Introduction
The majority of eukaryotic genes undergo alternative
splicing; once believed to be a peculiarity of a few genes, it is
now closer to being the rule rather than the exception. Alternative
splicing is a proteome-diversifying process through which several
mature RNA messengers are obtained from a single gene, each of
these transcripts potentially coding for a different protein
isoform with a potentially different function (1).
The specialized molecular machinery that performs
splicing is known as the spliceosome, a macromolecular complex
composed of four small nuclear ribonucleoproteins (snRNPs) (U1, U2,
U4/U6 and U5) and >100 non-snRNP splicing factors. The
spliceosome identifies individual splice sites along immature
transcripts through the recognition of consensus sequences located
in the exon/intron boundaries, which are complementary with the RNA
present in the snRNPs. Next, through a series of spatial
rearrangements, it facilitates two trans-esterification reactions
that result in the excision of the intronic sequence; the process
takes place at each intron to yield a mature mRNA (1).
On average, human genes have 8.8 introns (2) and ~90% of them produce more than one
mature transcript (3). Alternative
splicing can remove introns from a pre-mRNA in a number of
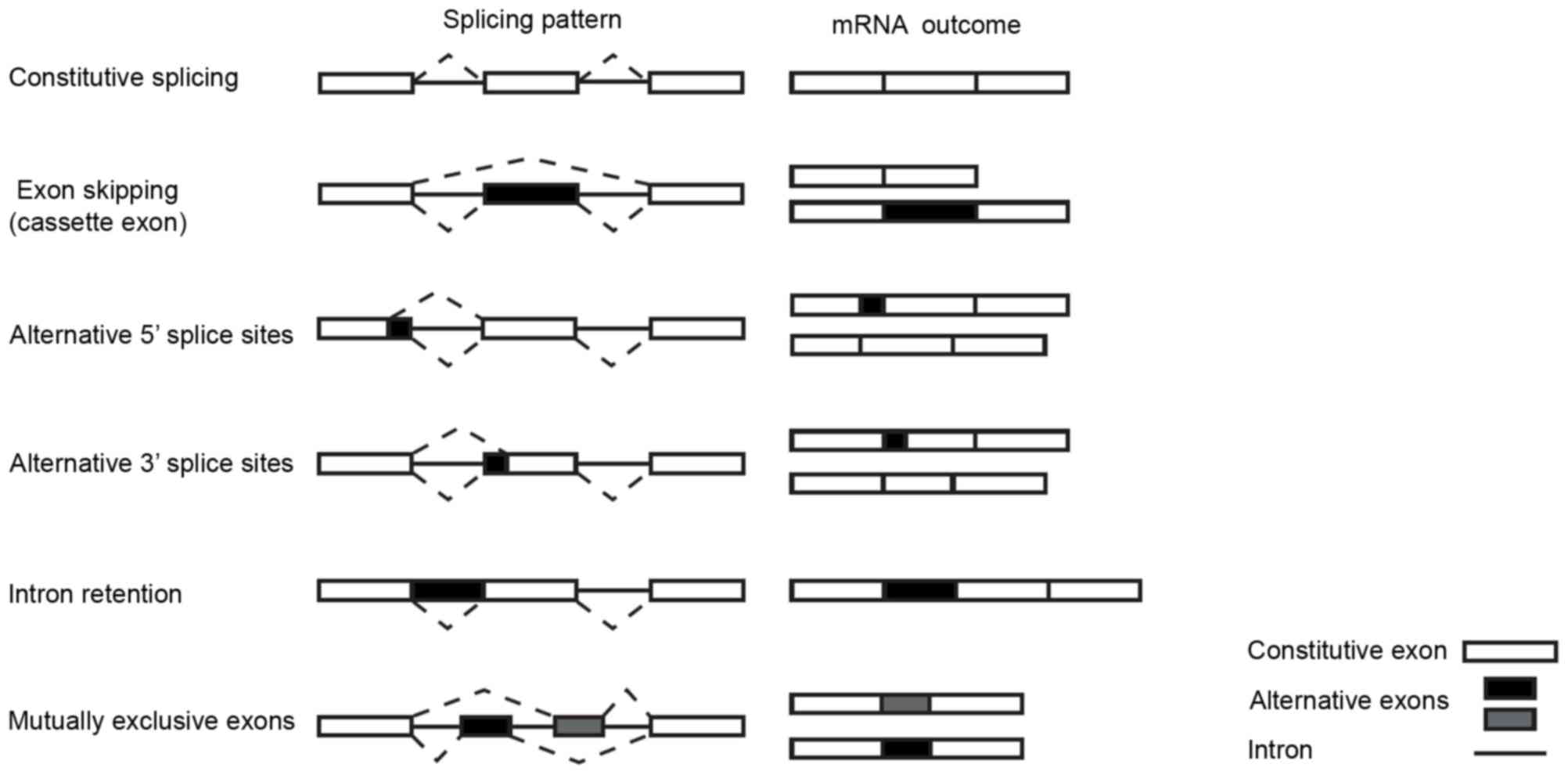
different combinations, giving rise to different mRNAs. Known
patterns of alternative splicing include exon skipping (removal of
an exon along with the surrounding introns), usage of alternative
intron donor (5′) or acceptor (3′) sites, intron retention and
mutually exclusive exon splicing (Fig.
