Introduction
Pancreatic ductal adenocarcinoma (PDAC; Online
Mendelian Inheritance in Man no. 260350) ranks fourth in the
leading causes of cancer-associated mortality in Western countries
(1). Despite diagnostic and
therapeutic advances, the prognosis of PDAC remains poor. Only 20%
of patients present with potentially resectable disease at the time
of diagnosis, while due to the high propensity for tumor
recurrence, the 5-year overall survival (OS) rate in patients
undergoing surgery with radical intent is usually <25%. Although
a number of prospective clinical trials have demonstrated that
adjuvant systemic therapy improves the patient outcome following
surgery, adjuvant chemotherapy appears to be effective only in a
minority of patients, and the majority of the patients ultimately
succumb to the disease. The prognosis of metastatic patients is
extremely poor, with a median OS time of <1 year (2). Consequently, novel regimens of adjuvant
treatment are being investigated and there is currently no
definitive standard of adjuvant therapy.
In PDAC, mutations in the V-Ki-ras2 Κirsten rat
sarcoma viral oncogene homolog (KRAS) gene occur in 75–90%
of cases, representing the most frequent, as well as the earliest,
genetic alteration. KRAS mutations, specifically in codons
12 and 13, lead to constitutive activation of downstream signaling
pathways that are important for tumor initiation, development and
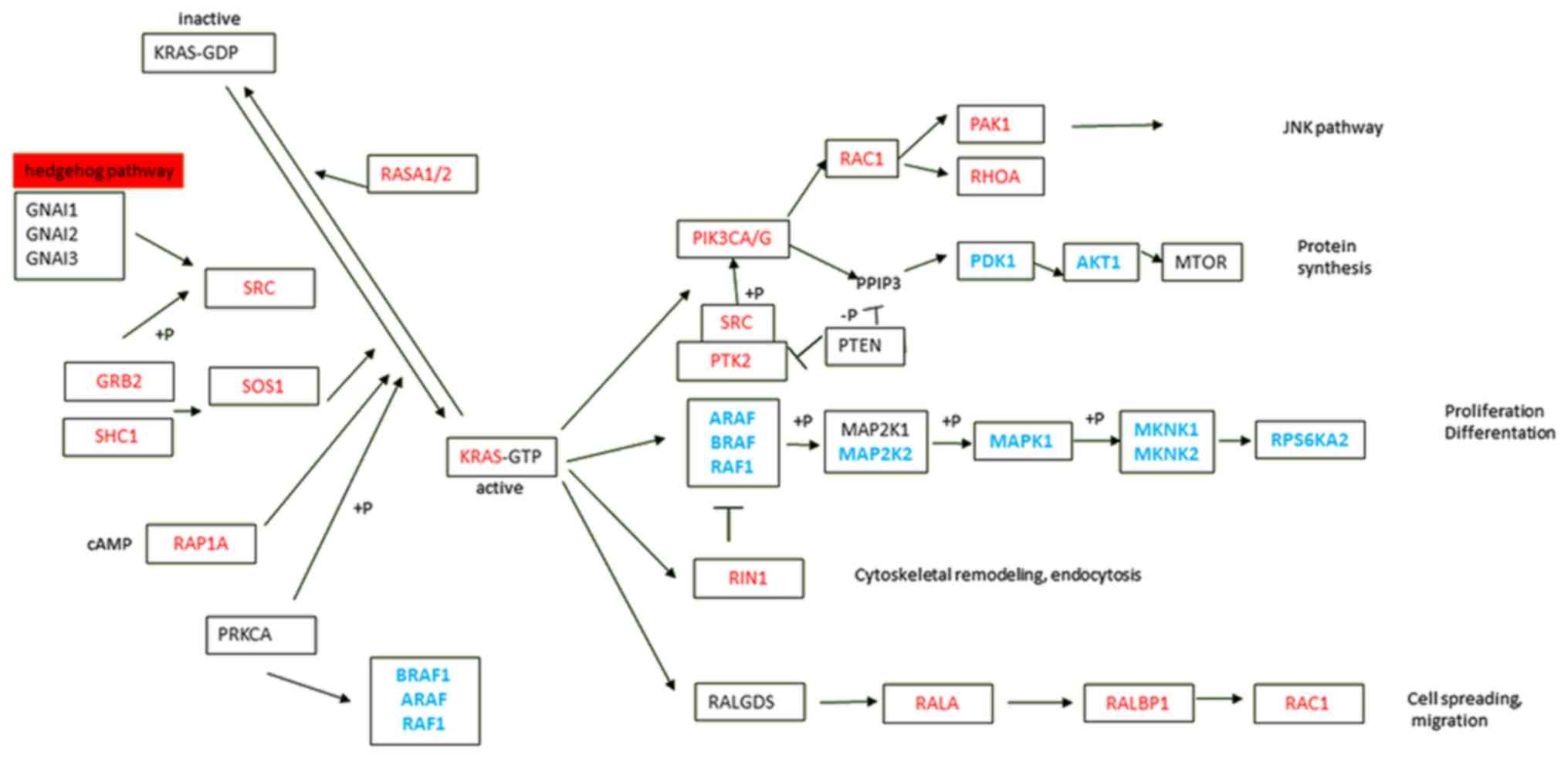
spread (3,4). KRAS signaling is highly complex and
dynamic, with various downstream effector pathways interconnected
at different levels by cross-signaling and feedback loops (5). The four major KRAS downstream pathways
reported in PDAC are RAF proto-oncogene serine/threonine-protein
kinase (RAF)/mitogen-activated protein kinase (MAPK),
phosphoinositide 3-kinase (PI3K)/3-phosphoinositide-dependent
protein kinase 1 (PDK1)/ABL proto-oncogene-1, non-receptor
tyrosine kinase (ABL), RAL guanine nucleotide exchange factor, and
Ras and Rab interactor 1 (RIN1)/ABL pathways (Fig. 1) (6–10). This
multiplicity of downstream pathways may partly explain the failure
of existing efforts to target epidermal growth factor receptor,
KRAS or serine/threonine-protein kinase B-raf (BRAF) using specific
inhibitors, underlining the complexity of genetic changes and the
resistance of the cancer cells.
