Introduction
Brain glioma is the most common type of primary
tumor in the central nervous system and is one of the leading
causes of mortality in patients with cancer (1,2). Currently
an integrated therapeutic method of surgery, radiotherapy and
chemotherapy has been adopted in clinical high-grade glioma
treatment (3). Patients receiving
radiotherapy with concomitant and adjuvant temozolomide (TMZ),
demonstrated certain advances in progression-free survival (PFS)
and 5-year survival; however, the rate of total survival has not
improved (4). TMZ is an oral
administered alkylating chemotherapeutic drug, capable of crossing
the blood-brain barrier and has been widely used to treat
refractory anaplastic astrocytoma and newly diagnosed glioblastoma
multiforme (5,6). The therapeutic benefits of TMZ treatment
primarily depend on its ability to methylate the O-6 positions of
guanine residues (7). This
methylation induces irreversible damage to the DNA and triggers
abnormal activation of the repair system, leading to cycle arrest
and cell death (7). However, certain
tumor cells are able to repair this type of DNA damage by
expression of the protein, O6-alkylguanine DNA alkyltransferase
(AGT), encoded in humans by the O-6-methylguanine-DNA
methyltransferase (MGMT) gene (8). A previous study revealed that the
presence of MGMT protein diminishes the therapeutic efficacy of TMZ
in brain tumors; the study also indicated that high MGMT
expression predicts poor response to TMZ and little benefit from
chemotherapy with TMZ (9).
Conversely, in specific cases, suppressed synthesis of MGMT due to
methylation of the MGMT gene promoter is considered a good
prognostic factor in TMZ treated patients with glioma (10). At the molecular level, several
mechanisms, including nuclear factor-κB (NF-κB) (11), tumor protein 53 (12), specificity protein 1 (12), clone of myelocytomatosis viral
oncogene in cancer (Myc) (13) and
c-Jun N-terminal kinase (JNK) (14)
mediated signaling pathways, have been suggested to be involved in
the transcription regulation of MGMT. As a consequence,
modifying MGMT expression via its transcription factors has
been proposed as a means to sensitize tumor cells to TMZ (15).
NF-κB represents a family of ubiquitous
transcription factors that modulate the expression levels of genes
by binding to specific κB sites (16). The activity of NF-κB is regulated by
the NF-κB inhibitory protein (IκB) (17). In the inactive state, IκB binds to and
sequesters NF-κB family members in the cytoplasm (17). Following NF-κB signaling pathway
activation by various stimuli, including hypoxia, cytokines and
chemotherapeutic drugs, IκB is phosphorylated by IκB kinase (IKK)
(17); phosphorylated IκB is
subjected to ubiquitination and kinase proteasome-mediated
degradation, which results in the activation and translocation of
NF-κB to the nucleus (17). Excluding
its roles in innate immunity and inflammation, the NF-κB signaling
pathway was revealed to regulate a number of cellular processes,
including cell proliferation, differentiation and apoptosis
(16–18). Furthermore, it has been reported that
activation of the NF-κB signaling pathway may also contribute to
tumor initiation, progression and resistance to radiotherapy or
chemotherapy (19,20). High constitutive NF-κB activity has
been observed in numerous lymphoid and myeloid tumors (21), in addition to various solid tumors,
including pancreatic cancer (22),
glioblastoma (23) and breast cancer
(24). Certain recent studies
demonstrated that hypoxia may activate the NF-κB signaling pathway,
induce the epithelial-mesenchymal transition of breast cancer cells
(24), cause invasion of pancreatic
cancer cells and contribute to gemcitabine mediated resistance
(25).
The present study focused on the association between
NF-κB activity and glioma cell progression and chemotherapy. The
critical roles which NF-κB serves in the progression and
chemoresistance of gliomas have been demonstrated by accumulating
experimental evidence (26). Similar
to MGMT, NF-κB also contributes to the development of glioma
resistance to alkylating agents (27). Furthermore, certain studies indicated
that NF-κB associated chemoresistance was partially mediated by the
NF-κB/MGMT signaling pathway, which may be activated by
alkylating drugs (28,29). In order to determine the effect of
inhibiting NF-κB activity on glioma cell viability and sensitivity
to alkylating drugs, in addition to clarifying the underlying
molecular mechanisms, the present study compared the proliferation
inhibiting, cell apoptosis inducing and cell cycle arresting
effects of TMZ, pyrrolidine dithiocarbamate (PDTC) and TMZ + PDTC
combined. This was followed by determining the expression level
alterations of MGMT and other associated genes, including
B-cell lymphoma extra large (BCL-XL) and survivin.
The present study aimed to further current understanding of the
effects and the underlying molecular mechanisms of inhibiting NF-κB
activity in glioma therapy, and aid the development of a clinical
strategy with combined TMZ and NF-κB inhibitors.
Materials and methods
Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS) and TRIzol® were obtained from
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA);
temozolomide and dimethyl sulfoxide (DMSO) were purchased from
Sigma Aldrich (Merck Millipore, Darmstadt, Germany); the MTT, cell
cycle and apoptosis analysis kit, Annexin V-fluorescein
isothiocyanate (FITC) kit, Bradford protein assay kit,
radioimmunoprecipitation assay, lysis buffer, phenyl
methanesulfonyl fluoride and BeyoECL Plus were all purchased from
Beyotime Institute of Biotechnology (Haimen, China). A Prime
Script™ RT Master Mix was obtained from Takara Bio, Inc. (Otsu,
Japan); a KAPA SYBR Fast qPCR kit was purchased from Kapa
Biosystems, Inc. (Wilmington, MA, USA). The primary antibodies used
for western blot analysis were mouse anti MGMT,
BCL-XL, survivin and β-actin, and the secondary
antibodies used were horseradish peroxidase-conjugated goat
anti-mouse IgG (H + L), all supplied by Abcam (Cambridge, UK).
Cell lines and cell culture
The U251 human glioblastoma cell line was purchased
from the National Institute of Biological Sciences (Beijing, China)
and cultured in DMEM (DMEM basic 1X 1199500) supplemented with 10%
FBS (10099141) (both from Gibco; Thermo Fisher Scientific, Inc.).
Cells were incubated for about 6 months at 37°C in a humidified
chamber with 5% CO2.
MTT assays for cell proliferation
Exponentially growing U251 cells were digested and
re-plated into 96-well plates (4×103 cells; 100 µl/well;
six repeat wells in each column). These plates were randomly
divided into four groups: TMZ group, PDTC group, TMZ + PDTC group
and control group. Following an incubation of 24 h, the groups were
cultured as follows: Medium of the TMZ group was replaced with
fresh medium (DMEM basic 1X with 10% FBS) containing 200 µmol/l
TMZ; PDTC group was divided into three subgroups and medium of the
three subgroups were replaced with fresh medium (DMEM basic 1X with
10% FBS) containing 20, 50 and 80 µmol/l PDTC; TMZ + PDTC group was
divided into three groups and medium of the three subgroups were
replaced with fresh medium (DMEM basic 1X with 10% FBS) containing
200 µmol/l TMZ and 20 µmol/l PDTC, 200 µmol/l TMZ and 50 µmol/l
PDTC or 200 µmol/l TMZ and 80 µmol/l PDTC. DMSO was added into the
control group. A column from each group was evaluated every 24 h
and 10 µl/well MTT (5 mg/ml) was added to six wells in one column.
Following incubation for 4 h, 100 µl/well formazan solution was
added and incubated for an additional 4 h. Absorbance was
determined at 570 nm, using an Ultra multi-functional microplate
reader (Tecan Group, Ltd., Durham, NC, USA). The evaluation was
performed for five consecutive days and the cell inhibition rates
were calculated as follows: Rate of proliferation inhibition =
[mean optical density (OD) value of control cells - mean OD value
of treated cells]/mean OD value of control cells. The results were
confirmed by ≥3 repetitions.