1). Alternative exons may possess splice sites with diverging
sequences, thus are less likely to be recognized through RNA-RNA
interaction; such sites are considered sub-optimal and their
recognition is aided by the recruitment of trans-acting
splicing factors that bind to cis regulatory elements in the
vicinity of splice sites and recruit the main spliceosome
components. These cis elements are classified in
exonic/intronic splicing enhancers/silencers (ESE, ESS, ISE and
ISS) according to their positions and functions, while the factors
that bind to them mainly belong to two families, namely the SR and
heterogeneous (hn)RNP proteins, which usually promote and prevent
splice site recognition, respectively (Fig. 2). This way, the alternative splicing
of a given precursor (pre)-mRNA is regulated by the abundance of
the pre-mRNA itself, its sequence (presence/absence of splicing
factor recognition sites) and the relative abundance of the
splicing factors (4–6). So, it is now understood that the main
components of the spliceosome remain largely unchanged and that the
majority of the regulatory nature of alternative splicing relies on
protein-protein, protein-nucleic acid interactions and their
kinetics (7). Furthermore, RNA
splicing is coupled to transcription physically and dynamically.
Spliceosome factors are recruited to the carboxy-terminal domain of
the RNA polymerase during RNA elongation, from where they are in
turn recruited to the splice sites along the nascent transcript as
they become available, thus making splicing dependent on the
processivity of the RNA polymerase elongation complex (8).
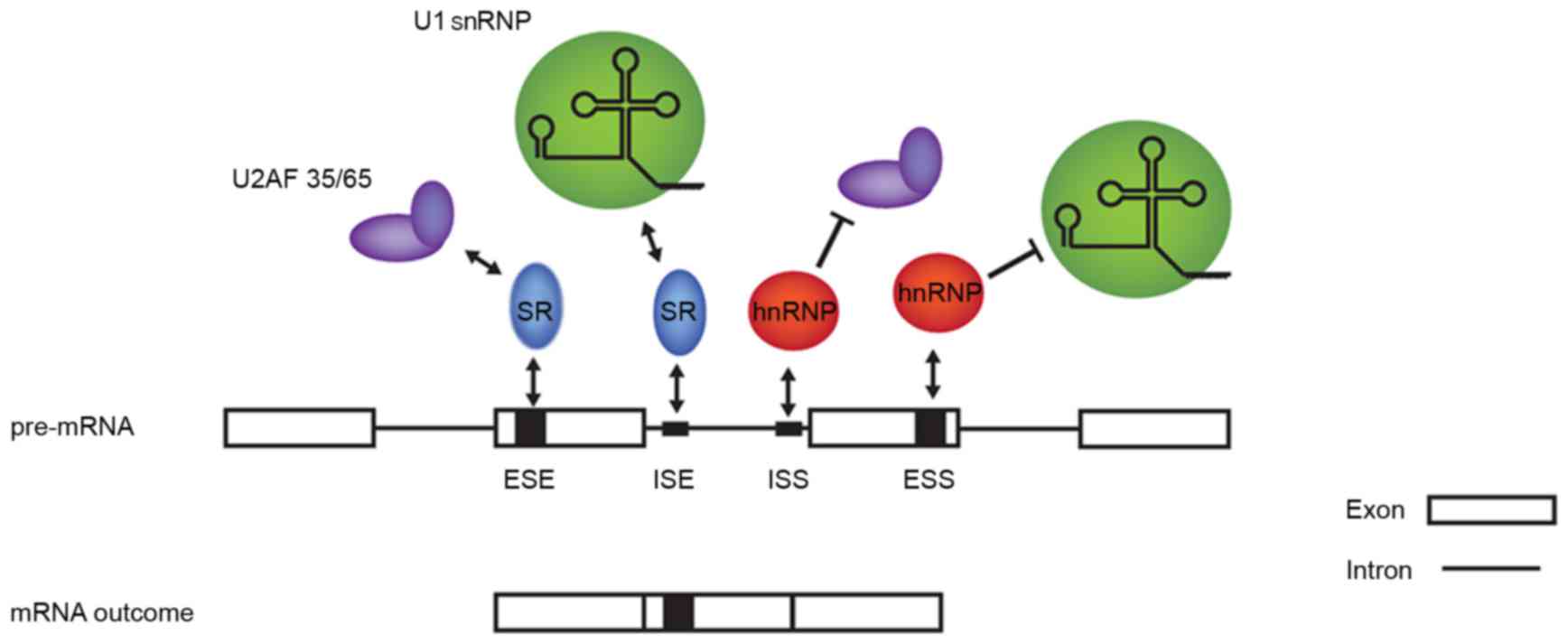 | Figure 2.Splicing cis elements
influence splice site recognition through recruitment of different
spliceosome components. Generally, splicing enhancers recruit SR
proteins, which in turn recruit the splicing machinery; splicing
supressors recruit hnRNP proteins, which hinder these interactions.
ESE, exonic splicing enhancer; ISE, intronic splicing enhancer;
ISS, intronic splicing suppressor; ESS, exonic splicing suppressor;
hnRNP, heterogeneous ribonucleoproteins; pre-mRNA, precursor-mRNA;
snRNP, small nuclear RNP. |
Alternative splicing has been observed to have an
effect on disease. There are a considerable number of hereditary
diseases caused by point mutations, which in general, disrupt
splice sites or ESE/ISEs and modify the aforementioned interactions
preventing the expression of one particular isoform of a protein
(9,10). The role that alternative splicing
plays in the generation and/or maintenance of complex pathological
conditions, such as inflammation, is not as straightforward, since
these conditions are the result of several alterations in pathways
that often comprise of dozens of proteins with diverse functions,
each of which is prone to regulation through alternative splicing.
