protein synthesis in the patients with
gastric cancer. These findings indicated that COX-2, which elevates
PGE2 levels and 15-PGDH, which decreases PGE2
levels, are significantly altered in gastric cancer, and that H.
pylori may modulate COX-2 and 15-PGDH mRNA expression and
protein synthesis.
References
|
1
|
Houghton J and Wang TC: Helicobacter
pylori and gastric cancer: A new paradigm for
inflammation-associated epithelial cancers. Gastroenterology.
128:1567–1578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ernst PB, Peura DA and Crowe SE: The
translation of Helicobacter pylori basic research to patient care.
Gastroenterology. 130:188–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu N, Wu Q, Wang Y, Sui H, Liu X, Zhou N,
Zhou L, Wang Y, Ye N, Fu X, et al: Helicobacter pylori promotes
VEGF expression via the p38 MAPKmediated COX2PGE2 pathway in MKN45
cells. Mol Med Rep. 10:2123–2129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu S, Ramanujam KS, Wong A, Fantry GT,
Drachenberg CB, James SP, Meltzer SJ and Wilson KT: Increased
expression and cellular localization of inducible nitric oxide
synthase and cyclooxygenase 2 in Helicobacter pylori gastritis.
Gastroenterology. 116:1319–1329. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Greenhough A, Smartt HJ, Moore AE, Roberts
HR, Williams AC, Paraskeva C and Kaidi A: The COX-2/PGE2 pathway:
Key roles in the hallmarks of cancer and adaptation to the tumour
microenvironment. Carcinogenesis. 30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fujino H, Xu W and Regan JW: Prostaglandin
E2 induced functional expression of early growth response factor-1
by EP4, but not EP2, prostanoid receptors via the
phosphatidylinositol 3-kinase and extracellular signal-regulated
kinases. J Biol Chem. 278:12151–12156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheng H, Shao J, Washington MK and DuBois
RN: Prostaglandin E2 increases growth and motility of colorectal
carcinoma cells. J Biol Chem. 276:18075–18081. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pai R, Szabo IL, Soreghan BA, Atay S,
Kawanaka H and Tarnawski AS: PGE(2) stimulates VEGF expression in
endothelial cells via ERK2/JNK1 signaling pathways. Biochem Biophys
Res Commun. 286:923–928. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tai HH: Prostaglandin catabolic enzymes as
tumor suppressors. Cancer Metastasis Rev. 30:409–417. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Na HK, Park JM, Lee HG, Lee HN, Myung SJ
and Surh YJ: 15-Hydroxyprostaglandin dehydrogenase as a novel
molecular target for cancer chemoprevention and therapy. Biochem
Pharmacol. 82:1352–1360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryu YM, Myung SJ, Park YS, Yang DH, Song
HJ, Jeong JY, Lee SM, Song M, Kim DH, Lee HJ, et al: Inhibition of
15-hydroxyprostaglandin dehydrogenase by Helicobacter pylori in
human gastric carcinogenesis. Cancer Prev Res (Phila). 6:349–359.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Myung SJ, Rerko RM, Yan M, Platzer P, Guda
K, Dotson A, Lawrence E, Dannenberg AJ, Lovgren AK, Luo G, et al:
15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of
colon tumorigenesis. Proc Natl Acad Sci USA. 103:12098–12102. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sitas F: Twenty five years since the first
prospective study by Forman et al (1991) on Helicobacter pylori and
stomach cancer risk. Cancer Epidemiol. 41:159–164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Wu WK, Gallo RL, Fang EF, Hu W,
Ling TK, Shen J, Chan RL, Lu L, Luo XM, et al: Critical role of
antimicrobial peptide cathelicidin for controlling Helicobacter
pylori survival and infection. J Immunol. 196:1799–1809. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parsonnet J, Friedman GD, Vandersteen DP,
Chang Y, Vogelman JH, Orentreich N and Sibley RK: Helicobacter
pylori infection and the risk of gastric carcinoma. N Engl J Med.