Flow cytometry detecting cell
apoptosis and cell cycle distribution
U251 cells (8×105) were re-plated in
6-well plates (9.6 cm2). Fresh medium (DMEM basic 1X
with 10% FBS) was added with the following supplements: 200 µmol/l
TMZ into the TMZ group; 20 µmol/l PDTC, 50 µmol/l PDTC and 80
µmol/l PDTC into the three subgroups of the PDTC group,
respectively; 200 µmol/l TMZ and 20 µmol/l PDTC, 200 µmol/l TMZ and
50 µmol/l PDTC or 200 µmol/l TMZ and 80 µmol/l PDTC, into the three
subgroups of the TMZ + PDTC group, respectively; 200 µmol/l TMZ and
100 µmol/l PDTC into the fourth subgroup of the TMZ + PDTC group
(only added in this part of the experiment); and the solvent DMSO
was added into control group. Following incubation for 24 h, cells
were digested by trypsinization and detached cells in the medium
were collected. Cells were harvested and washed with cold PBS,
5×105 cells were collected in 1.5 ml microtubes
(MCT-150-C; Axygen, Union City, CA, USA). Subsequently, cells were
gently resuspended in 100 µl 1X binding buffer with 5 µl Annexin
V-FITC and 5 µl propidium iodide (PI) staining solution. Following
incubation for 15 min in the dark at 25°C, cells were mixed gently
with 400 µl 1X binding buffer and filtered with a 0.45 µm filter
unit Millex-HV (Merck Millipore), prior to flow cytometry analysis
(FACSCalibur™; BD Biosciences, San Jose, CA, USA).
Subsequent analyses of flow cytometry data were performed using
CellQuest Pro software (version 5.1; BD Biosciences). For cell
cycle distribution evaluation, cells (groups with 100 µmol/l PDTC
were removed) were collected and resuspended with cold ethanol
(70%) and fixed in ethanol for 12 h at 4°C, prior to being
harvested. Following washing with cold PBS twice, cells were mixed
gently with 500 µl PI and incubated for 30 min in the dark at 35°C.
Cell cycle distribution was detected using flow cytometry.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Cells (5×106 cells initially seeded) were
cultured in 6-well plates and the following treatment was
administered for 48 h at 37°C, in a humidified chamber containing
5% CO2: 80 µmol/l PDTC to the PDTC group, 200 µmol/l TMZ
to the TMZ group, 200 µmol/l TMZ and 50 µmol/l PDTC to the TMZ +
PDTC group, and DMSO to the control groups. Total RNA was extracted
using TRIzol® reagent, according to the manufacturer's
instructions. First-strand cDNA was reverse transcribed from 1 mg
total RNA using the Prime Script™ RT Master Mix, and target gene
mRNA was amplified by the KAPA SYBR Fast qPCR kit, using a Bio-Rad
CFX96™ real-time system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The reaction system (15 µl), PCR conditions and gene specific
primers are presented in Tables I and
II.
 | Table I.Reaction system and conditions for
RT-qPCR analysis. |
Table I.
Reaction system and conditions for
RT-qPCR analysis.
| Reaction
system | Reaction
conditions |
|---|
|
|
|---|
| Reagents | Amount added,
µl | Step | Action |
|---|
| 2X KAPA SYBR Fast
qPCR |
|
|
|
| Master Mix
Universal | 7.50 | 1. | 95°C for 2 min |
| 10 µM forward
primer | 0.15 | 2. | 95°C for 5 sec |
| 10 µM reverse
primer | 0.15 | 3. | 60°C for 20 sec,
value read |
| cDNA template | 1.00 (50.00
ng) | 4. | Repeat 2 and 3 for
39 repetitions |
| PCR-grade
water | 6.20 | 5. | Melt curve 65°C to
95°C in increments of 0.5°C, for 5 sec each, value read, end. |
| Table II.Gene primers for RT-qPCR
analysis. |
Table II.
Gene primers for RT-qPCR
analysis.
| Gene | Primer |
|---|
| MGMT | F:
5′-GGGTCTGCACGAAATAAAGC-3′ |
|
| R:
5′-CTCCGGACCTCCGAGAAC-3′ |
| BCL-XL | F:
5′-AAAAGATCTTCCGGGGGCTG-3′ |
|
| R:
5′-CCCGGTTGCTCTGAGACATT-3′ |
|
Survivin | F:
5′-TTCTCAAGGACCACCGCATC-3′ |
|
| R:
5′-AATGGGGTCGTCATCTGGCT-3′ |
| β-actin | F:
5′-GTACCACTGGCATCGTGATGGACT-3′ |
|
| R:
5′-ATCCACACGGAGTACTTGCGCTCA-3′ |
Western blot analysis
Cells were cultured in 6-cm dishes, following
grouping and treatment as in RT-qPCR analysis; the cell lysates
were harvested and protein level was evaluated using Bradford
Protein Assay kit (P0006C; Beyotime Institute of Biotechnology).
Equal amounts of total protein (80–200 µg) were separated using 10%
SDS-PAGE and transferred to polyvinylidene difluoride membranes.
Subsequent to blocking with 5% fat-free milk and 0.1% Tween-20 in
PBS-T for 1 h at room temperature, the membranes were incubated
with a dilution of anti-MGMT (mouse monoclonal MT3.1 to
MGMT; ab39253; 1:500), anti-BCL-XL (mouse monoclonal MT3.1
to MGMT; ab39253; 1:500), anti-survivin (mouse monoclonal
60.11 to survivin; ab93274; 1:800) and anti-β-actin (anti-β-actin
antibody mAbcam 8226, ab8226; 1:600) (all from Abcam) primary
antibodies. Horseradish peroxidase-conjugated anti-mouse secondary
antibodies (goat anti-mouse IgG H&L horseradish peroxidase
pre-adsorbed; ab97040; 1:1200; Abcam) were used, and bound
antibodies were detected using the BeyoECL system (P0018; Beyotime
Institute of Biotechnology).
Statistical analysis
Data are presented as the mean ± standard deviation.
All statistical analyses were performed using SPSS 13.0 software
(SPSS, Inc., Chicago, IL, USA). The Student's t-test (one sample or
independent-samples t-test) was used to analyze the difference
between the means of the treatment group and the control group.
One-way analysis of variance (ANOVA) was used to analyze the
significance among ≥3 groups and Fisher's least significant
difference method for multiple comparisons was used when the
probability for ANOVA was statistically significant. Methods of
nonparametric statistical analysis including the Mann-Whitney U
Test and Kruskal-Wallis ANOVA, were used when the variances did not
pass the Levene test for normality or homogeneity. P<0.05 was
considered to indicate a statistically significant difference.
Results
Combining TMZ and PDTC induces the
highest proliferation inhibition rate
TMZ, PDTC and TMZ + PDTC treatments all
significantly suppressed the proliferation of U251 cells
(P<0.05). Cell OD values decreased with increasing PDTC
concentrations (P<0.05; Fig. 1A).
The TMZ and PDTC treatment combination exhibited a greater
significant inhibiting effect and lower OD values compared with the
corresponding PDTC group (P<0.05; TMZ + PDTC vs. PDTC). However,
TMZ + 80 µmol/l PDTC did not induce lower OD values compared with
TMZ + 50 µmol/l PDTC (P<0.05; Fig.
1A). The proliferation inhibition rates in the TMZ + PDTC 50
µmol/l PDTC group (61.56% at 72 h) were higher, compared with in
the TMZ group (29.88% at 72 h) and 80 µmol/l PDTC group (44.02% at
72 h; P<0.05; Fig. 1B).