High-throughput sequencing and bioinformatics have made it possible
to assess alternative splicing modifications on a global scale
(3,11); however, these methods are not yet
without limitations and require complementing with particular,
functional studies. The present review summarizes the impact of
alternative splicing in the core components of the TNF signaling
pathway.
Tumor necrosis factor (TNF) pathway
TNF-a (also known as cachectin) is a strong
pro-inflammatory cytokine, which plays an important role in certain
processes, including inflammation, cell proliferation,
differentiation and apoptosis. Inflammation is an extremely
important part of innate immunity and is regulated in a number of
steps (12).
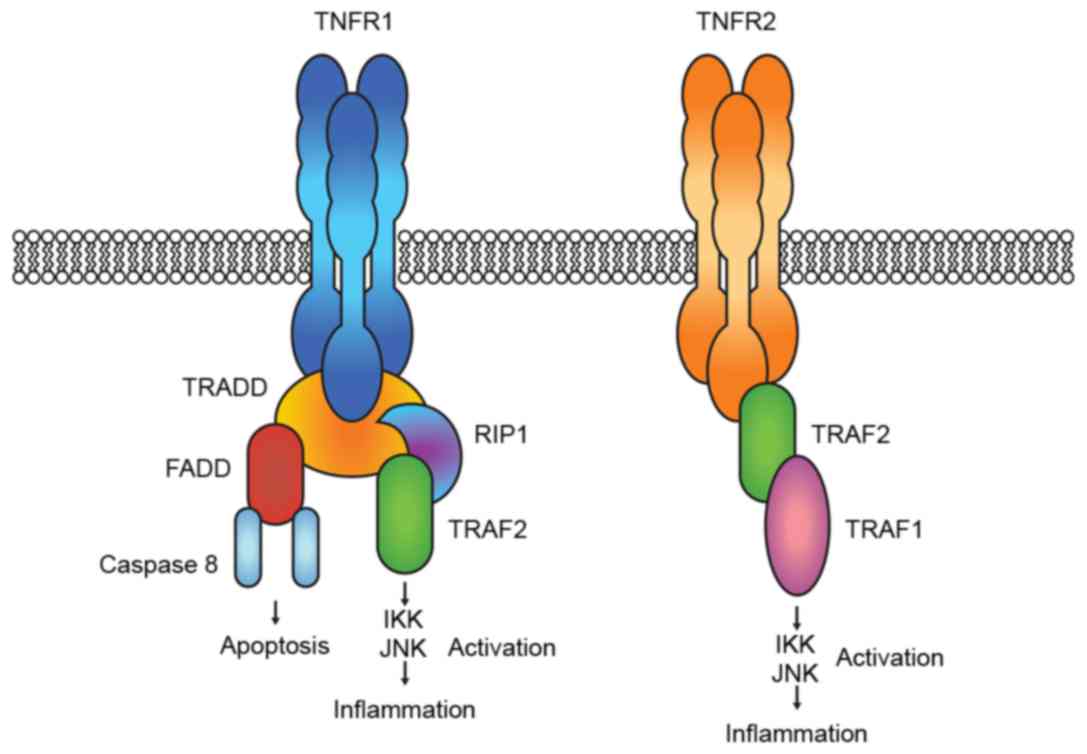
TNF-a binds two distinct receptors: TNFR1 and TNFR2.
Activation of TNFR1 leads to the formation of signal complexes that
activate pathways that lead to: i) Expression of pro-inflammatory
genes through the recruitment of receptor-interacting protein 1
(RIP1) and TNF-receptor-associated factor 2 (TRAF2); and ii)
apoptosis and cell death by recruiting Fas-associated death domain
protein (FADD) and caspase 8. Binding to TNFR2 induces only
inflammation through the direct recruitment of TRAF2, which in turn
recruits TRAF1. The two pro-inflammatory complexes lead to IKK
(inhibitor of nuclear factor κ-B kinase and mitogen-activated
protein kinase (JNK and p38) activation (Fig. 3) (12).
The balance between TNF-activated inflammation and
apoptosis is regulated on several levels, including signal
strength, expression of signaling molecules and regulating
proteins, and crosstalk with other cell signals (13), all of which can be heavily influenced
by the alternative splicing of each element.
Known isoforms in TNF signaling
A search on the Ensmbl database (14) for the splice variants originated from
the transcripts of the receptor-proximal elements of TNF signaling
shows that all but FADD have at least 2 potential protein variants
(Table I). Experimental evidence is
not available for all of them; and thus far, it comprises mostly
association data, rather than molecular evidence that offers an
understanding of their regulation. In the following section,
experimental evidence concerning alternative splicing of the key
players in TNF signaling is reviewed.
 | Table I.Splicing isoforms of the main
receptor-proximal elements of the TNF signaling pathway. |
Table I.