325:1127–1131. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peek RM Jr and Blaser MJ: Helicobacter
pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer.
2:28–37. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tanigawa T, Watanabe T, Hamaguchi M,
Sasaki E, Tominaga K, Fujiwara Y, Oshitani N, Matsumoto T, Higuchi
K and Arakawa T: Anti-inflammatory effect of two isoforms of COX in
H. pylori-induced gastritis in mice: Possible involvement of PGE2.
Am J Physiol Gastrointest Liver Physiol. 286:G148–G156. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pai R, Soreghan B, Szabo IL, Pavelka M,
Baatar D and Tarnawski AS: Prostaglandin E2 transactivates EGF
receptor: A novel mechanism for promoting colon cancer growth and
gastrointestinal hypertrophy. Nat Med. 8:289–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Society of Stomach Cancer of Chinese
Anti-Cancer Association, ; Society of Pathology of Chinese
Anti-Cancer Association; Chinese Society of Clinical Oncology, .
Chinese expert consensus on the molecular-targeted therapy for
HER-2-positive advanced gastric cancer. Zhonghua Zhong Liu Za Zhi.
35:315–319. 2013.(In Chinese). PubMed/NCBI
|
|
20
|
Ding Y, Tong M, Liu S, Moscow JA and Tai
HH: NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH)
behaves as a tumor suppressor in lung cancer. Carcinogenesis.
26:65–72. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wolf I, O'Kelly J, Rubinek T, Tong M,
Nguyen A, Lin BT, Tai HH, Karlan BY and Koeffler HP:
15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of
human breast cancer. Cancer Res. 66:7818–7823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pham H, Chen M, Li A, King J, Angst E,
Dawson DW, Park J, Reber HA, Hines OJ and Eibl G: Loss of
15-hydroxyprostaglandin dehydrogenase increases prostaglandin E2 in
pancreatic tumors. Pancreas. 39:332–339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tseng-Rogenski S, Gee J, Ignatoski KW,
Kunju LP, Bucheit A, Kintner HJ, Morris D, Tallman C, Evron J, Wood
CG, et al: Loss of 15-hydroxyprostaglandin dehydrogenase expression
contributes to bladder cancer progression. Am J Pathol.
176:1462–1468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song HJ, Myung SJ, Kim IW, et al:
15-hydroxyprostaglandin dehydrogenase is downregulated and exhibits
tumor suppressor activity in gastric cancer. Cancer Invest.
29:257–265. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oshima H, Oshima M, Inaba K and Taketo MM:
Hyperplastic gastric tumors induced by activated macrophages in
COX-2/mPGES-1 transgenic mice. EMBO J. 23:1669–1678. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sierra JC, Hobbs S, Chaturvedi R, Yan F,
Wilson KT, Peek RM Jr and Polk DB: Induction of COX-2 expression by
Helicobacter pylori is mediated by activation of epidermal growth
factor receptor in gastric epithelial cells. Am J Physiol
Gastrointest Liver Physiol. 305:G196–G203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Slomiany BL and Slomiany A: Induction in
gastric mucosal prostaglandin and nitric oxide by Helicobacter
pylori is dependent on MAPK/ERK-mediated activation of IKK-beta and
cPLA2: Modulatory effect of ghrelin. Inflammopharmacology.
21:241–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun WH, Yu Q, Shen H, Ou XL, Cao DZ, Yu T,
Qian C, Zhu F, Sun YL, Fu XL and Su H: Roles of Helicobacter pylori
infection and cyclooxygenase-2 expression in gastric
carcinogenesis. World J Gastroenterol. 10:2809–2813. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arber N, Eagle CJ, Spicak J, Rácz I, Dite
P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, et al:
Celecoxib for the prevention of colorectal adenomatous polyps. N
Engl J Med. 355:885–895. 2006. View Article : Google Scholar : PubMed/NCBI
|