Introduction
Hepatocarcinoma is a tumor with a high degree of
malignancy. Hepatocarcinoma is the second most prevalent cause of
cancer-associated mortality in males and the sixth leading cause in
female patients (1). Typically,
treatment for hepatocarcinoma is not administered in a timely
manner due to late diagnosis. The majority of patients are
diagnosed when effective surgery is not possible and in other
patients, metastases are identified during surgery. Therefore
patient prognosis is typically poor (2). Increasing attention has focused on
identifying efficient therapeutic targets for hepatocarcinoma
treatment.
Endocan expression has been reported in numerous
types of tumor and therefore may represent a target for carcinoma
treatment (3–8). In particular, endocan is overexpressed
in tumor cells where it promotes tumor growth (9). In addition, the circulating levels of
endocan increase over time and have been positively correlated with
tumor size (9). Endocan was
identified to be significantly overexpressed in endothelial cells
isolated from hepatocellular carcinoma tissue compared with
corresponding non-cancerous liver tissue (10). Endocan is also expressed at the
periphery of tumor cells in hepatocellular carcinoma tissue
(11). Furthermore, the mRNA
expression of endocan in tissue samples has been associated with
the tumor node metastasis stage, tumor vascular invasion and
metastasis in patients with hepatocarcinoma (11). Further studies have demonstrated that
the endocan expression in tumors is associated with their
angiogenic and invasive properties (12,13). In
vascular endothelial cells, certain factors promote the expression
of endocan, including cultured hepatocyte growth factors, scatter
factors, fibroblast growth factor-2 and vascular endothelial growth
factor (14).
In the present study, endocan silencing experiments
were performed using small interfering (si)RNA. In particular, cell
proliferation and invasion were examined following endocan siRNA
treatment in SK-HEP-1 cells, and cell survival was evaluated using
flow cytometry and western blot analysis.
Materials and methods
Cell culture
Human SK-HEP-1 hepatocarcinoma cells were obtained
from the American Type Culture Collection (Manassas, VA, USA).
Cells were cultured in minimal essential medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
bovine serum (FBS; Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA). All cells were incubated at 37°C in an atmosphere containing
5% CO2. The inoculum size was 3×103
cells/well in a 96-multiwell plate, 2×104 cells/well in
a 24-multiwell plate or 3×105 cells/well in a
6-multiwell plate (BD Biosciences, Franklin Lakes, NJ, USA). The
medium was replaced every 2 days. The dose of pyrrolidine
dithiocarbamate (PDTC) was 10 µM and the duration of treatment was
24 h at 37°C. The controls used dimethyl sulfoxide (DMSO).
siRNA knockdown of endocan gene
expression of human SK-HEP-1 hepatocarcinoma cells
In 6-well plates, 8 µl Lipofectamine
2000® (Thermo Fisher Scientific, Inc.). +2 µg
pRNA-H1.1+2 ×106 cells (70–80% confluence) were placed
in every well. Endocan-targeting and control siRNAs
(5′-TTCTCCGAACGTGTCACGT-3′) were purchased from Sangon Biotech Co.,
Ltd. (Shanghai, China). pRNA-H1.1 plasmids were constructed and
used as the siRNA vector according to the protocol of the
manufacturer (Jinsirui Science and Technology Biology Corp.,
Nanjing, China), and it was transfected into cells using
Lipofectamine 2000®. The plasmid connection system
contains 0.03 pmol pRNA-H1.1, 20 nmol Insert DNA, 2.5 µl 10×T4 DNA
Ligase Buffer, 1 µl T4 DNA Ligase and ddH2O up to 25 µl.
The target sequence of endocan was 5′-GGTCTCCCGTAATGAGGAA-3′. A
total of 2 µg siRNA vector was used to transfect into SK-HEP-1
cells with Lipofectamine 2000 kit (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Western blot analysis
Western blotting was performed 48 h following
transfection. SDS-PAGE (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) was conducted using 10 and 14% gels, according to the
manufacturer's protocol. Protein extraction was performed with 2 ml
radioimmunoprecipitation lysate (Beyotime Institute of
Biotechnology, Haimen, China) on ice for 5 min. Centrifugation was
performed at 24,000 × g and 4°C for 10 min. The protein
quantification method used was BCA, 200 µl of BCA at 37°C for 20
min. A total of 40 µg protein was loaded per lane. Proteins were
transferred onto polyvinylidene difluoride membranes. The membrane
was blocked in a solution of TBS-Tween 20 containing 5% non-fat dry
milk for 1 h at 25°C with constant agitation. Proteins were probed
with a number of primary antibodies (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). The primary antibodies were as follows:
Endocan antibody (sc-515304; Santa Cruz Biotechnology, Inc.; 1:200)
at 4°C overnight; LC3 antibody (AL221; Beyotime Institute of
Biotechnology; 1:500) at 4°C overnight; ATG5 antibody (PA2260;
Boster Biological Technology, Pleasanton, CA, USA; 1:400,) at 4°C
overnight; ATG7 antibody (PB9479; Boster Biological Technology;
1:400) at 4°C overnight; DRAM antibody (bs-4233R, BIOSS, Beijing,
China; 1:500) at 4°C overnight; Beclin-1 antibody (AB123; Beyotime
Institute of Biotechnology; 1:1,000) at 4°C overnight and NF-κB p65
antibody (AF0246; Beyotime Institute of Biotechnology; 1:1,000) at
4°C overnight. The membranes were incubated with secondary
horseradish peroxidase-conjugated anti-Immunoglobulin G antibody
(A0181; Beyotime Institute of Biotechnology; 1:5,000) at 37°C for
45 min. β-actin was used as a loading control. Immunolabeled
proteins were detected following incubation with ECL substrate
(Beyotime Institute of Biotechnology) at room temperature for 50
sec, followed by exposure of the membrane to autoradiographic film.