The aim of the present study was to assess the
association between gene expression from the four major
KRAS-effective pathways in PDAC and the clinical features of the
patients, and to evaluate the potential predictive and prognostic
significance.
Materials and methods
Patients
A cohort of 45 consecutive patients with PDAC who
underwent surgery with curative intent was recruited from two
oncology centers in the Czech Republic (Institute of Clinical and
Experimental Medicine, Prague; and University Hospital, Masaryk
University, Brno, Czech Republic) between August 2008 and January
2012. Inclusion criteria were: i) Adult operable patients with
suspected pancreatic carcinoma based on clinical imaging methods;
ii) patients who provided informed consent; and iii) pancreatic
carcinoma diagnosis was verified by collaborating pathologist. None
of the patients had received prior chemotherapy. Characteristics of
the patient cohort are summarized in Table I. The tissue specimen collection and
processing, and the data retrieval were as described previously
(11).
 | Table I.Characteristics of the patient
cohort. |
Table I.
Characteristics of the patient
cohort.
| Variables | 20 |
|---|
| Mean age (range),
years | 63.9 (46–80) |
| Sex, n (%) |
|
|
Male | 20 (44.4) |
|
Female | 25 (55.6) |
| Histological grade,
n (%) |
|
| G1+G2
(well to moderate) | 30 (66.7) |
| G3+G4
(poor) | 15 (33.3) |
| Primary tumor
extent of invasion, n (%) |
|
| pT1
tumor limited to the pancreas ≤2 cm | 1 (2.2) |
| pT2
tumor limited to the pancreas >2 cm | 5 (11.1) |
| pT3
tumor extending beyond the pancreas | 39 (86.7) |
| Regional lymph
nodes, n (%) |
|
|
pN0 | 17 (37.8) |
|
pN1 | 28 (62.2) |
| KRAS mutations in
codons 12 and 13a, n
(%) |
|
|
Wild-type (GGTGGC) | 9 (20.0) |
| G12V
(GTTGGC) | 10 (22.2) |
| G12D
(GATGGC) | 15 (33.3) |
| G12R
(CGTGGC) | 6 (13.3) |
| Other
(G13D, Q61R, Q61H) | 3 (6.7) |
| Not
assessed | 2 (4.4) |
| BRAF
mutationsa, n (%) |
|
|
Wild-type (GTG) | 43 (95.6) |
| V600E
(GAG) | 0 (0.0) |
| Not
assessed | 2 (4.4) |
| Patient status at
the data cut off, n (%) |
|
|
Deceased | 37 (82.2) |
|
Alive | 8 (17.8) |
All patients signed an informed consent form, in
accordance with the requirements for ethical approval, which was
provided by the Institutional Review Boards of the Institute of
Clinical and Experimental Medicine and University Hospital, Masaryk
University, Brno.