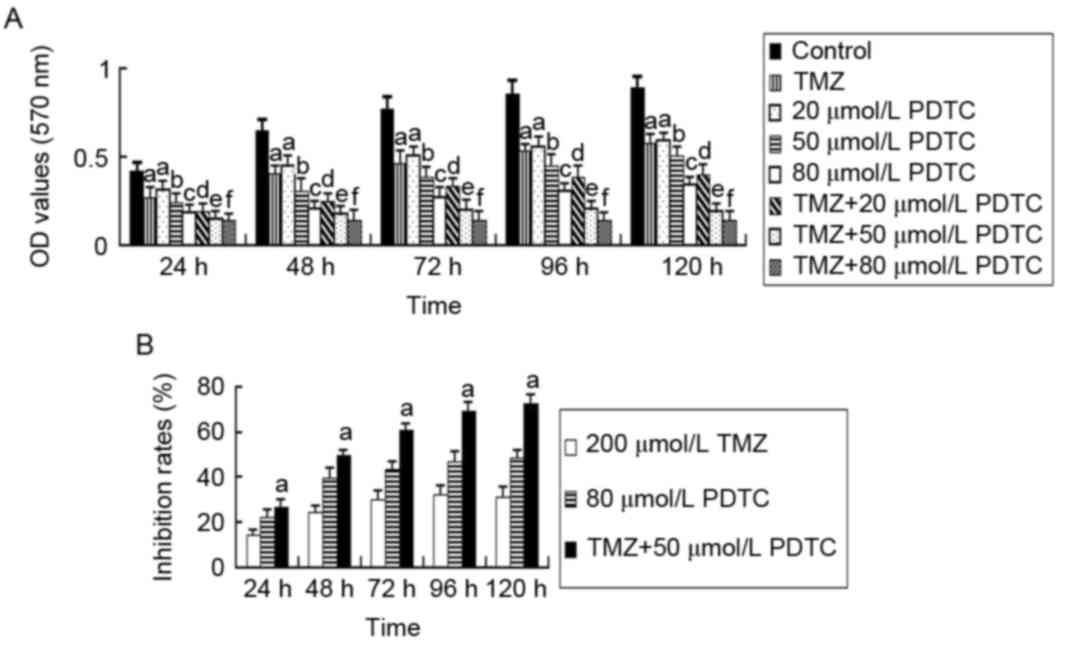 | Figure 1.MTT assays detecting the viability of
U251 cells treated with TMZ, PDTC and TMZ + PDTC. (A) OD values of
cells in alternate groups. Cell OD values of PDTC groups were
markedly lower than the control group and decreased with increasing
PDTC concentrations (a: P<0.05, compared with the control group;
b: P<0.05, compared with the control group and 20 µmol/l PDTC
group; c: P<0.05, compared with the control group and 50 µmol/l
PDTC group; independent-samples t test and one-way ANOVA followed
by LSD analysis or Kruskal-Wallis test). Cell OD values of TMZ +
PDTC groups were significantly lower than those of the TMZ group
and the corresponding PDTC groups (d: P<0.05, compared with the
TMZ group and 20 µmol/l PDTC group; e: P<0.05, compared with the
TMZ group, 50 µmol/l PDTC group and TMZ + 20 µmol/l PDTC group; f:
P<0.05, compared with TMZ group and 80 µmol/l PDTC group;
one-way ANOVA followed by LSD analysis or the Kruskal-Wallis test).
(B) Cell proliferation inhibition rates in three representative
groups. The inhibition rates of the TMZ + 50 µmol/l PDTC treatment
group were significantly higher, compared with 200 µmol/l TMZ or 80
µmol/l PDTC treatments at 24–120 h (a: P<0.05, compared with the
TMZ and PDTC group, one-way ANOVA followed by LSD analysis). OD,
optical density; TMZ, temozolomide; PDTC, pyrrolidine
dithiocarbamate; ANOVA, analysis of variance; LSD, least
significant difference. |
Combination of TMZ and PDTC induces
the most significant cell apoptosis
Following an incubation of 24 h, TMZ, PDTC and TMZ +
PDTC markedly induced cell apoptosis compared with the control
(Fig. 2). TMZ + PDTC induced higher
apoptosis rates as compared with TMZ and the corresponding
concentrations of PDTC (Table III).
The rate of cell apoptosis increased with increasing PDTC
concentrations (P<0.05; Table
III). However, when treated with 100 µmol/l PDTC or TMZ + ≥50
µmol/l PDTC, cell apoptosis rates reached >93%, and no
significant difference had been identified among these groups
(P<0.05; Table III).
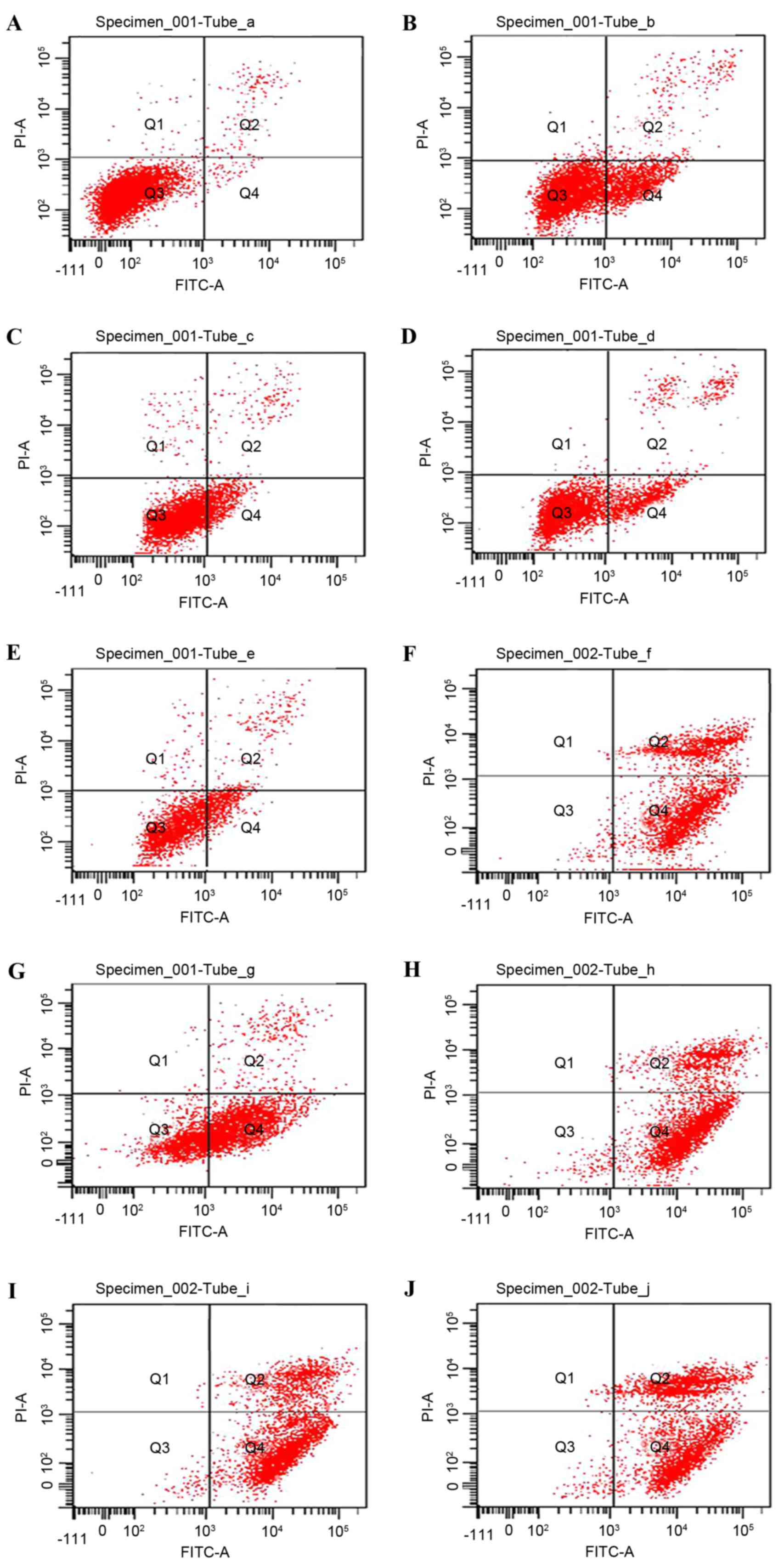 | Figure 2.Flow cytometric analysis with Annexin
V-FITC/PI staining detecting apoptosis of U251 cells in the
control, TMZ, PDTC and TMZ + PDTC groups. (A) Control, (B) 200
µmol/l TMZ, (C) 20 µmol/l PDTC, (D) TMZ + 20 µmol/l PDTC, (E) 50
µmol/l PDTC, (F) TMZ + 50 µmol/l PDTC, (G) 80 µmol/l PDTC, (H) TMZ
+ 80 µmol/l PDTC, (I) 100 µmol/l PDTC and (J) TMZ + 100 µmol/l
PDTC. FITC, fluorescein isothiocyanate; PI, propidium iodide; TMZ,
temozolomide; PDTC, pyrrolidine dithiocarbamate. |
| Table III.Apoptosis rates of U251 cells in
distinct groups. |
Table III.