Density analysis was performed using a Gel Doc system (Gel Doc XR+;
Bio-Rad Laboratories, Inc.). The nucleus protein and cell plasma
protein extraction kit (P0028, contain reagent A and reagent B) was
purchased from Beyotime Institute of Biotechnology and used to
separate the cytoplasm and nuclear fractions. Reagent A digested
the cell membrane and Reagent B digested the cell nucleus.
Cell viability measured using an MTT
assay
Following varying durations following transfection
(24, 48, 72 or 96 h), the culture medium was replaced with a medium
containing 5 mg/ml MTT (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), and the cells were incubated for 24 h at 37°C. The
supernatant was discarded and DMSO was used to dissolve the purple
crystals. The absorbance at a wavelength of 490 nm was measured
using a microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
Cell invasion assays
After 48-h of endocan siRNA treatment in Dulbecco's
modified Eagle medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS, the SK-HEP-1 cells were harvested (24,000 × g
for 5 min at room temperature) using trypsin. Then the cells were
washed in DMEM with a soybean trypsin inhibitor and without serum.
The cells were suspended in serum-free DMEM at a density of
1×105 cells/ml. Matrigel (BD Biosciences) was put into
Transwell. The cells (2×104/well) were allowed to
migrate towards DMEM containing 20% FBS in the bottom chamber of
the Transwell. The 24-well plates were incubated for 24 h at 37°C.
The invaded cells on the bottom surface of the membrane were
stained with crystal violet at room temperature for 5 min. The
number of migrated cells was counted in 5 randomly selected fields
using a light microscope at a magnification of ×200. Data presented
are representative of the average of 3 individual wells.
Apoptosis detection using flow
cytometry
The SK-HEP-1 cells were cultured for 48 h following
treatment with endocan siRNA in DMEM containing 10% FBS. The cells
were harvested (24,000 × g for 5 min at room temperature) by
trypsinization and washed in DMEM with a soybean trypsin inhibitor
and without serum. The cells were suspended in 500 µl of binding
buffer (556547; BD Biosciences), and 5 µl Annexin V-FITC was added,
together with 5 µl propidium iodide. The cells ware incubated for
15 min. The flow cytometer and software (WinMDI v2.8) used was
FACSCalibur (BD Biosciences).
mRFP-GFP-LC3 fluorescence system
assays
Following the appropriate cell culture period
(density 80%), adenovirus infection was performed according to the
manufacturer's protocol (mRFP-GFP-LC3; Hanbio, Shanghai, China;
adenovirus number: cell number, 15:1). The plates were incubated
for 4 h at 37°C with 5% CO2 in a humidified atmosphere.
Stationary infected cells, or those cells that were able to stably
present fluorescent signals, were incubated in 4% paraformaldehyde
for 0.5 h at 4°C. The level of fluorescence in the cells was
observed using a fluorescence microscope. Cells in every 5 fields
of view were counted.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA extraction buffer was purchased from BioTek
Instruments, Inc. (RP5611). TIANScript RT kit (KR104) was supplied
by Tiangen Biotech Co., Ltd. (Beijing, China). The primer sequences
were as follows: Endocan forward, 5′-CTGGGAAACATGAAGAGCG-3′ and
reverse, 5′-GCCTGAGACTGTGCGGTAG-3′ and β-actin forward,
5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT-3′. The fluorophore used Gold View
(Beijing Solarbio Science and Technology, Co., Ltd, Beijing,
China). The RT reaction was performed with 1 µl oligo (dT)15, 1 µl
random primer, 2 µl dNTP (2.5 mM each), 10.5 µl ddH2O
(2), and the thermocycler conditions
were as follows: 70°C for 5 min, cooling on ice for 2 min, and then
4 µl 5X First-Strand Buffer, 0.5 µl RNasin, 1 µl (200 U) TIANScript
M-MLV were placed in each tube at 42°C for 50 min, then at 95°C for
5 min. The thermocycler conditions were: 95°C for 5 min, 95°C for
20 sec, 52°C for 20 sec, 72°C for 30 sec for 40 cycles. The
experiment was repeated 3 times. The method of quantification was
gel electrophoresis, using 1.5% sepharose gel. The gel mixture was
heated in the microwave oven until boiling. When cooled to 50–60°C,
Gold View (Beyotime Institute of Biotechnology, Shanghai, China)
dye (0.01%) was blended. The DNA ladder was purchased from Beyotime
Institute of Biotechnology.
NF-κB inhibitor treatment
Cell cultures were performed as aforementioned. The
four groups prepared as follows: the NC + DMSO group, siRNA + DMSO
group, NC + PDTC group, siRNA + PDTC group. DMSO and 10 µM NF-κB
pathway inhibitor ammonium PDTC (Beyotime Institute of
Biotechnology) were added to the culture for 24 h at room
temperature.
Statistical analysis
Data were assessed using the Student's t-test or
one-way analysis of variance, as appropriate, followed by the
Turkey's post-hoc test. All statistics were calculated using SPSS
software (version 17.0; SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference. Data
are presented as the mean ± standard deviation. The number of
replications was three.