Isolation of nucleic acids and cDNA
synthesis
Tissue samples were homogenized and total RNA and
DNA was isolated as previously described (12,13). cDNA
was synthesized using 0.5 µg total RNA and characterized as
previously described (14). cDNA was
then pre-amplified by TaqMan® PreAmp Master mix to
enrich the specific targets for gene expression analysis using
TaqMan Gene Expression assays (Life Technologies; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) (Table II). The cDNA pre-amplification was
performed with 5 µl cDNA using 14 pre-amplification cycles (10 min
at 95°C and 14 cycles of 15 sec at 95°C), and the pre-amplification
uniformity of cDNA was checked according to the procedure
recommended by the manufacturer (Thermo Fisher Scientific,
Inc.).
| Table II.List of TaqMan gene expression assays
used in the study. |
Table II.
List of TaqMan gene expression assays
used in the study.
| Gene
abbreviation | Gene name | Assay ID |
|---|
| AKT1 | V-akt murine
thymoma viral oncogene homolog 1 | Hs00178289_m1 |
| AKT2 | V-akt murine
thymoma viral oncogene homolog 2 | Hs01086102_m1 |
| ARAF | V-raf murine
sarcoma viral oncogene homolog 1 | Hs00176427_m1 |
| BRAF | V-raf murine
sarcoma viral oncogene homolog Β1 | Hs00269944_m1 |
| GRB2 | Growth factor
receptor-bound protein 2 | Hs00257910_s1 |
| GSK3B | Glycogen synthase
kinase 3-β | Hs00275656_m1 |
| KRAS | V-ki-ras2 Κirsten
rat sarcoma viral oncogene homolog | Hs00364284_g1 |
| MAP2K1 | Mitogen-activated
protein kinase kinase 1 | Hs00983247_g1 |
| MAP2K2 | Mitogen-activated
protein kinase kinase 2 | Hs04194606_gH |
| MAP2K7 | Mitogen-activated
protein kinase kinase 7 | Hs00178198_m1 |
| MAP3K1 | Mitogen-activated
protein kinase kinase kinase 1 | Hs00394890_m1 |
| MAP3K2 | Mitogen-activated
protein kinase kinase kinase 2 | Hs01109981_m1 |
| MAP3K4 | Mitogen-activated
protein kinase kinase kinase 4 | Hs00245958_m1 |
| MAP3K7 | Mitogen-activated
protein kinase kinase kinase 7 | Hs01105682_m1 |
| MAPK1 | Mitogen-activated
protein kinase 1 | Hs01046830_m1 |
| MAPK10 | Mitogen-activated
protein kinase 10 | Hs00373455_m1 |
| MAPK14 | Mitogen-activated
protein kinase 14 | Hs01051152_m1 |
| MAPK3 | Mitogen-activated
protein kinase 3 | Hs00946872_m1 |
| MAPK7 | Mitogen-activated
protein kinase 7 | Hs00611114_g1 |
| MAPK8 | Mitogen-activated
protein kinase 8 | Hs00177083_m1 |
| MAPK9 | Mitogen-activated
protein kinase 9 | Hs00177102_m1 |
| MKNK1 | Mitogen-activated
protein kinase-interacting serine/threonine kinase 1 | Hs00374376_m1 |
| MKNK2 | Mitogen-activated
protein kinase-interacting serine/threonine kinase 2 | Hs01046586_g1 |
| MTOR | Mechanistic target
of rapamycin | Hs00234508_m1 |
| PAK1 | p21
protein-activated kinase 1 | Hs00176815_m1 |
| PDPK1 |
3-phosphoinositide-dependent protein
kinase 1 | Hs00176884_m1 |
| PIK3CA |
Phosphatidylinositol 3-kinase, catalytic,
α | Hs00907966_m1 |
| PIK3CG |
Phosphatidylinositol 3-kinase, catalytic,
γ | Hs00277090_m1 |
| PLK3 | Polo-like kinase
3 | Hs00177725_m1 |
| PRKACA | Protein kinase,
camp-dependent, catalytic, α | Hs00427274_m1 |
| PRKCA | Protein kinase c,
α | Hs00925195_m1 |
| PTEN | Phosphatase and
tensin homolog | Hs02621230_s1 |
| PTK2 | Protein-tyrosine
kinase, cytoplasmic | Hs01056457_m1 |
| PTK2B | Protein-tyrosine
kinase 2, β | Hs01559708_m1 |
| RAC1 | Ras-related C3
botulinum toxin substrate 1 | Hs01025984_m1 |