Apoptosis rates of U251 cells in
distinct groups.
| Group | Concentration,
µmol/l | Early stage, % | Late stage, % | Total apoptosis,
% |
|---|
| Control |
| (1.6±0.54) | (0.8±0.03) | (2.4±1.03) |
| TMZ | 200 |
(22.8±2.24)a |
(3.1±0.75)a |
(25.9±3.15)a |
| PDTC | 20 |
(2.0±1.41)a |
(2.7±0.43)a |
(17.9±1.26)a |
|
| 50 |
(25.8±0.85)b |
(5.8±0.91)b |
(31.6±1.75)b |
|
| 80 |
(59.8±2.09)c | (4.1±1.46) |
(63.9±4.24)c |
|
| 100 |
(71.7±2.76)d |
(22.5±2.07)d |
(93.2±4.73)d |
| TMZ + PDTC | 20 |
(26.6±2.27)e |
(6.7±0.85)e |
(33.3±2.44)e |
|
| 50 |
(70.2±5.12)f |
(21.5±2.57)f |
(91.7±5.22)f |
|
| 80 | (64.1±3.04) |
(33.2±2.06)g |
(97.3±4.46)g |
|
| 100 | (57.7±3.55) |
(39.2±2.89)h | (96.9±5.10) |
PDTC enhances the cell cycle arresting
effect of TMZ treatment
The combination of TMZ and PDTC led to a significant
G0/G1 cell cycle arresting effect in U251
cells (Fig. 3). The percentage of
cells in the G0/G1 stage in TMZ + PDTC groups
was markedly higher than in the TMZ group (42.4±1.39%) and the
corresponding PDTC groups (P<0.05), whereas the percentage of
cells in the G2 stage in TMZ + PDTC groups were markedly
lower compared with that in the TMZ group (25.1±1.14%) and the
corresponding PDTC groups (P<0.05; Table IV). A higher concentration of PDTC
induced a higher proportion of cells in the
G0/G1 stage; however, TMZ + 80 µmol/l PDTC
induced no more significant G0/G1 arrest than
TMZ + 50 µmol/l PDTC (P<0.05; Table
IV).
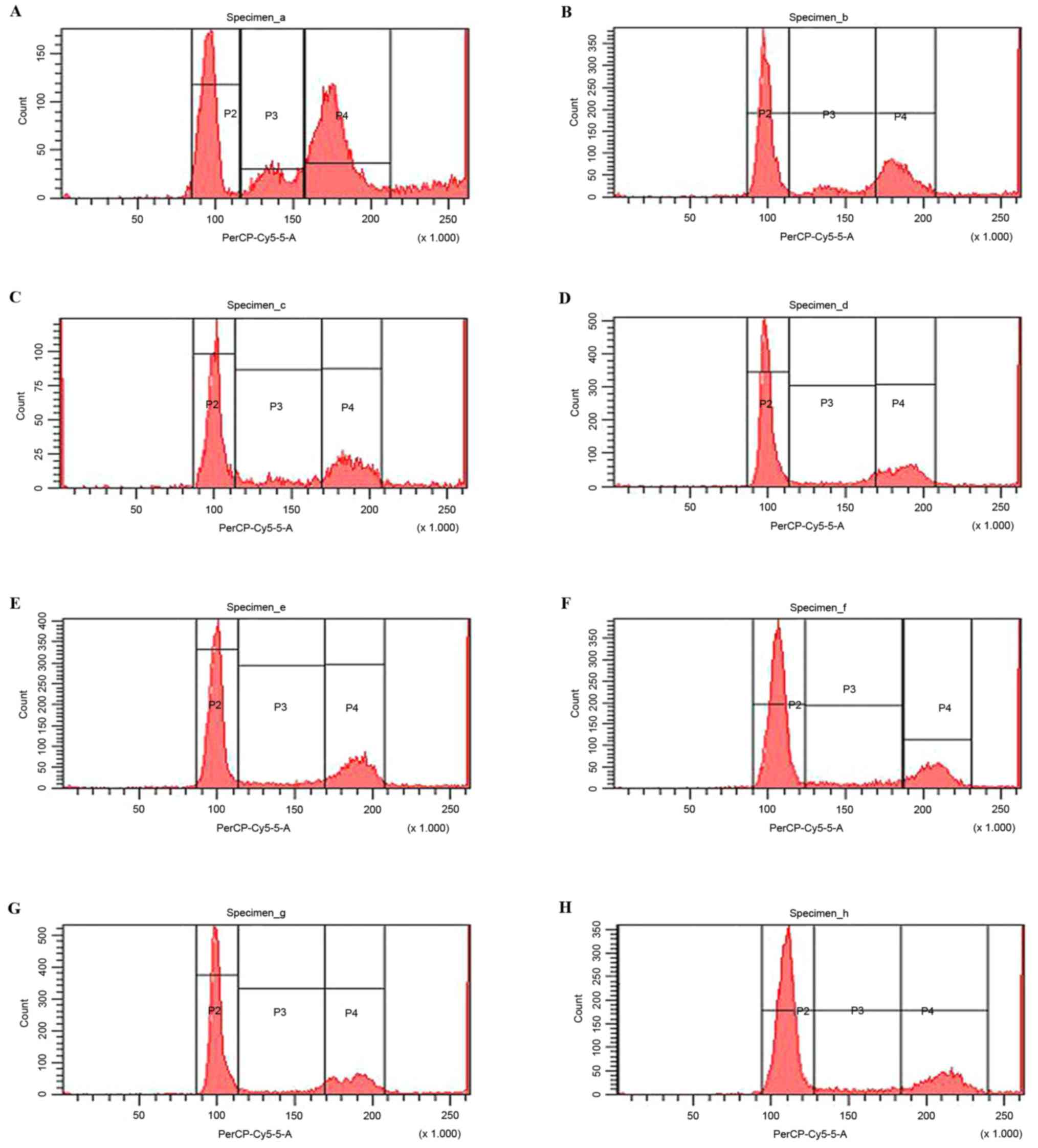 | Figure 3.Flow cytometry analysis of U251 cells
in G0/G1, S and G2 phases
following treatment with PDTC in various concentrations. (A)
Control, (B) 200 µmol/l TMZ, (C) 20 µmol/l PDTC, (D) TMZ + 20
µmol/l PDTC, (E) 50 µmol/l PDTC, (F) TMZ + 50 µmol/l PDTC, (G) 80
µmol/l PDTC, (H) TMZ + 80 µmol/l PDTC. (P1)
G0/G1 phase, (P2) S phase, (P3) G2
phase. PDTC, pyrrolidine dithiocarbamate; TMZ, temozolomide. |
| Table IV.Cell cycle distribution of U251 cells
in distinct groups. |
Table IV.
Cell cycle distribution of U251 cells
in distinct groups.