Results
Gene silencing of endocan decreases
cell proliferation and cell invasion
In the present study, SK-HEP-1 cells treated with
endocan siRNA were monitored for cell proliferation and invasion
using an MTT and a Transwell assay, respectively. The following 3
groups were established: Untreated control; negative control (NC)
siRNA; and endocan siRNA groups. To determine siRNA efficiency, the
endocan mRNA transcript and protein level were evaluated using
RT-PCR and western blotting, respectively (Fig. 1A and B). The level of endocan
expression was decreased in the endocan siRNA compared with the NC
and control groups (Fig. 1A and B).
Cell proliferation was evaluated using the MTT assay and was
significantly reduced in siRNA-treated cells compared with the two
control groups (Fig. 1C). In the
Transwell assay, the invasive ability of endocan siRNA-treated
cells was significantly reduced compared with the NC and control
groups (Fig. 1D).
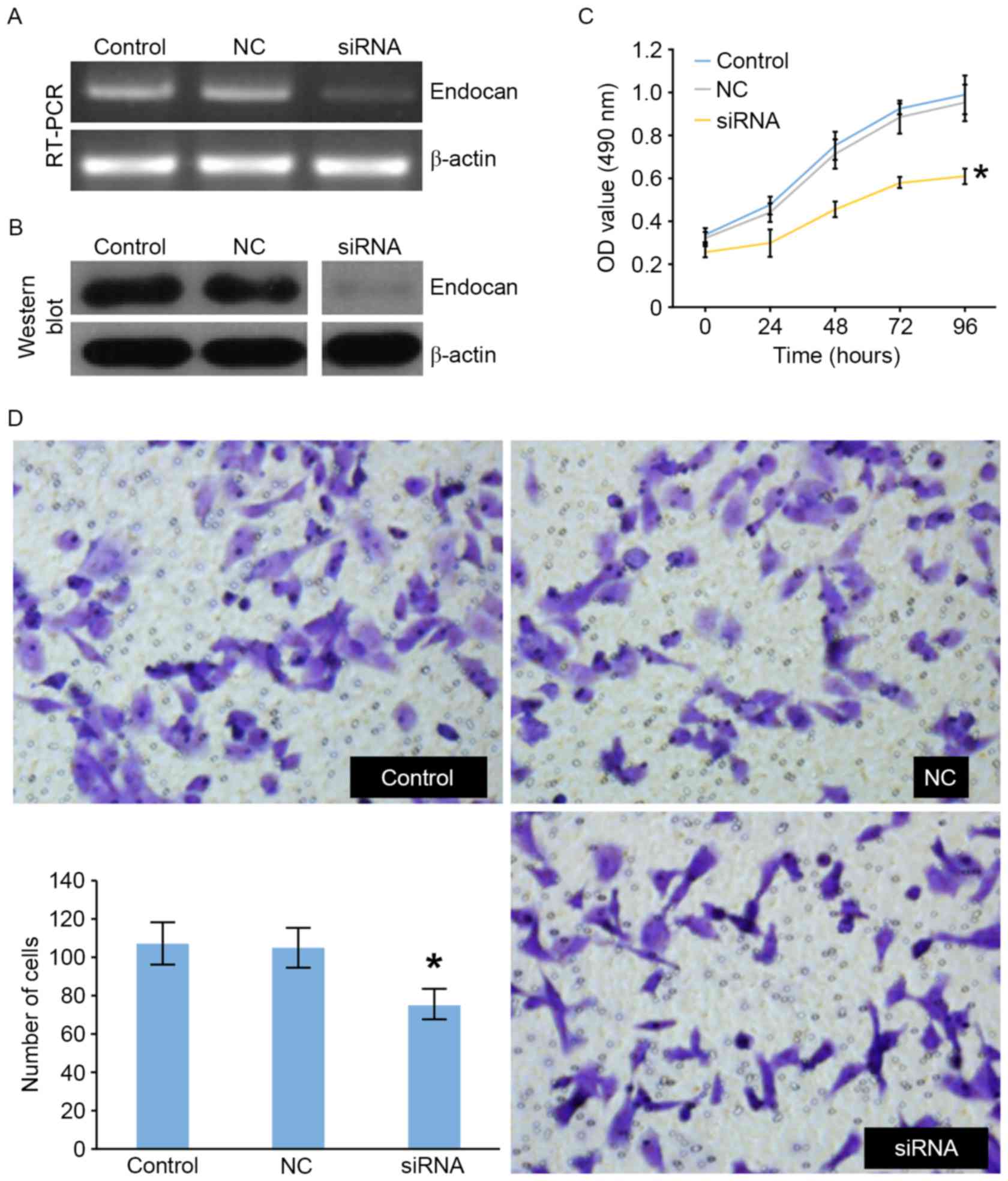 | Figure 1.Endocan siRNA and NC siRNA constructs
were transfected in SK-HEP-1 cells. siRNA against endocan
suppressed its expression, evaluated using (A) RT-PCR and (B)
western blotting (normalized to β-actin). (C) SK-HEP-1 cells
cultured in 96-well plates were transfected with endocan siRNA, and
cells were analyzed 0, 24, 48, 72 and 96 h following transfection.