| RAF1 | V-raf-1 murine
leukemia viral oncogene homolog 1 | Hs00234119_m1 |
| RALA | V-ral simian
leukemia viral oncogene homolog A | Hs01564991_g1 |
| RALBP1 | RalA-binding
protein 1 | Hs01034988_g1 |
| RALGDS | Ral guanine
nucleotide dissociation stimulator | Hs00325141_m1 |
| RAP1A | Ras-related protein
1A | Hs01092205_g1 |
| RASA1 | Ras p21 protein
activator 1 | Hs00963555_m1 |
| RASA2 | Ras p21 protein
activator 2 | Hs01003325_m1 |
| RHOA | Ras homolog gene
family, member A | Hs00357608_m1 |
| RIN1 | Ras and rab
interactor 1 | Hs00182870_m1 |
| RPS6KA2 | Ribosomal protein
S6 kinase, 90-kd, 2 | Hs00179731_m1 |
| RPS6KA4 | Ribosomal protein
S6 kinase, 90-kd, 4 | Hs00177670_m1 |
| RPS6KA5 | Ribosomal protein
S6 kinase, 90-kd, 5 | Hs01046594_m1 |
| SHC1 | SHC transforming
protein | Hs01050699_g1 |
| SOS1 | Son of sevenless,
Drosophila, homolog 1 | Hs00362316_m1 |
| SOS2 | Son of sevenless,
Drosophila, homolog 2 | Hs00412876_g1 |
| SRC | V-src avian sarcoma
(Schmidt-Ruppin A-2) viral oncogene | Hs01082238_g1 |
| STAT3 | Signal transducer
and activator of transcription 3 | Hs01047580_m1 |
|
ELF1a | E74-like factor
1 | Hs00152844_m1 |
|
EIF2B1a | Eukaryotic
translation initiation factor 2B, subunit 1 | Hs00426752_m1 |
|
MRPL19a | Mitochondrial
ribosomal protein l19 | Hs00608519_m1 |
| POP4 a | Processing of
precursor 4, S. cerevisiae, homolog of | Hs00198357_ml |
Quantitative polymerase chain reaction
(qPCR)
qPCR was performed using the ViiA7 Real-Time PCR
system using TaqMan Gene Expression assays (Table I), with optimized primer and probe
sets and TaqMan Gene Expression Master mix (Thermo Fisher
Scientific, Inc.). Processing of precursor 4, S. cerevisiae,
homolog of, mitochondrial ribosomal protein L19, E74-like factor 1
and eukaryotic translation initiation factor 2B subunit 1 were used
as reference genes for studies of gene expression in human
pancreatic carcinoma based on our previously published data
(15). Determination of transcript
levels was performed exactly as previously described (10) and the qPCR study adhered to the
Minimum Information for Publication of Quantitative Real-Time PCR
Experiments Guidelines (16).
KRAS and BRAF mutation status
KRAS and BRAF gene mutation analysis
was performed using the
KRAS/BRAF/phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit α (PIK3CA) (KBP) Array
(EV3799A/B; Randox Laboratories Ltd., Crumlin, Northern Ireland)
according to the manufacturer's instructions. The assay is based on
a combination of multiplex PCR and biochip array hybridization for
high discrimination between multiple wild-type and mutant DNA
regions in the KRAS (mutations in codons 12, 13 and 61),
BRAF (V600E mutation) and PIK3CA (mutations in codons
542, 545 and 1,047) genes. Providing there are enough copies of DNA
present, ~1% of mutants can readily be detected in a background of
wild-type genomic DNA. A unique primer set is designed for each
mutation target (and control), which will hybridize to a
complementary discrete test region (DTR) on the biochip array. Each
DTR corresponds to a particular mutation target. One of the
target-specific primers in each pair contains a biotin label, which
on addition of streptavidin-horseradish peroxidase conjugate
permits chemiluminescence detection of hybridized products on the
biochip array. Dedicated software processes produced automatic
results.