| Group | Concentration,
µmol/l |
G0/G1, % | S, % | G2, % |
|---|
| Control |
| (26.0±1.42) | (10.4±1.36) | (34.2±1.85) |
| TMZ | 200 |
(42.4±1.39)a | (9.8±1.71) |
(25.1±1.14)a |
| PDTC | 20 |
(38.3±2.52)a | (9.5±1.83) |
(21.5±0.94)a |
|
| 50 |
(47.7±3.32)b |
(7.9±1.53)b | (21.6±3.04) |
|
| 80 |
(50.8±4.05)c | (6.8±1.32) | (21.3±2.16) |
| TMZ + PDTC | 20 |
(49.1±4.02)d | (8.1±0.47) |
(17.7±2.06)d |
|
| 50 |
(58.2±2.17)e | (7.8±1.42) | (16.7±3.22) |
|
| 80 | (59.3±2.92) | (6.3±2.57) | (18.6±2.58) |
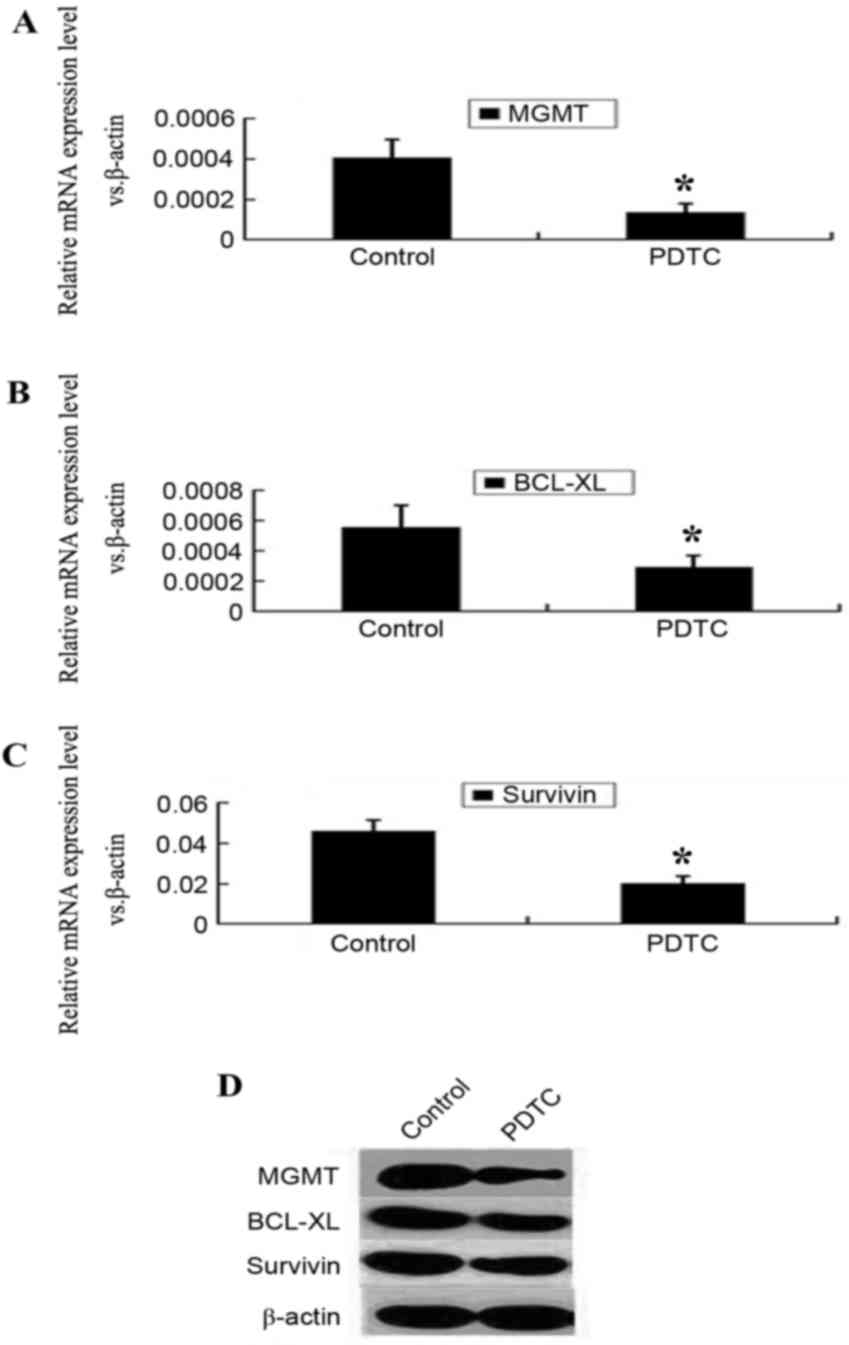
PDTC suppresses the expression of
MGMT, BCL-XL and survivin
Following incubation with PDTC for 48 h, the mRNA
levels of MGMT, BCL-XL and survivin were
significantly reduced (P<0.05; Fig.
4A-C). The protein levels of MGMT, BCL-XL and
survivin were also reduced following PDTC treatment
(Fig. 4D).
PDTC counteracts MGMT and BCL-XL
upregulation induced by TMZ
MGMT, BCL-XL and survivin
expression levels increased significantly, subsequent to TMZ
treatment for 24 h (P<0.05; Fig.
5). However, this upregulation was abrogated by simultaneous
treatment with PDTC. MGMT and BCL-XL expression
levels in the PDTC + TMZ group were markedly lower compared with
that in the TMZ group, whereas survivin expression was not
altered significantly (P<0.05; Fig.
5).
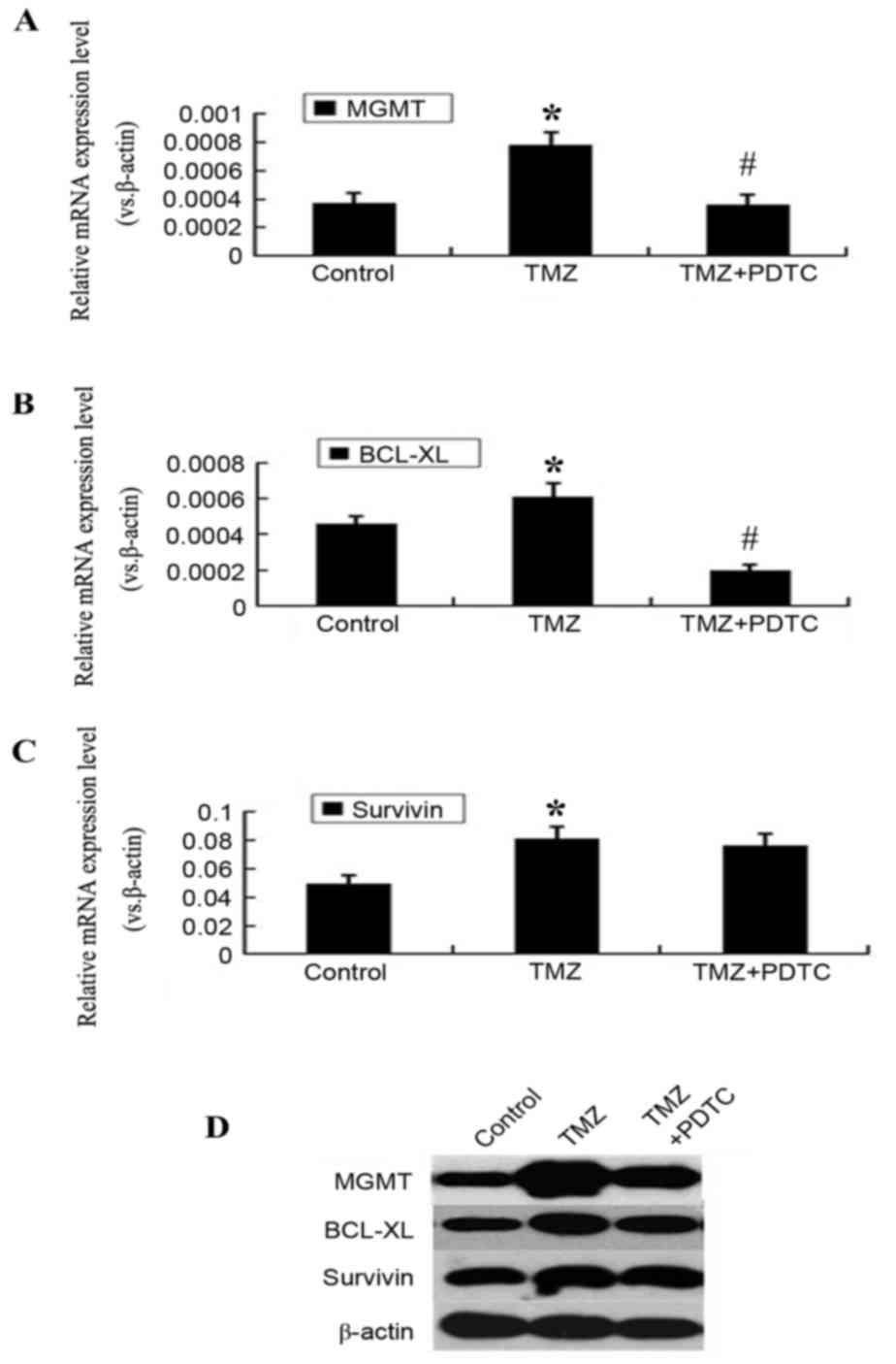 | Figure 5.PDTC counteracts the upregulation of
MGMT and BCL-XL induced by TMZ treatment. (A) RT-qPCR
results revealed that mRNA level of MGMT in TMZ-treated
cells was significantly higher than that in control cells and TMZ +
PDTC-treated cells. *P<0.05 compared with control cells,
#P<0.05 compared with TMZ-treated cells. (B) The mRNA
level of BCL-XL in TMZ-treated cells was higher than that in
control cells and TMZ + PDTC-treated cells. *P<0.05 compared
with control cells, #P<0.05 compared with TMZ-treated
cells. (C) The survivin mRNA level in TMZ-treated cells was
much higher than that in control cells, but showed no statistical
difference between TMZ-treated cells and TMZ + PDTC treated cells.