The cell proliferation was determined with an MTT assay. Data were
presented as the mean ± standard deviation (n=3). The average OD
value from each sample was obtained from 5 replicates. (D)
Transwell assays were used to investigate the migratory and
invasive abilities of the control, NC and siRNA groups. Invading
cells were stained with crystal violet and counted in ≥5 fields
using a light microscope (magnification, ×200). *P<0.05 in siRNA
groups vs. NC groups. OD, optical density; NC, negative control;
siRNA, small interfering RNA; RT-PCR, reverse transcription
polymerase chain reaction. |
Gene silencing of endocan enhances
apoptosis in SK-HEP-1 cells
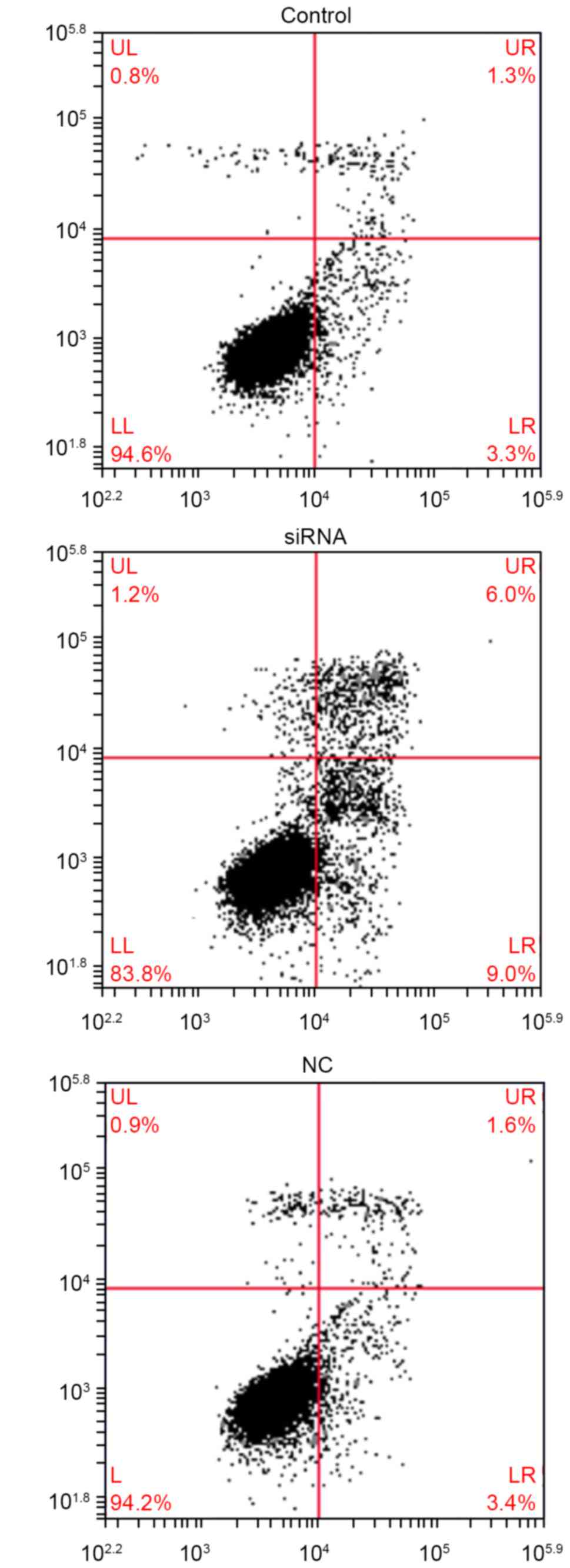
The apoptosis assay demonstrated that endocan
silencing resulted in a marked increase in the apoptosis rate
(Fig. 2). Overall, these results
suggest that endocan silencing promotes cell apoptosis.
Gene silencing of endocan enhances
autophagy in SK-HEP-1 cells
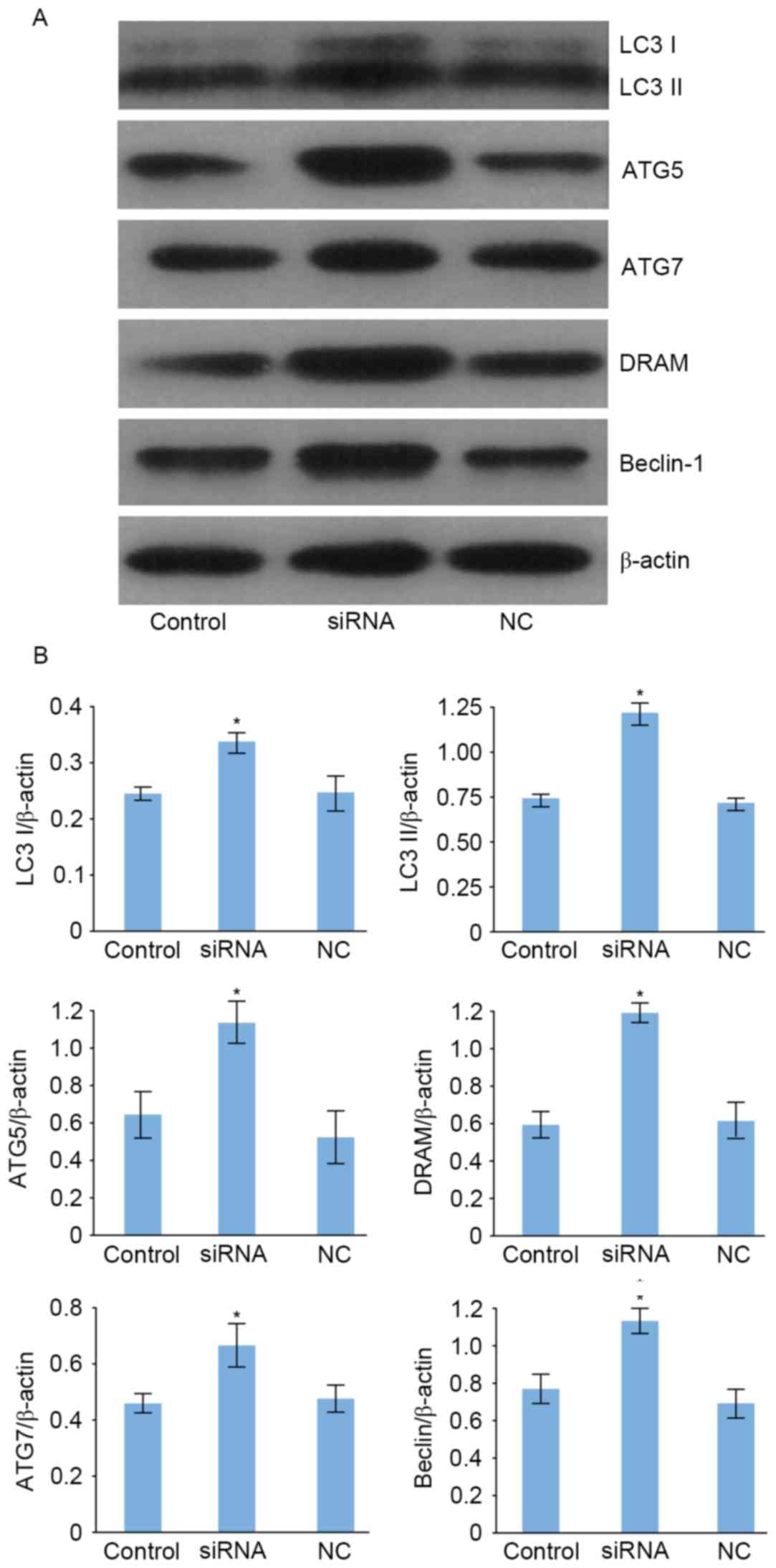