Statistical analysis
Differences in gene expression profiles between
tumor and paired non-neoplastic control tissues and between
wild-type and KRAS-mutated samples were evaluated using the
RT2 Profiler PCR Assay Data Analysis v3.5 program
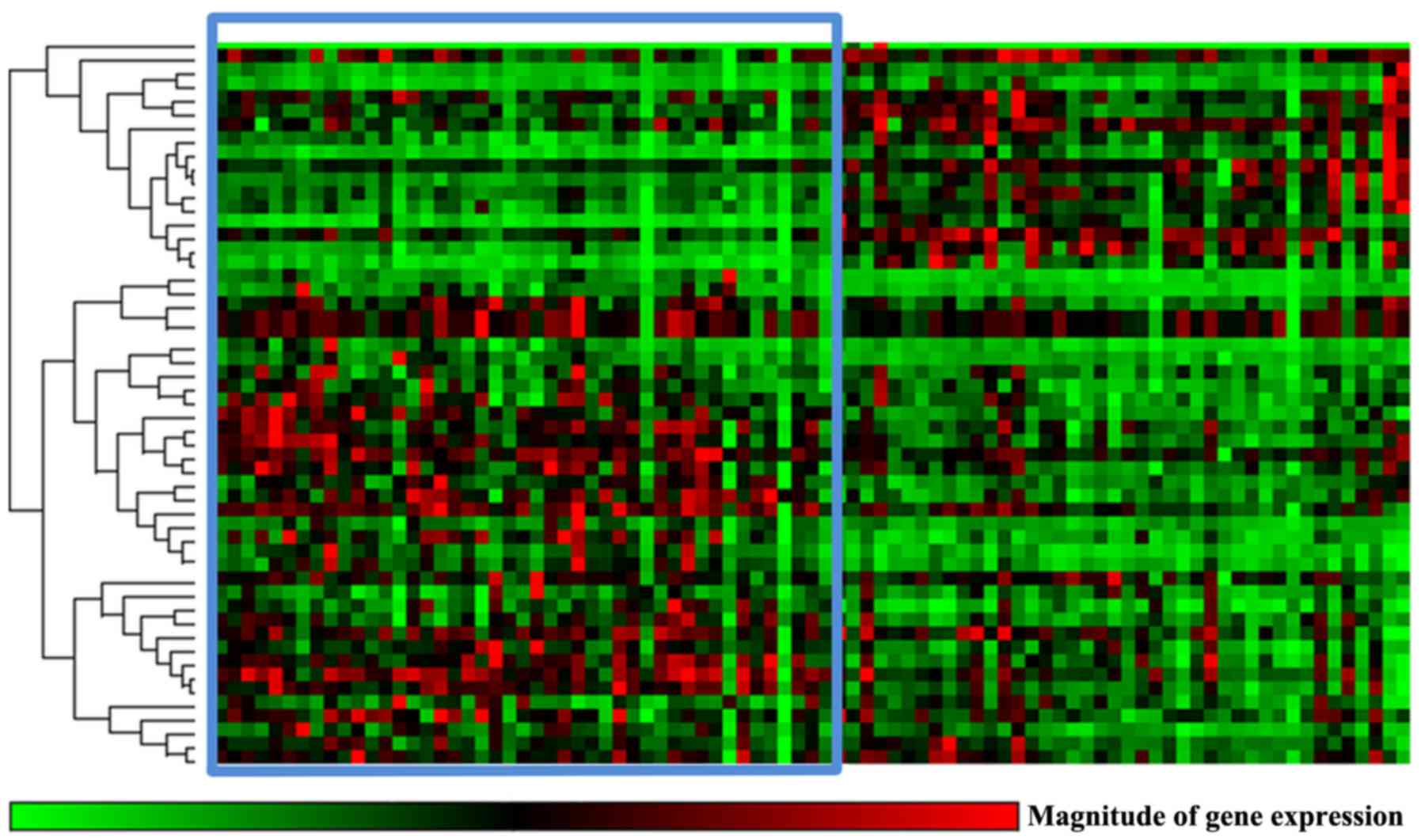
(Qiagen GmbH, Hilden, Germany). This gene expression analysis suite
performs fold-change calculations from raw quantification cycle
values for reference and target genes based on the ΔΔCq method
described by Livak and Schmittgen (17), and enables hierarchical clustering of
gene expression profiles between the compared groups of patients
and data. Differences in intratumoral gene expression levels
between patients stratified by clinical data were evaluated by the
Kruskal-Wallis test.
OS was defined as the time between the date of
surgery and all-cause mortality. Surviving patients were censored
at the last follow-up in December 2015. Patients were divided into
two groups by the median intratumoral gene expression levels of
individual genes and the survival functions were computed by the
Kaplan-Meier method, with statistical significance evaluated by the
Breslow test using SPSS v16.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically
significant difference. All P-values are departures from two-sided
tests. The correction for multiple testing was applied according to
the Bonferroni and the false discovery rate (FDR) methods.