*P<0.05 compared with control cells. (D) Western blotting
results demonstrated that MGMT, BCL-XL and
survivin protein levels in TMZ treated cells were higher
compared with the control cells; MGMT and BCL-XL
protein levels of TMZ + PDTC treated cells were low compared with
TMZ treated cells; however, the survivin protein level
between TMZ + PDTC treated and TMZ treated cells demonstrated no
obvious difference. PDTC, pyrrolidine dithiocarbamate; MGMT,
O-6-methylguanine-DNA methyltransferase; BCL-XL,
B-cell lymphoma extra-large; TMZ, temozolomide; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction. |
Discussion
NF-κB serves a number of critical roles in the
development, invasion, recurrence and chemoresistance of malignant
brain glioma (30). Abnormal
constitutive activation of the NF-κB signaling pathway may be
identified in a large number of clinical glioma specimens (31). Wang et al (32) revealed that >90% of the 259 human
diffuse gliomas included in the study exhibited high activation of
NF-κB, and the extent of activation represented by the protein
expression level of the p65 subunit was positively associated with
glioma grade and malignancy. Highly activated NF-κB may lead to
increased expression levels of matrix metalloproteinases
(MMPs) (33) and vascular
endothelial growth factor (VEGF), which facilitates
microvascular invasion and distal metastases of glioblastoma stem
cells (GSCs) (34). In conjunction
with the high level of constitutive activity, it has previously
been demonstrated that numerous alkylating agents may activate
NF-κB in glioma cells (35).
Contributing to the anti-apoptotic effect of the NF-κB signaling
pathway and blocking constitutive or stimulated NF-κB activation
may be a potential approach to suppressing glioma cell viability
and enhance the efficacy of alkylating drugs (36).
Furthermore, the inhibition of NF-κB by genetic or
chemical inhibitors induces apoptosis of various glioma cells and
restores the apoptotic response following treatment with ionizing
radiation or chemotherapeutic agents, thus reversing NF-κB linked
radioresistance or chemoresistance in a number of models (37). For example, when NF-κB function was
profoundly suppressed in U87 and U251 glioma cells via the
overexpression of non-degradable IκB, chemical agents including
carmustine (BCNU), carboplatin, SN38 glucuronide and tumor necrosis
factor-α, induced significant proliferation inhibition and
apoptosis in these refractory cell lines (29). SN50, a specific NF-κB inhibitor, may
induce differentiation and reduce malignant characters (including
neuro-sphere formation, motility, invasion and tumor initiation
in vivo) of GSCs. The GSCs sensitivity to TMZ and
radiotherapy was markedly enhanced by a low dose of SN50 (38). In the present study, another specific
NF-κB inhibitor, PDTC, was used: PDTC is membrane permeable and an
antioxidant (39). PDTC may reduce
the phosphorylation and degradation of IκB and subsequently block
NF-κB activation and nuclear translocation (40). In conjunction with previous studies
(28,41,42), the
results of the MTT assay and flow cytometry conducted in the
present study demonstrated that a low concentration of PDTC may
markedly inhibit proliferation and induce apoptosis in U251 cells.
Furthermore, the combined treatment of TMZ and PDTC led to a
significant increase in proliferation-inhibition rate, apoptosis
rate and a more significant cell cycle arrest, as compared with TMZ
or PDTC treatment alone (Figs.
1–3). These results indicated a
synergistic effect between TMZ and NF-κB inhibitors; a
comprehensive understanding of the molecular mechanisms underlying
this synergistic action may facilitate the exploration of more
efficacious NF-κB inhibitors and a practical combination strategy
of NF-κB inhibitors and alkylating agents.
As previously described, MGMT is a key factor
of chemoresistance in gliomas (10)
and inhibiting the NF-κB signaling pathway may markedly enhance TMZ
chemotherapeutic efficacy in U251 cells (37). The results of current study suggest
that NF-κB inhibitors mediate their killing effects by inhibiting a
potential NF-κB/MGMT signaling pathway. In order to further
support this suggestion, evidence is required which indicates that
an NF-κB/MGMT signaling pathway exists in glioma cells and
that NF-κB inhibitors, including PDTC, significantly reduce
MGMT expression in glioma cell lines following treatment
with alkylating agents. A number of studies have previously
demonstrated the existence of an NF-κB/MGMT signaling
pathway in glioma cells (15,43,44). Two
putative NF-κB binding sites within the MGMT promoter region
have been discovered by Lavon et al (28), demonstrated a specific and direct
interaction of NF-κB at each of these sites. They further revealed
that forced expression of the NF-κB subunit p65 in HEK293 cells and
glioma cells induced an increase in MGMT expression levels,
whereas the addition of the NF-κB inhibitor IκB completely
abrogated this induction (28).
MGMT expression was revealed to be attenuated by fluoxetine
via disrupting the NF-κB signaling pathway and consequently
sensitized 98 G, SF767 and U251 glioma cells to TMZ treatment
(44). Similarly, the present study
identified markedly lower MGMT expression levels in
PDTC-treated U251 cells (Fig. 4). As
aforementioned, alkylating agents may activate the NF-κB signaling
pathway, and NF-κB serves an important role in MGMT
regulation. Also, it has previously been revealed that alkylating
drugs stimulate MGMT expression in glioma cells (45). Therefore, an NF-κB/MGMT
signaling pathway may be activated by alkylating drugs, in addition
to stimulating MGMT expression. Other anti-apoptotic
mechanisms may also serve major roles in NF-κB-mediated
chemoresistance to alkylating agents (31). In order to confirm these suggestions,
the present study treated U251 cells with 100 µmol/l TMZ. As a
notable result, significant increases in MGMT expression
levels and in two additional NF-κB downstream genes, BCL-XL
and survivin, were demonstrated.
Additional evidence derives from a previous study by
Lavon et al (28), which
revealed that the suppression of MGMT activity with
O6-benzylguanine eradicates the chemoresistance acquired by glioma
cell lines, with ectopic p65 or high constitutive NF-κB activity.
Despite the understanding that high MGMT expression in
resistant glioma cells is stimulated by constitutive or
drug-activated high NF-κB activity (29,32),
evidence from previous studies supports the present study's
hypothesis of the existence and the contribution of an
NF-κB/MGMT signaling pathway towards chemoresistance
(28,43,44).
However, the questions remain about whether this signaling pathway
may be exploited to combat fatal gliomas, and if the efficacy of
combined TMZ and PDTC treatment is a result of the downregulation
of MGMT. In order to investigate these considerations, the
present study compared the expression levels of MGMT in
cells treated with TMZ + PDTC and in cells treated with TMZ alone,
it was revealed that the expression level of MGMT was
significantly reduced by the addition of PDTC (Fig. 5). These results are supported by a
number of similar previous studies (28,44). For
instance, in glioblastoma initial cells, resveratrol was identified
to block MGMT upregulation induced by TMZ treatment via
inhibiting NF-κB activation (15).
Therefore, downregulation of the NF-κB/MGMT signaling
pathway is a feasible strategy in order to overcome TMZ-resistance.
For example, BAY 11–7082, a specific IκB kinase (IKK) inhibitor
leading to the de-activation of the NF-κB signaling pathway,
sensitized selected U251 TMZ-resistant cells (TR/U251) to TMZ
treatment, resulting in substantial cell death (37). However, Bay 11–7082 may induce cell
death independent from inhibiting the activation of the NF-κB
signaling pathway (46), and this is
the reason for the selection of PDTC, instead of Bay 11–7082 in the
present study.
In addition to the regulation of MGMT
expression, the impact of NF-κB activity on its canonical target
genes, including BCL-XL, survivin, BCL-2,
inhibitor of apoptosis 1/2 (IAP1/IAP2), TNF
receptor associated factor 1/2, VEGF, matrix
metallopeptidase 9 (MMP9) and cyclin D1, are also
worth further study for glioma therapy. BCL-XL,
BCL-2, survivin and IAP1/IAP2 are key anti-apoptotic
factors contributing to cell survival, and are closely associated
with the progression and chemotherapy or radiotherapy resistance of
various types of cancer (47–49). BCL-XL upregulation by signal
transducer and activator of transcription 3, contributes to mutant
Kirsten rat sarcoma-mediated apoptosis resistance in colorectal
cancer (49). In a recent study with
117 ovarian epithelial neoplasms, survivin overexpression
was identified in the majority of malignant cases and was
associated with patient prognosis, indicating a crucial role in the
development of epithelial ovarian neoplasms (50). Furthermore, it has previously been
revealed that survivin expression may be upregulated by
treatment in certain lymphoid or myeloid tumors (48). As a consequence, a large number of
novel antagonist and inhibitors of these anti-apoptotic factors
have been developed, and a number of them demonstrated good
clinical results (47,51). The overexpression of these
anti-apoptotic factors may be induced by highly activated upstream
NF-κB; therefore, inhibiting NF-κB activity may be a potential
method by which to suppress tumors with overexpressed
BCL-XL, BCL-2 or survivin (52). In conjunction, the present study
identified that the expression of BCL-XL and survivin
are significantly inhibited by PDTC treatment in U251 cells
(Fig. 4). Furthermore, NF-κB activity
and, consequently, cyclin D1, COX-2, BCL-XL
and BCL-2 expression levels may be suppressed in vivo
in prostate tumors by Apigenin, which has been administrated to
transgenic adenocarcinoma mouse prostates (53). Similarly, MGMT, BCL-XL,
BCL-2 and survivin serve important roles in NF-κB
mediated therapeutic resistance (54). Conversely, combining traditional
agents with novel inhibitors of these survival and anti-apoptotic
factors may be a promising cancer therapy strategy (55). Downregulated anti-apoptotic factors
may be an additional mechanism underlying PDTC-enhanced TMZ
efficacy in the present study, particularly considering that NF-κB
and its downstream anti-apoptotic factors are often stimulated by
radiotherapy and chemotherapy (54).