To determine whether the gene silencing of endocan
is able to induce autophagy, autophagy-associated protein
expression levels were evaluated using western blotting (Fig. 3A). The expression levels of
microtubule associated protein 1 light chain 3 α (LC3), autophagy
related (ATG)5, ATG7, DNA damage regulated autophagy modulator 1
(DRAM) and Beclin-1 protein following transfection with endocan
siRNA were significantly increased compared with those of the NC,
and control groups (Fig. 3B). These
results suggest that endocan silencing promotes autophagy in
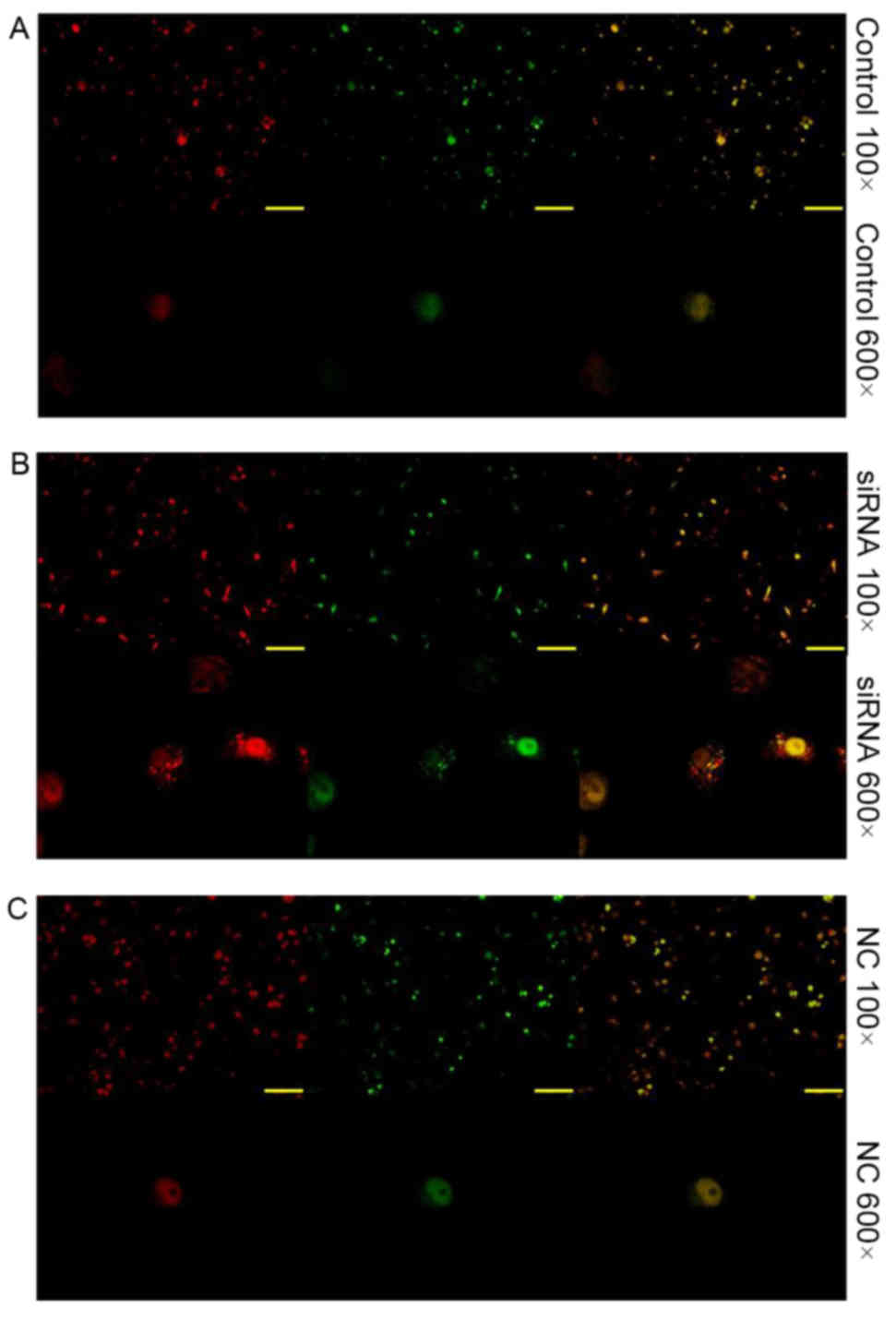
hepatocarcinoma cells. In Fig. 4A, no
increased level red fluorescence was observed compared with in
Fig. 4B or C. In Fig. 4B, an increased level red fluorescence
was observed compared with in Fig. 4A or
C, this revealed that there was an increased level of autophagy
in Fig. 4B. In Fig. 4C, no increased level red fluorescence
was observed compared with in Fig. 4A or
B.