Splicing isoforms of the main
receptor-proximal elements of the TNF signaling pathway.
| Gene | Ensembl ID | No. of exons | No. of
transcripts | No. of
protein-coding transcripts |
|---|
| TNFR1 |
ENSG00000067182 | 10 | 16 | 9 |
| TNFR2 |
ENSG00000028137 | 10 | 4 | 2 |
| TRADD |
ENSG00000102871 | 5 | 5 | 2 |
| FADD |
ENSG00000168040 | 2 | 1 | 1 |
| TRAF1 |
ENSG00000056558 | 8 | 3 | 3 |
| TRAF2 |
ENSG00000127191 | 11 | 10 | 6 |
| TRAF3 |
ENSG00000131323 | 11 | 9 | 7 |
| RIP1 |
ENSG00000017797 | 10 | 5 | 5 |
TNFR
Two recent studies suggested that the TNF receptor
superfamily member 1A (TNFRSF1A) transcript balance may depend on
TNFRSF1A alleles. Gregory et al (15) investigated a single nucleotide
polymorphism (rs1800693, c.625+10A>G) in the TNFRSF1A gene that
was previously identified as a susceptibility marker for multiple
sclerosis through genome-wide association studies. In in
vitro splicing assays, only the G allele resulted in skipping
of exon 6. TNFR1 exon 6 skipping results in a frameshift and a
premature stop codon, which translates into a soluble form of TNFR
[D6-TNFR, comprising only the amino-terminus of TNFR1, followed by
a novel 45-amino acid (aa) sequence] for which a TNFR-antagonistic
role was suggested. Another detailed study described a TNFR1
transcript lacking exon 2 (TNFR1-d2) and its association with a
specific haplotype at 3 single nucleotide polymorphisms (SNPs)
previously associated with TNF-receptor-associated periodic
syndrome (TRAPS). These SNPs have distant locations along the
TNFR1A gene: rs4149570 lies at the promoter region (c.610G>T),
rs767455 at exon 1 (c.36A>G) and rs1800692 at exon 4
(c.473-33C>T), so it is plausible that an interplay of
transcription and splicing dynamics determines transcript outcome
(16). This evidence strongly
suggests that these SNPs disrupt splicing enhancers/silencers along
intronic or exonic sequences, an aspect yet to be studied for other
reported SNPs associated with TRAPS (17) or other inflammatory conditions,
including Crohn's disease (18).
TNFR2 isoforms have been described as well. Seitz
et al (19) characterized
hicpTNFR, a TNFR2 splice variant with an alternate exon 1 sequence
that results from the usage of an alternate transcription start
site and alternative splicing. This variant is mostly retained in
the trans-Golgi network and in endosomes where it could function as
a storage pool of preformed p75TNFR that is not affected by
shedding. Upon emerging on the cell surface, hicp75TNFR is
functionally no longer distinguishable from p75TNFR; furthermore,
hicpTNFR colocalizes with endogenous TNF, hinting at intracellular
activation of the hicp75TNFR by endogenous TNF (20). There are a number of studies on a
soluble TNFR2 isoform lacking exons 7 and 8, known as
differentially spliced (DS)-TNFR; as a consequence of splicing, it
lacks the transmembrane and cytoplasmic domains. The data gathered
suggests that it regulates TNFR function by antagonizing its
biological activity. This soluble receptor was detected at
increased levels in patients with sepsis and at higher
concentrations in patients with rheumatoid arthritis, relative to
the levels detected in the sera of healthy individuals (21). In another study, performed in
insulin-resistant patients, this same isoform was found in 26% of
samples from patients with type 2 diabetes and in 44% of
non-diabetic subjects. An increase in waist size was associated
with a progressive decrease in DS-TNFR2 concentration. Moreover, it
was suggested that DS-TNFR2 exhibits anti-inflammatory activity,
based on observations of the correlation between DS-TNFR2 and
circulating adiponectin (22). This
novel isoform and its potential anti-inflammatory properties have
been associated with markers of liver injury (23) and with a favorable outcome in patients
with rheumatoid arthritis (24),
although no information is currently available on the dynamics of
this splicing event.
RIP
Alternative splicing has been described in the RIP
family only in the RIP3 gene; two splice variants (RIP3-β and
RIP3-γ) with truncated N-terminal kinase domains and novel, shorter
C termini have been found. These variants abrogate
nucleocytoplasmic shuttling and are therefore not able to induce
apoptosis. Additionally, they downregulate RIP3 pro-apoptotic
activity (25). Suppression of
RIP3-dependent apoptotic TNF signaling could potentially upregulate
RIP1-dependent pro-inflammatory pathways.
Splice variants of the human RIP1 gene should not be
ruled out, since it has been demonstrated that the associated
Xenopus RIP1 gene produces at least one alternative
splicing-derived isoform (26).
According to the Ensembl database annotations, human RIP1 10
exon-pre-mRNA potentially produces 5 alternative transcripts, which
are yet to be experimentally described.
TRAF
A splice variant of TRAF2 was described as early as
1998, this variant (TRAF2A) contains a 7-aa insertion within the
RING finger domain, presumably produced by alternative usage of
splice donor sites present at the 3′ end of exon 1. TRAF2A is
incapable of mediating the activation of nuclear factor (NF)-κB,
thus it can act as a dominant inhibitor of TNFR2-mediated NF-κB
activation; this way, cells can regulate NF-κB activation through
TNF family receptors by modifying alternative splicing of primary
transcripts from the TRAF2/TRAF2A gene to produce different ratios
of TRAF2A and TRAF2 mRNAs (27). In a
comparison of the structure of the human, murine and
Drosophila TRAF genes, the TRAF2A transcript was only found
to be expressed in mice, and not in humans or rats, although only
Ramos (human B lymphoma) and HEK293 (human embryonic kidney) cells
were assayed (28).