The present study also demonstrated TMZ induced the upregulation of
BCL-XL and survivin in U251 cells. Additionally, a
lower level of BCL-XL, not survivin, was demonstrated
in PDTC + TMZ treated cells, as compared with TMZ only treated
cells (Fig. 5). Therefore, a
reduction BCL-XL expression may serve a role in PDTC induced
U251 cell sensitivity to TMZ, whereas the expression level of
survivin may not be altered significantly in TMZ treated
cells by PDTC, at a low concentration of ~50 µmol/l.
The results of the present study in regards to
survivin were in accordance with the findings of Elhag et
al (56), which revealed that the
ability of silibinin, a natural plant component, to potentiate the
cytotoxic efficacy of TMZ in human LN229, U87 and A172 glioblastoma
cells was unrelated to survivin protein levels, whereas
silibilin may attenuate metastatic processes by suppressing NF-κB
and the downstream gene, MMP9. Additionally, it was revealed
that expression levels of cyclin D1 were not significantly
altered by PDTC treatment, although the addition of PDTC led to an
enhanced G0/G1 phase arresting effect
(56).
The present study demonstrated that PDTC, a widely
tested inhibitor of NF-κB activation as well as an antioxidant and
metal chelator, may inhibit cell proliferation, induce apoptosis
and cell cycle arrest and enhance sensitivity to TMZ in U251 glioma
cells. It was also revealed that PDTC sensitized U251 cells to TMZ,
mainly via blocking the NF-κB and NF-κB/BCL-XL signaling
pathways that were activated by TMZ treatment. Currently, a large
number of anti-NF-κB drugs have been developed for cancer therapy
(57); the results of the present
study may provide specific aid in furthering the understanding of
the chemotherapeutic effects, and the underlying mechanisms
involved in inhibiting NF-κB in glioma cells, thus providing an
important theoretical basis for developing NF-κB inhibitors with
greater efficacy and improved methods of cancer treatment.
Acknowledgements
The present study was supported by The National
Natural Science Foundation of China (grant no. NSFC: 81272783) and
The National Key Technology Research and Development Program of the
Ministry of Science and Technology of China (grant no.
2014BAI04B02). The authors thank Mrs Xiaohong Yang (The National
Institute of Biological Sciences, Beijing, China) for her
contributions and Dr Weiliang Jiang (Shanghai Jiaotong University
School of Medicine, Shanghai, China) for his comments on the
manuscript.
References
|
1
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Perry A, Burger P, Ellison DW,
Reifenberger G, von Deimling A, Aldape K, Brat D, Collins VP,
Eberhart C, et al: International society of neuropathology-haarlem
consensus guidelines for nervous system tumor classification and
grading. Brain Pathol. 24:429–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carlsson SK, Brothers SP and Wahlestedt C:
Emerging treatment strategies for glioblastoma multiforme. EMBO Mol
Med. 6:1359–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gilbert MR, Friedman HS, Kuttesch JF,
Prados MD, Olson JJ, Reaman GH and Zaknoen SL: A phase II study of
temozolomide in patients with newly diagnosed supratentorial
malignant glioma before radiation therapy. Neuro Oncol. 4:261–267.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Villano JL, Seery TE and Bressler LR:
Temozolomide in malignant gliomas: Current use and future targets.
Cancer Chemother Pharmacol. 64:647–655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion O6-methylguanine.
Oncogene. 26:186–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jacinto FV and Esteller M: MGMT
hypermethylation: A prognostic foe, a predictive friend. DNA Repair
(Amst). 6:1155–1160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Spiegl-Kreinecker S, Pirker C, Filipits M,
Lötsch D, Buchroithner J, Pichler J, Silye R, Weis S, Micksche M,
Fischer J and Berger W: O6-Methylguanine DNA methyltransferase
protein expression in tumor cells predicts outcome of temozolomide
therapy in glioblastoma patients. Neuro Oncol. 12:28–36. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bobola MS, Alnoor M, Chen JY, Kolstoe DD,
Silbergeld DL, Rostomily RC, Blank A, Chamberlain MC and Silber JR:
O6-methylguanine-DNA methyltransferase activity is associated with
response to alkylating agent therapy and with MGMT promoter
methylation in glioblastoma and anaplastic glioma. BBA Clin.
3:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vlachostergios PJ, Hatzidaki E and
Papandreou CN: MGMT repletion after treatment of glioblastoma cells
with temozolomide and O6-benzylguanine implicates NFκB and mutant
p53. Neurol Res. 35:879–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bocangel D, Sengupta S, Mitra S and Bhakat
KK: p53-mediated down-regulation of the human DNA repair gene
O6-methylguanine-DNA methyltransferase (MGMT) via interaction with
Sp1 transcription factor. Anticancer Res. 29:3741–3750.
2009.PubMed/NCBI
|
|
13
|
Pyko IV, Nakada M, Sabit H, Teng L,
Furuyama N, Hayashi Y, Kawakami K, Minamoto T, Fedulau AS and
Hamada J: Glycogen synthase kinase 3β inhibition sensitizes human
glioblastoma cells to temozolomide by affecting O6-methylguanine
DNA methyltransferase promoter methylation via c-Myc signaling.
Carcinogenesis. 34:2206–2217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okada M, Sato A, Shibuya K, Watanabe E,
Seino S, Suzuki S, Seino M, Narita Y, Shibui S, Kayama T and
Kitanaka C: JNK contributes to temozolomide resistance of stem-like
glioblastoma cells via regulation of MGMT expression. Int J Oncol.
44:591–599. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang H, Lin H, Zhang X and Li J:
Resveratrol reverses temozolomide resistance by downregulation of
MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway.
Oncol Rep. 27:2050–2056. 2012.PubMed/NCBI
|
|
16
|
Senftleben U and Karin M: The
IKK/NF-kappaB pathway. Crit Care Med. 30 (1 Supp):S18–S26. 2002.
View Article : Google Scholar
|
|
17
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayden MS and Ghosh S: Regulation of NF-κB
by TNF family cytokines. Semin Immunol. 26:253–266. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rayet B and Gélinas C: Aberrant rel/nfkb
genes and activity in human cancer. Oncogene. 18:6938–6947. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Annunziata CM, Davis RE, Demchenko Y,
Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W,
et al: Frequent engagement of the classical and alternative
NF-kappaB pathways by diverse genetic abnormalities in multiple
myeloma. Cancer Cell. 12:115–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM,
Zhou HX, Jia G, Wang YW, Kong R, Pan SH, et al: Nuclear
factor-κB-dependent epithelial to mesenchymal transition induced by
HIF-1α activation in pancreatic cancer cells under hypoxic
conditions. PLoS One. 6:e237522011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garner JM, Fan M, Yang CH, Du Z, Sims M,
Davidoff AM and Pfeffer LM: Constitutive activation of signal
transducer and activator of transcription 3 (STAT3) and nuclear
factor κB signaling in glioblastoma cancer stem cells regulates the
Notch pathway. J Biol Chem. 288:26167–26176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huber MA, Azoitei N, Baumann B, Grünert S,
Sommer A, Pehamberger H, Kraut N, Beug H and Wirth T: NF-kappaB is
essential for epithelial-mesenchymal transition and metastasis in a
model of breast cancer progression. J Clin Invest. 114:569–581.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng ZX, Wang DW, Liu T, Liu WX, Xia WB,
Xu J, Zhang YH, Qu YK, Guo LQ, Ding L, et al: Effects of the HIF-1α
and NF-κB loop on epithelial-mesenchymal transition and
chemoresistance induced by hypoxia in pancreatic cancer cells.