Association between endocan gene
expression and the nuclear factor (NF)-κB signaling pathway
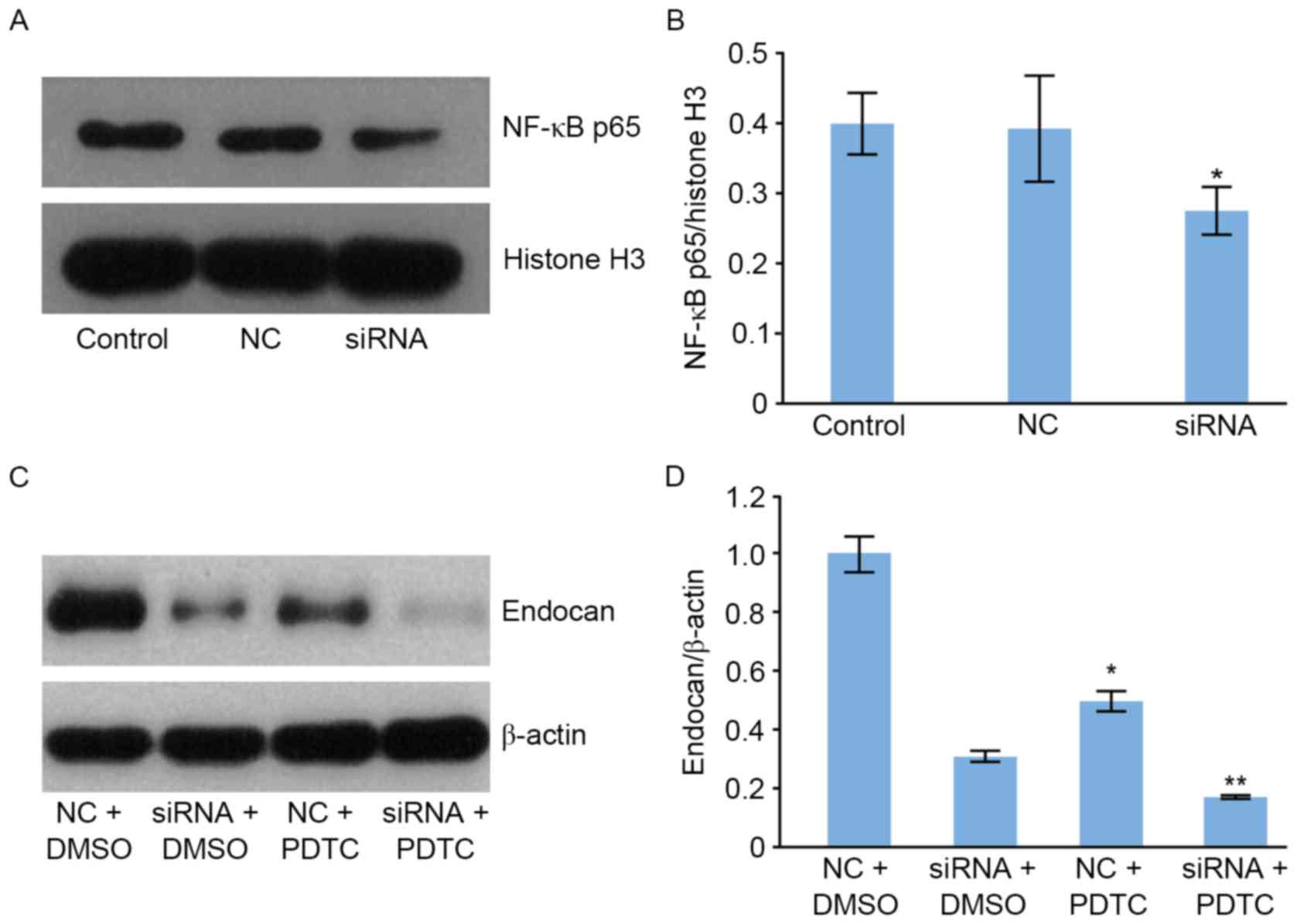
To determine whether the gene silencing of endocan
is able to influence the NF-κB signaling pathway, NF-κB p65 protein
expression was evaluated using western blotting (Fig. 5A). The level of NF-κB p65 protein
within the nucleus following transfection with endocan siRNA was
significantly reduced compared with that of the cells in the NC and
control groups (Fig. 5A and B).