TRAF3 is another member of the TRAF family of
proteins, (currently six genes have been identified, TRAF1 to 6).
Three isoforms produced though alternative splicing of this gene
were initially identified; they differ in the number of Zn fingers
remaining from the five contained in the full-length TRAF3. The
TRAF3b isoform (∆25 aa) contains four Zn fingers with a C-terminal
finger formed by the fusion of the N-terminal half of the 3rd and
the C-terminal half of the 4th finger, the TRAF3c isoform (∆52 aa)
contains three Zn fingers with a C-terminal finger formed by the
fusion of the 2nd and the 4th finger, while the TRAF3d isoform (∆56
aa) contains three complete Zn fingers and the N-terminal portion
of the 5th finger. Additionally, the study detected three variant
5′-untranslated regions (UTRs) and two variant 3′ UTRs among the
isolated clones, although they did not establish whether they
corresponded to particular isoform-coding open reading frames
(29).
A further study identified five additional TRAF-3
protein isoforms with alterations in the Zn finger domains (∆27aa,
∆83aa, ∆103aa, ∆130aa and ∆221aa). TRAF3 splice-deletion variants,
including ∆25 aa, ∆52 aa and ∆56 aa, were found to induce NF-κB
activation in 293T cells, while full-length TRAF3 and TRAF3 ∆221
failed to do so. Unexpectedly, when full-length TRAF3 was
co-expressed with each of the 7 TRAF3 splice variants capable of
activating NF-κB alone, a 1.4-to 5-fold augmentation of their NF-κB
activation was observed (30). All
but the ∆130 aa were found to be expressed in four different
lymphoma cell lines (Jurkat D1.1, BJAB, Daudi and Raji).
Overexpression experiments of individual isoforms revealed that
only the ∆27 aa, ∆103 aa or ∆130 aa isoforms are able to induce
NF-κB activation in BJAB cells in contrast to full-length TRAF-3 or
∆221aa, which could not; notably, TRAF-3 ∆25 aa, ∆52 aa, ∆56 aa and
∆83 aa variants also failed to induce NF-κb activity, contrary to
their activity in 293T cells (31).
The difference in the TRAF-3 splice variants activation properties
in different cell lines suggests an association between TRAF-3
isoforms and the cellular environment. The two examples provide
evidence of the regulation of NF-κB activation in TRAF family genes
through alternative splicing: The TRAF2 and TRAF3 shorter splice
variants have this ability, while the full-length proteins lack
them.
Splicing potential
The demonstration by Rittore et al (16) that SNPs can have an effect on the
alternative splicing pattern of the TNFR1 gene may be the
cornerstone that joins two groups of association studies, namely,
transcript diversity and SNPs, and sheds light on their
association. The Rittore group identified that SNPs rs4149570,
rs767455 and rs1800692 have a combined effect on the TNFR1
transcript output through regulation of alternative splicing. When
considering that the aforementioned TNFR1 SNPs have already been
found to be associated with inflammation-related conditions,
including susceptibility to develop invasive pulmonary
aspergillosis (32), the prognosis of
peripheral T-cell non-Hodgkin lymphoma (33), radiation-induced toxicity following
treatment for non-small cell lung cancer (34), the risk of breast cancer (35), inflammatory demyelinating diseases
(36), the response to Crohn's
disease treatment (37) and adult
onset Still's disease (38), it is
plausible to consider whether it is the differential balance of
TNFR1 isoforms produced by alternative splicing that causes these
associations.
There is conclusive evidence that supports the fact
that SNPs can modify the outcome of alternative splicing by
disrupting canonical splice sites (39), or by modifying cis elements
(enhancers or silencers) along both exonic and intronic sequences,
as Pagani et al (40) found in
a number of synonymous mutations that cause exon 12 skipping of the
cystic fibrosis transmembrane conductance regulator. Curated
repositories that gather SNP association data, such as SNPedia
(41), pinpoint relevant SNPs worth
analyzing for their potential effect on alternative splicing. A
search in SNPedia reveals several disease-associated polymporphisms
in virtually all of the receptor-proximal elements of TNF signaling
(Table II).
| Table II.Number of disease-associated SNPs of
the main receptor-proximal elements of the TNF signaling
pathway. |
Table II.
Number of disease-associated SNPs of
the main receptor-proximal elements of the TNF signaling
pathway.
| Gene | SNPs with
association data available on SNPedia |
|---|
| TNFR1 | Rs2234649,
Rs4149570, Rs767455, Rs104895217, Rs104895218, Rs104895219,
Rs104895220, Rs104895221, Rs104895222 |
| TNFR2 | Rs3397, Rs1061624,
Rs652625 |
| TRADD | Rs9939768, Rs6979,
Rs9033, Rs868213 |
| FADD | Rs387906839 |
| TRAF1 | Rs2476601,
Rs3761847, Rs6457617, Rs7574865, Rs6920220, Rs10818488, Rs2416808,
Rs10499194, Rs660895, Rs2416804, Rs7026551, Rs1930780, Rs1953126,
Rs2900180, Rs2395148, Rs292001, Rs7021206 |
| TRAF2 | Rs7852970 |
| TRAF3 | Rs10133111,
Rs11160706, Rs1131877 |
Regarding human cancer, evidence points out that
specific functions of certain genes regulating the TNF signaling
pathway can be affected by alternative splicing, with an impact on
tumor phenotype. In this sense, the TNF-apoptosis inhibitor c-FLIP
(cellular FLICE inhibitory protein) has distinct alternative
splicing variants (c-FLIPL and c-FLIPs) that
have distinct roles in the TNFα-induced signaling cascade. Park
et al (42) demonstrated that
c-FLIP alternative variants activate either Erk or NF-κB through
the association with Raf and TRAF2, which contributes to
TNF-induced cell cycle promotion. For instance, c-FLIPL
showed stronger affinity to Raf in order to activate Erk and PI3K.