Oncol Rep. 31:1891–1898. 2014.PubMed/NCBI
|
|
26
|
Puliyappadamba VT, Hatanpaa KJ,
Chakraborty S and Habib AA: The role of NF-kappaB in the
pathogenesis of glioma. Mol Cell Oncol. 1:e9634782014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caporali S, Levati L, Graziani G, Muzi A,
Atzori MG, Bonmassar E, Palmieri G, Ascierto PA and D'Atri S: NF-κB
is activated in response to temozolomide in an AKT-dependent manner
and confers protection against the growth suppressive effect of the
drug. J Transl Med. 10:2522012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lavon I, Fuchs D, Zrihan D, Efroni G,
Zelikovitch B, Fellig Y and Siegal T: Novel mechanism whereby
nuclear factor kappaB mediates DNA damage repair through regulation
of O(6)-methylguanine-DNA-methyltransferase. Cancer Res.
67:8952–8959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weaver KD, Yeyeodu S, Cusack JC Jr,
Baldwin AS Jr and Ewend MG: Potentiation of chemotherapeutic agents
following antagonism of nuclear factor kappa B in human gliomas. J
Neurooncol. 61:187–196. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee DW, Ramakrishnan D, Valenta J, Parney
IF, Bayless KJ and Sitcheran R: The NF-κB RelB protein is an
oncogenic driver of mesenchymal glioma. PLoS One. 8:e574892013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang L, Wei B, Hu G, Wang L, Bi M, Sun Z
and Jin Y: Screening of differentially expressed genes associated
with human glioblastoma and functional analysis using a DNA
microarray. Mol Med Rep. 12:1991–1996. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Wang H, Zhang W, Huang HJ, Liao WS
and Fuller GN: Analysis of the activation status of Akt, NFkappaB,
and Stat3 in human diffuse gliomas. Lab Invest. 84:941–951. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun P, Mu Y and Zhang S: A novel
NF-κB/MMP-3 signal pathway involves in the aggressivity of glioma
promoted by Bmi-1. Tumour Biol. 35:12721–12727. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Z, Banerjee S, Li Y, Rahman KM, Zhang
Y and Sarkar FH: Down-regulation of notch-1 inhibits invasion by
inactivation of nuclear factor-kappaB, vascular endothelial growth
factor, and matrix metalloproteinase-9 in pancreatic cancer cells.
Cancer Res. 66:2778–2784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang CY, Cusack JC Jr, Liu R and Baldwin
AS Jr: Control of inducible chemoresistance: Enhanced anti-tumor
therapy through increased apoptosis by inhibition of NF-kappaB. Nat
Med. 5:412–417. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim HJ, Hawke N and Baldwin AS: NF-kappaB
and IKK as therapeutic targets in cancer. Cell Death Differ.
13:738–747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Jia L, Jin X, Liu Q, Cao W, Gao X,
Yang M and Sun B: NF-κB inhibitor reverses temozolomide resistance
in human glioma TR/U251 cells. Oncol Lett. 9:2586–2590.
2015.PubMed/NCBI
|
|
38
|
Zhang L, Ren X, Cheng Y, Liu X, Allen JE,
Zhang Y, Yuan Y, Huang SY, Yang W, Berg A, et al: The NFκB
inhibitor, SN50, induces differentiation of glioma stem cells and
suppresses their oncogenic phenotype. Cancer Biol Ther. 15:602–611.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qin JD, Cao ZH, Li XF, Kang XL, Xue Y, Li
YL, Zhang D, Liu XY and Xue YZ: Effect of ammonium pyrrolidine
dithiocarbamate (PDTC) on NF-κB activation and CYP2E1 content of
rats with immunological liver injury. Pharm Biol. 52:1460–1466.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kan S, Zhou H, Jin C and Yang H: Effects
of PDTC on NF-κB expression and apoptosis in rats with severe acute
pancreatitis-associated lung injury. Int J Clin Exp Med.
8:3258–3270. 2015.PubMed/NCBI
|
|
41
|
Zhang JJ, Xu ZM, Zhang CM, Dai HY, Ji XQ,
Wang XF and Li C: Pyrrolidine dithiocarbamate inhibits nuclear
factor-κB pathway activation, and regulates adhesion, migration,
invasion and apoptosis of endometriotic stromal cells. Mol Hum
Reprod. 17:175–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gu JW, Young E, Busby B, Covington J and
Johnson JW: Oral administration of pyrrolidine dithiocarbamate
(PDTC) inhibits VEGF expression, tumor angiogenesis, and growth of
breast cancer in female mice. Cancer Biol Ther. 8:514–521. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lan F, Yang Y, Han J, Wu Q, Yu H and Yue
X: Sulforaphane reverses chemo-resistance to temozolomide in
glioblastoma cells by NF-κB-dependent pathway downregulating MGMT
expression. Int J Oncol. 48:559–568. 2016.PubMed/NCBI
|
|
44
|
Song T, Li H, Tian Z, Xu C, Liu J and Guo
Y: Disruption of NF-κB signaling by fluoxetine attenuates MGMT
expression in glioma cells. Onco Targets Ther. 8:2199–2208.
2015.PubMed/NCBI
|
|
45
|
Fritz G, Tano K, Mitra S and Kaina B:
Inducibility of the DNA repair gene encoding O6-methylguanine-DNA
methyltransferase in mammalian cells by DNA-damaging treatments.
Mol Cell Biol. 11:4660–4668. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rauert-Wunderlich H, Siegmund D, Maier E,
Giner T, Bargou RC, Wajant H and Stühmer T: The IKK inhibitor Bay
11–7082 induces cell death independent from inhibition of
activation of NFκB transcription factors. PLoS One. 8:e592922013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Opferman JT: Attacking cancer's Achilles
heel: Antagonism of anti-apoptotic BCL-2 family members. FEBS J.
283:2661–2675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wall NR, Beck FW, Al-Katib AM and Mohammad
RM: Treatment-induced expression of anti-apoptotic proteins in
WSU-CLL, a human chronic lymphocytic leukemia cell line. J Drug
Target. 9:329–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zaanan A, Okamoto K, Kawakami H, Khazaie
K, Huang S and Sinicrope FA: The mutant KRAS Gene Up-regulates
BCL-XL protein via STAT3 to confer apoptosis resistance that is
reversed by BIM protein induction and BCL-XL antagonism. J Biol
Chem. 290:23838–23849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Plewka D, Jakubiec-Bartnik B, Morek M,
Bogunia E, Bienioszek M, Wolski H, Kotrych D, Dziekan K,
Seremak-Mrozikiewicz A and Plewka A: Survivin in ovary tumors.
Ginekol Pol. 86:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou W, Xu J, Gelston E, Wu X, Zou Z, Wang
B, Zeng Y, Wang H, Liu A, Xu L and Liu Q: Inhibition of Bcl-xL
overcomes polyploidy resistance and leads to apoptotic cell death
in acute myeloid leukemia cells. Oncotarget. 6:21557–21571. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vallianou NG, Evangelopoulos A, Schizas N
and Kazazis C: Potential anticancer properties and mechanisms of
action of curcumin. Anticancer Res. 35:645–651. 2015.PubMed/NCBI
|
|
53
|
Shukla S, Shankar E, Fu P, MacLennan GT
and Gupta S: Suppression of NF-κB and NF-κB-regulated gene
expression by apigenin through IκBα and IKK pathway in TRAMP mice.
PLoS One. 10:e01387102015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bravatà V, Minafra L, Russo G, Forte GI,
Cammarata FP, Ripamonti M, Casarino C, Augello G, Costantini F,
Barbieri G, et al: High-dose ionizing radiation regulates gene
expression changes in the MCF7 breast cancer cell line. Anticancer
Res. 35:2577–2591. 2015.PubMed/NCBI
|
|
55
|
Kunami N, Katsuya H, Nogami R, Ishitsuka K
and Tamura K: Promise of combining a Bcl-2 family inhibitor with
bortezomib or SAHA for adult T-cell leukemia/lymphoma. Anticancer
Res. 34:5287–5294. 2014.PubMed/NCBI
|
|
56
|
Elhag R, Mazzio EA and Soliman KF: The
effect of silibinin in enhancing toxicity of temozolomide and
etoposide in p53 and PTEN-mutated resistant glioma cell lines.
Anticancer Res. 35:1263–1269. 2015.PubMed/NCBI
|
|
57
|
Olivier S, Robe P and Bours V: Can
NF-kappaB be a target for novel and efficient anti-cancer agents?
Biochem Pharmacol. 72:1054–1068. 2006. View Article : Google Scholar : PubMed/NCBI
|