Furthermore, the level of endocan protein was significantly reduced
following treatment with NF-κB pathway inhibitor ammonium PDTC
(Fig. 5C and D) compared with DMSO.
These results demonstrated the synergistic effects of endocan gene
expression on the NF-κB signaling pathway. The expression level of
the endocan protein were as follows: NC+DMSO group>siRNA+DMSO
group; siRNA+DMSO group> siRNA+PDTC group; NC+PDTC group>
siRNA+PDTC group.
Discussion
Effective tumor treatment remains a challenge for
clinicians and researchers. There are currently 2 types of
non-surgical therapy for hepatocarcinoma; One therapy targets the
tumor and the other targets the tumor microenvironment. Previous
studies have demonstrated that endocan may represent a target for
tumor-targeted therapy (3–8). Although there is insufficient data
regarding whether endocan may be an effective tumor marker similar
to α-fetoprotein, a number of previous studies have reported that
endocan is associated with tumor cell survival (9,10,12,14).
Indeed, endocan gene silencing was utilized to inhibit tumor cell
survival in human colon carcinoma (14), concordant with the results of the
present study. Using MTT and Transwell assays, the results of the
current study further demonstrate that endocan silencing inhibits
SK-HEP-1 cell proliferation and invasion, which indicates that
endocan may serve as a treatment target in hepatocarcinoma.
The induction of programmed cell death (PCD) is the
preferred result for tumor treatment. Specifically, tumor-cell PCD
may be targeted without injuring healthy cells. Apoptosis
represents type I PCD and involves a series of morphological and
biochemical processes, including alterations in the mitochondrial
membrane potential, opening of mitochondrial pores, phosphoserine
shifts and the gathering of nuclear chromatin (15). An Annexin V-FITC kit was used with
flow cytometry analysis to determine alterations in apoptotic
rates, and the results identified that apoptosis was induced in
siRNA-treated SK-HEP-1 cells. The low apoptotic cell ratio may be
due to the short observation time; therefore, if the observation
time had been extended, the ratio may have increased. Autophagy
serves an important role in cell survival and death, and a certain
degree of autophagy occurs during normal cellular metabolism. The
purpose of autophagy is to digest metabolic waste or remove
misfolded proteins. Furthermore, autophagy leads to cell death,
specifically type II PCD (16).
Through autophagy-associated protein determination, evidence of
autophagy was observed in endocan siRNA-treated cells. The
autophagy-associated proteins investigated include LC3 (17,18), ATG5
(19), ATG7, Beclin-1 (20) and DRAM (21). The significant increase in the
expression of these proteins in siRNA-treated cells compared with
NC and control cells indicate the increased level of autophagy in
the endocan siRNA-treated cells. Furthermore, red and green
fluorescent dyes were used for the detection of LC3, as observed
through the lack of green fluorescence due to the formation of
autophagic lysosomes, it is suggested that endocan silencing
induces autophagy.
NF-κB serves an important role in cell survival and
this transcription factor may be phosphorylated to regulate gene
expression in the nucleus (22,23). NF-κB
is also an important regulator of gene promoters. In the process of
cell apoptosis, NF-κB may regulate the function of apoptotic
proteins (24,25), anti-apoptotic proteins (26–28) and
tumor-suppressor proteins (29–31). To
investigate the underlying mechanism of endocan gene silencing in
hepatocarcinoma cell apoptosis and autophagy, the NF-κB signaling
pathway was used as an indicator. In the current study, it was
demonstrated that following endocan gene silencing, NF-κB
expression decreased significantly in the cell nucleus compared
with NC and control cells that were not treated with endocan siRNA,
which may have contributed to the impaired survival of
hepatocarcinoma cells. In addition, the influence of the NF-κB
signaling pathway on endocan expression was demonstrated as
following inhibition of the NF-κB signaling pathway through
treatment of cells with PDTC, endocan expression was significantly
decreased compared with untreated cells.
In conclusion, the results of the present study
demonstrated that endocan silencing induced PCD in SK-HEP-1 cells.
Thus, endocan may be a treatment target for hepatocarcinoma.
Furthermore, these results identified an association between
endocan gene expression and the NF-κB signaling pathway, suggesting
that combined treatments may improve the efficiency of inhibiting
hepatocarcinoma progression.
Acknowledgements
The authors would like to thank Dr Xihe Yu for
technical support and the China-Japan Union Hospital (Changchun,
China) for its support.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song MJ and Bae SH: Newer treatments for
advanced hepatocellular carcinoma. Korean J Intern Med. 29:149–155.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abid MR, Yi XJ, Yano K, Shih SC and Aird
WC: Vascular endocan is preferentially expressed in tumor
endothelium. Microvasc Res. 72:136–145. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maurage CA, Adam E, Minéo JF, Sarrazin S,
Debunne M, Siminski RM, Baroncini M, Lassalle P, Blond S and
Delehedde M: Endocan expression and localization in human
glioblastomas. J Neuropathol Exp Neurol. 68:633–641. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ziol M, Sutton A, Calderaro J, Barget N,
Aout M, Leroy V, Blanc JF, Sturm N, Bioulac-Sage P, Nahon P, et al:
ESM-1 expression in stromal cells is predictive of recurrence after
radiofrequency ablation in early hepatocellular carcinoma. J
Hepatol. 59:1264–1270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oltean S, Pullerits R, Flodén A, Olausson
M and Oltean M: Increased resistin in brain dead organ donors is
associated with delayed graft function after kidney
transplantation. J Transl Med. 11:2332013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leroy X, Aubert S, Zini L, Franquet H,
Kervoaze G, Villers A, Delehedde M, Copin MC and Lassalle P:
Vascular endocan (ESM-1) is markedly overexpressed in clear cell
renal cell carcinoma. Histopathology. 56:180–187. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zuo L, Zhang SM, Hu RL, Zhu HQ, Zhou Q,
Gui SY, Wu Q and Wang Y: Correlation between expression and
differentiation of endocan in colorectal cancer. World J
Gastroenterol. 14:4562–4568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scherpereel A, Gentina T, Grigoriu B,
Sénéchal S, Janin A, Tsicopoulos A, Plénat F, Béchard D, Tonnel AB
and Lassalle P: Overexpression of endocan induces tumor formation.