Meanwhile, c-FLIPs showed strong affinity to TRAF2 to
mediate activation of the TRAF-JNK pathway. Moreover, each splicing
variant is regulated differentially at transcriptional level, that
is, after TNF-α stimulation, a delayed c-FLIPL response
is induced, whereas c-FLIPs shows a rapid response to
the stimulation (42). Thus,
alternative splicing enables protein diversity, generating
structurally and functionally distinct proteins from the same gene.
Later, Haag et al (43)
reported the expression of these splicing variants in pancreatic
cancer tissue. It was observed that c-FLIP is underexpressed in
pancreatic intraepithelial neoplasm and pancreatic ductal
adenocarcinomas compared with normal pancreatic tissues. Moreover,
in pancreatic cancer cell line ULA-PaC, the downregulation of these
isoforms by RNA interference enhances apoptosis, indicating that
c-FLIP is an important regulator of death receptor-induced
apoptosis (43).
In another study, in neoplastic and more notably, in
non-neoplastic cells, two novel TNF-associated apoptosis-inducing
ligand (TRAIL) splice variants were reported: TRAIL-β, lacking exon
3, corresponding to loss of 98 aa, and TRAIL-γ, lacking exons 2 and
3, corresponding to loss of 52 aa. These two variants result in a
truncation of the extracellular domain, which is important for
trimeric stability and ligand-receptor binding; consequently, these
splice variants fail to trigger apoptosis signaling. The study
suggested that these novel TRAIL variants may have implications for
deepening our understanding of TRAIL-mediated apoptosis in
neoplastic and non-neoplastic human cells (44). Recently, Krieg et al (45) reported the first study that describes
the expression of TRAIL splice variants in 41 gastric carcinoma
tissue samples by reverse transcription-quantitative polymerase
chain reaction. Notably, all three TRAIL-splice variants could be
detected in non-malignant and malignant tissues, but only TRAIL-γ
had a prognostic value, since it was associated with a
significantly higher survival rate (45).
Concluding remarks
TNF-mediated inflammation, as with numerous other
signaling pathways, involves the concerted expression of a fairly
well known number of genes; however, the human transcriptome has
turned out to be far more complex than initially conceived, and the
contribution of alternative splicing to the transcriptomic
variability through the diversification of mature transcripts
produced from the same gene, is becoming clearer and more important
(3). A considerable amount of
research is currently being conducted into the search for
transcriptomic signatures associated with inflammation-related
diseases (among numerous other conditions), including
osteoarthritis (46), atherosclerotic
plaque progression (47), multiple
sclerosis, systemic lupus erythematosus, juvenile rheumatoid
arthritis, Crohn's disease, ulcerative colitis and type 1 diabetes
(48), to name a few. Incorporation
of alternative splicing data into this information can shed light
on how these transcriptomic signatures come to be, and on the
possible scenarios that they lead to (49).
The present study has reviewed the literature
showing that protein isoforms derived from alternative transcripts
of the genes involved in TNF signaling can have different, mainly
antagonistic, functions. We therefore find it reasonable to
hypothesize that, although TNF signaling ultimately leads to an
inflammatory response, the pathway itself can be subject to
regulation through alternative splicing of its elements,
potentially modifying its end result.
References
|
1
|
Sharp PA: Split genes and RNA splicing.
Cell. 77:805–815. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al:
Initial sequencing and analysis of the human genome. Nature.
409:860–921. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pan Q, Shai O, Lee LJ, Frey BJ and
Blencowe BJ: Deep surveying of alternative splicing complexity in
the human transcriptome by high-throughput sequencing. Nat Genet.
40:1413–1415. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Black DL: Mechanisms of alternative
pre-messenger RNA splicing. Annu Rev Biochem. 72:291–336. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stamm S, Ben-Ari S, Rafalska I, Tang Y,
Zhang Z, Toiber D, Thanaraj TA and Soreq H: Function of alternative
splicing. Gene. 344:1–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith CW and Valcárcel J: Alternative
pre-mRNA splicing: The logic of combinatorial control. Trends
Biochem Sci. 25:381–388. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matlin AJ, Clark F and Smith CW:
Understanding alternative splicing: Towards a cellular code. Nat
Rev Mol Cell Biol. 6:386–398. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kornblihtt AR, Schor IE, Alló M, Dujardin
G, Petrillo E and Muñoz MJ: Alternative splicing: A pivotal step
between eukaryotic transcription and translation. Nat Rev Mol Cell
Biol. 14:153–165. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cáceres JF and Kornblihtt AR: Alternative
splicing: Multiple control mechanisms and involvement in human
disease. Trends Genet. 18:186–193. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baralle D and Baralle M: Splicing in
action: Assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang ET, Sandberg R, Luo S, Khrebtukova I,
Zhang L, Mayr C, Kingsmore SF, Schroth GP and Burge CB: Alternative
isoform regulation in human tissue transcriptomes. Nature.