Cancer Res. 63:6084–6089. 2003.PubMed/NCBI
|
|
10
|
Kang YH, Ji NY, Lee CI, Lee HG, Kim JW,
Yeom YI, Kim DG, Yoon SK, Kim JW, Park PJ and Song EY: ESM-1
silencing decreased cell survival, migration, and invasion and
modulated cell cycle progression in hepatocellular carcinoma. Amino
Acids. 40:1003–1013. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen LY, Liu X, Wang SL and Qin CY:
Over-expression of the Endocan gene in endothelial cells from
hepatocellular carcinoma is associated with angiogenesis and tumour
invasion. J Int Med Res. 38:498–510. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang GW, Tao YM and Ding X: Endocan
expression correlated with poor survival in human hepatocellular
carcinoma. Dig Dis Sci. 54:389–394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gerritsen ME, Tomlinson JE, Zlot C, Ziman
M and Hwang S: Using gene expression profiling to identify the
molecular basis of the synergistic actions of hepatocyte growth
factor and vascular endothelial growth factor in human endothelial
cells. Br J Pharmacol. 140:595–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim JH, Park MY, Kim CN, Kim KH, Kang HB,
Kim KD and Kim JW: Expression of endothelial cell-specific
molecule-1 regulated by hypoxia inducible factor-1α in human colon
carcinoma: Impact of ESM-1 on prognosis and its correlation with
clinicopathological features. Oncol Rep. 28:1701–1708. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kepp O, Galluzzi L, Lipinski M, Yuan J and
Kroemer G: Cell death assays for drug discovery. Nat Rev Drug
Discov. 10:221–237. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jardon MA, Rothe K, Bortnik S, Vezenkov L,
Jiang X, Young RN, Lum JJ and Gorski SM: Autophagy: From structure
to metabolism to therapeutic regulation. Autophagy. 9:2180–2182.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang R and Liu W: Identifying an
essential role of nuclear LC3 for autophagy. Autophagy. 11:852–853.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lai SC and Devenish RJ: LC3-associated
phagocytosis (LAP): Connections with host autophagy. Cells.
1:396–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Codogno P and Meijer AJ: Atg5: More than
an autophagy factor. Nat Cell Biol. 8:1045–1047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang X, Qi Q, Hua X, Li X, Zhang W, Sun
H, Li S, Wang X and Li B: Beclin 1, an autophagy-related gene,
augments apoptosis in U87 glioblastoma cells. Oncology Rep.
31:1761–1767. 2014. View Article : Google Scholar
|
|
21
|
Criollo A, Dessen P and Kroemer G: DRAM: A
phylogenetically ancient regulator of autophagy. Cell Cycle.
8:2319–2320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu X, Nesic-Taylor O, Qiu J, Rea HC,
Fabian R, Rassin DK and Perez-Polo JR: Activation of nuclear
factor-kappaB signaling pathway by interleukin-1 after
hypoxia/ischemia in neonatal rat hippocampus and cortex. J
Neurochem. 93:26–37. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karin M, Yamamoto Y and Wang QM: The IKK
NF-kappa B system: A treasure trove for drug development. Nat Rev
Drug Discov. 3:17–26. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tato CM and Hunter CA: Host-pathogen
interactions: Subversion and utilization of the NF-kappa B pathway
during infection. Infect Immun. 70:3311–3317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bai X, Feldman NE, Chmura K, Ovrutsky AR,
Su WL, Griffin L, Pyeon D, McGibney MT, Strand MJ, Numata M, et al:
Inhibition of nuclear factor-kappa B activation decreases survival
of Mycobacterium tuberculosis in human macrophages. PLoS One.
8:e619252013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Loeuillet C, Martinon F, Perez C, Munoz M,
Thome M and Meylan PR: Mycobacterium tuberculosis subverts innate
immunity to evade specific effectors. J Immunol. 177:6245–6255.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukuda M, Kusama K and Sakashita H:
Cimetidine inhibits salivary gland tumor cell adhesion to neural
cells and induces apoptosis by blocking NCAM expression. BMC
Cancer. 8:3762008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Djavaheri-Mergny M, Amelotti M, Mathieu J,
Besançon F, Bauvy C, Souquère S, Pierron G and Codogno P: NF-kappaB
activation represses tumor necrosis factor-alpha-induced autophagy.
J Biol Chem. 281:30373–30382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peri S, Devarajan K, Yang DH, Knudson AG
and Balachandran S: Meta-analysis identifies NF-κB as a therapeutic
target in renal cancer. PLoS One. 8:e767462013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Orlowski RZ and Baldwin AS Jr: NF-kappaB
as a therapeutic target in cancer. Trends Mol Med. 8:385–389. 2002.
View Article : Google Scholar : PubMed/NCBI
|