456:470–476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biology.
11:372–377. 2001. View Article : Google Scholar
|
|
13
|
Zelová H and Hošek J: TNF-α signalling and
inflammation: Interactions between old acquaintances. Inflamm Res.
62:641–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flicek P, Ahmed I, Amode MR, Barrell D,
Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S,
et al: Ensembl 2013. Nucleic Acids Res. 41:(Database Issue).
D48–D55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gregory AP, Dendrou CA, Attfield KE,
Haghikia A, Xifara DK, Butter F, Poschmann G, Kaur G, Lambert L,
Leach OA, et al: TNF receptor 1 genetic risk mirrors outcome of
anti-TNF therapy in multiple sclerosis. Nature. 488:508–511. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rittore C, Sanchez E, Soler S,
Barat-Houari M, Albers M, Obici L, McDermott MF, Touitou I and
Grandemange S: Identification of a new exon 2-skipped TNFR1
transcript: Regulation by three functional polymorphisms of the
TNFR-associated periodic syndrome (TRAPS) gene. Ann Rheum Dis.
73:290–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hull KM, Drewe E, Aksentijevich I, Singh
HK, Wong K, McDermott EM, Dean J, Powell RJ and Kastner DL: The TNF
receptor-associated periodic syndrome (TRAPS): Emerging concepts of
an autoinflammatory disorder. Medicine (Baltimore). 81:349–368.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Waschke KA, Villani AC, Vermeire S,
Dufresne L, Chen TC, Bitton A, Cohen A, Thomson AB and Wild GE:
Tumor necrosis factor receptor gene polymorphisms in crohn's
disease: Association with clinical phenotypes. Am J Gastroenterol.
100:1126–1133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seitz C, Muller P, Krieg RC, Mannel DN and
Hehlgans T: A novel p75TNF receptor isoform mediating NFkappa B
activation. J Biol Chem. 276:19390–19395. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scherübl C, Schneider-Brachert W, Schütze
S, Hehlgans T and Männel DN: Colocalization of endogenous TNF with
a functional intracellular splice form of human TNF receptor type
2. J Inflamm. 2:72005. View Article : Google Scholar
|
|
21
|
Lainez B, Fernandez-Real JM, Romero X,
Esplugues E, Cañete JD, Ricart W and Engel P: Identification and
characterization of a novel spliced variant that encodes human
soluble tumor necrosis factor receptor 2. Int Immunol. 16:169–177.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fernandez-Real JM, Straczkowski M, Lainez
B, Chacón MR, Kowalska I, López-Bermejo A, García-España A,
Nikolajuk A, Kinalska I and Ricart W: An alternative spliced
variant of circulating soluble tumor necrosis factor-alpha
receptor-2 is paradoxically associated with insulin action. Eur J
Endocrinol. 154:723–730. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Esteve E, Botas P, Delgado E,
López-Bermejo A, Lainez B, Engel P, Ricart W and Fernández-Real JM:
Soluble TNF-alpha receptor 2 produced by alternative splicing is
paradoxically associated with markers of liver injury. Clin
Immunol. 123:89–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cañete JD, Albaladejo C, Hernández MV,
Laínez B, Pinto JA, Ramírez J, López-Armada MJ, Rodríguez-Cros JR,
Engel P, Blanco FJ and Sanmartí R: Clinical significance of high
levels of soluble tumour necrosis factor-α receptor-2 produced by
alternative splicing in rheumatoid arthritis: A longitudinal
prospective cohort study. Rheumatology (Oxford). 50:721–728. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Hu W, Feng S, Ma J and Wu M: RIP3
beta and RIP3 gamma, two novel splice variants of
receptor-interacting protein 3 (RIP3), downregulate RIP3-induced
apoptosis. Biochem Biophys Res Commun. 332:181–187. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishizawa YH, Tamura K, Yamaguchi T,
Matsumoto K, Komiyama M, Takamatsu N, Shiba T and Ito M: Xenopus
death-domain-containing proteins FADD and RIP1 synergistically
activate JNK and NF-kappaB. Biol Cell. 98:465–478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brink R and Lodish HF: Tumor necrosis
factor receptor (TNFR)-associated factor 2A (TRAF2A), a TRAF2
splice variant with an extended RING finger domain that inhibits
TNFR2-mediated NF-kappaB activation. J Biol Chem. 273:4129–4134.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grech A, Quinn R, Srinivasan D, Badoux X
and Brink R: Complete structural characterisation of the mammalian
and Drosophila TRAF genes: Implications for TRAF evolution and the
role of RING finger splice variants. Mol Immunol. 37:721–734. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van Eyndhoven WG, Frank D, Kalachikov S,
Cleary AM, Hong DI, Cho E, Nasr S, Perez AJ, Mackus WJ, Cayanis E,
et al: A single gene for human TRAF-3 at chromosome 14q32.3 encodes
a variety of mRNA species by alternative polyadenylation, mRNA
splicing and transcription initiation. Mol Immunol. 35:1189–1206.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Eyndhoven WG, Gamper CJ, Cho E, Mackus
WJ and Lederman S: TRAF-3 mRNA splice-deletion variants encode
isoforms that induce NF-kappaB activation. Mol Immunol. 36:647–658.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gamper C, Omene CO, Van Eyndhoven WG,
Glassman GD and Lederman S: Expression and function of TRAF-3
splice-variant isoforms in human lymphoma cell lines. Hum Immunol.
62:1167–1177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sainz J, Salas-Alvadado I, López-Fernández
E, Olmedo C, Comino A, García F, Blanco A, Gómez-Lopera S, Oyonarte
S, Bueno P and Jurado M: TNFR1 mRNA expression level and TNFR1 gene
polymorphisms are predictive markers for susceptibility to develop
invasive pulmonary aspergillosis. Int J Immunopathol Pharmacol.
23:423–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heemann C, Kreuz M, Stoller I, Schoof N,
von Bonin F, Ziepert M, Löffler M, Jung W, Pfreundschuh M, Trümper
L and Kube D: Circulating levels of TNF receptor II are prognostic
for patients with peripheral T-cell Non-Hodgkin lymphoma. Clin
Cancer Res. 18:3637–3647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hildebrandt MA, Komaki R, Liao Z, Gu J,
Chang JY, Ye Y, Lu C, Stewart DJ, Minna JD, Roth JA, et al: Genetic
variants in inflammation-related genes are associated with
radiation-induced toxicity following treatment for non-small cell
lung cancer. PLoS One. 5:e124022010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Madeleine MM, Johnson L, Malkki M, Resler
AJ, Petersdorf EW, McKnight B and Malone KE: Genetic variation in
proinflammatory cytokines IL6, IL6R, TNF-region, and TNFRSF1A and
risk of breast cancer. Breast Cancer Res Treat. 129:887–899. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park TJ, Kim HJ, Kim JH, Bae JS, Cheong
HS, Park BL and Shin HD: Associations of CD6, TNFRSF1A and IRF8
polymorphisms with risk of inflammatory demyelinating diseases.
Neuropathol Appl Neurobiol. 39:519–530. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsukara H, Ikeda S, Yoshimura N, Takazoe
M and Muramatsu M: Genetic polymorphisms of tumour necrosis factor
receptor superfamily 1A and 1B affect responses to infliximab in
Japanese patients with Crohn's disease. Aliment Pharmacol Ther.
27:765–770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cosan F, Emrence Z, Erbag G, Azakli H,
Yilmazer B, Yazici A, Ekmekci SS, Abaci N, Ustek D and Cefle A: The
association of TNFRSF1A gene and MEFV gene mutations with adult
onset Still's disease. Rheumatol Int. 33:1675–1680. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shimada MK, Hayakawa Y, Takeda J, Gojobori
T and Imanishi T: A comprehensive survey of human polymorphisms at
conserved splice dinucleotides and its evolutionary relationship
with alternative splicing. BMC Evol Biol. 10:1222010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pagani F, Raponi M and Baralle FE:
Synonymous mutations in CFTR exon 12 affect splicing and are not
neutral in evolution. Proc Natl Acad Sci USA. 102:6368–6372. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cariaso M and Lennon G: SNPedia: A wiki
supporting personal genome annotation, interpretation and analysis.
Nucleic Acids Res. 40:(Database Issue). D1308–D1312. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park SJ, Kim YY, Ju JW, Han BG, Park SI
and Park BJ: Alternative splicing variants of c-FLIP transduce the
differential signal through the Raf or TRAF2 in TNF-induced cell
proliferation. Biochem Biophys Res Commun. 289:1205–1210. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haag C, Stadel D, Zhou S, Bachem MG,
Möller P, Debatin KM and Fulda S: Identification of c-FLIP(L) and
c-FLIP(S) as critical regulators of death receptor-induced
apoptosis in pancreatic cancer cells. Gut. 60:225–237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Krieg A, Krieg T, Wenzel M, Schmitt M,
Ramp U, Fang B, Gabbert HE, Gerharz CD and Mahotka C: TRAIL-beta
and TRAIL-gamma: Two novel splice variants of the human TNF-related
apoptosis-inducing ligand (TRAIL) without apoptotic potential. Br J
Cancer. 88:918–927. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Krieg A, Mersch S, Wolf N, Stoecklein NH,
Verde PE, Esch Am JS II, Heikaus S, Gabbert HE, Knoefel WT and
Mahotka C: Expression of TRAIL-splice variants in gastric
carcinomas: Identification of TRAIL-γ as a prognostic marker. BMC
Cancer. 13:3842013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ritter SY, Subbaiah R, Bebek G, Crish J,
Scanzello CR, Krastins B, Sarracino D, Lopez MF, Crow MK, Aigner T,
et al: Proteomic analysis of synovial fluid from the osteoarthritic
knee: Comparison with transcriptome analyses of joint tissues.
Arthritis Rheum. 65:981–992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nührenberg T, Langwieser N, Binder H, Kurz
T, Stratz C, Kienzle RP, Trenk D, Zohlnhöfer-Momm D and Neumann FJ:
Transcriptome analysis in patients with progressive coronary artery
disease: Identification of differential gene expression in
peripheral blood. J Cardiovasc Transl Res. 6:81–93. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tuller T, Atar S, Ruppin E, Gurevich M and
Achiron A: Common and specific signatures of gene expression and
protein-protein interactions in autoimmune diseases. Genes Immun.
14:67–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Frankish A, Mudge JM, Thomas M and Harrow
J: The importance of identifying alternative splicing in vertebrate
genome annotation. Database (Oxford). 2012:bas0142012. View Article : Google Scholar : PubMed/NCBI
|