Results
Study population
The study was performed on 45 patients with resected
(R0 resection in >90% of cases) PDAC who had not received any
prior neoadjuvant therapy. Overall, 80% (36/45) of patients
harbored KRAS mutations in the DNA of the tumor tissue,
while BRAF mutations were not found in any sample (Table II). The majority of patients (76%;
n=34) received adjuvant chemotherapy consisting of nucleoside
analogs (gemcitabine and/or 5-fluorouracil).
The median OS time was 23.7 months, with 18% of
patients (n=8) alive at the time of data cut off (December
2015).
Transcript levels of KRAS signaling
pathways genes in PDAC
Considering the pivotal role of KRAS oncogene in the
integration and transduction of mitogenic and metabolic signals,
the transcript levels of 52 genes covering four major pathways
downstream of KRAS were measured (Table I). The KRAS pathway was significantly
dysregulated in tumors compared with that in adjacent non-malignant
pancreatic tissues (Fig. 2; Table III). Significant overexpression of
genes of the PI3K/PDK1/AKT, RAL guanine nucleotide
exchange factor, and RIN1/ABL [phosphatidylinositol
3-kinase, catalytic, α/γ (PIK3CA/G), p21 protein-activated
kinase 1, V-ral simian leukemia viral oncogene homolog A,
RalA-binding protein 1, Ras-related C3 botulinum toxin substrate 1,
RIN1, protein-tyrosine kinase, cytoplasmic, and V-src avian
sarcoma (Schmidt-Ruppin A-2) viral oncogene] pathways were
observed, leading to cytoskeletal remodeling, endocytosis, cell
spreading and migration (Table III;
Fig. 1). By contrast, genes of the
RAF/MAPK pathway exhibited significantly lower expression in
tumors compared with that in the paired adjacent non-malignant
pancreatic tissues (particularly in genes ARAF, BRAF,
V-RAF-1 murine leukemia viral oncogene homolog 1
(RAF1), mitogen-activated protein kinase kinase,
mitogen-activated protein kinase 1, mitogen-activated protein
kinase-interacting serine/threonine kinase 1/2 (MKNK1/2) and
ribosomal protein S6 kinase, 90-kd, 2. All these results remained
significant after FDR adjustment for multiple testing and the
majority remained significant after Bonferroni correction (Table III; Fig.
1).
| Table III.Dysregulation of KRAS pathway genes
in pancreatic ductal adenocarcinoma tumors in comparison to paired
adjacent non-malignant tissues. |
Table III.
Dysregulation of KRAS pathway genes
in pancreatic ductal adenocarcinoma tumors in comparison to paired
adjacent non-malignant tissues.
| Gene |
Fold-changea (tumor vs. non-malignant
tissue) | 95% confidence
intervala |
P-valuea |
|---|
| AKT1 | 0.73 | (0.63–0.83) | <0.001b |
| ARAF | 0.72 | (0.63–0.81) |
<0.001b |
| BRAF | 0.84 | (0.74–0.93) | 0.001 |
|
GRB2c | 1.37 | (1.04–1.69) |
<0.001b |
|
KRASc | 2.04 | (1.67–2.41) |
<0.001b |
| MAP2K2 | 0.64 | (0.46–0.82) | <0.001b |
| MAP2K7 | 0.52 | (0.39–0.65) |
<0.001b |
| MAP3K1 | 0.85 | (0.75–0.95) | 0.010 |
|
MAP3K2c | 1.24 | (1.13–1.36) |
<0.001b |
|
MAP3K7c | 1.28 | (1.14–1.41) |
<0.001b |
| MAPK1 | 0.77 | (0.59–0.94) | <0.001b |
|
MAPK14c | 1.27 | (1.14–1.40) | <0.001b |
|
MAPK3c | 1.71 | (1.26–2.15) | <0.001b |
|
MAPK7c | 1.20 | (1.01–1.38) | 0.006 |
| MAPK8 | 0.81 | (0.74–0.88) | <0.001b |
| MAPK9 | 0.47 | (0.38–0.55) |
<0.001b |
| MKNK1 | 0.31 | (0.25–0.38) | <0.001b |
| MKNK2 | 0.35 | (0.26–0.44) |
<0.001b |
|
PAK1c | 1.27 | (1.08–1.45) | 0.001 |
| PDPK1 | 0.73 | (0.64–0.81) |
<0.001b |
|
PIK3CAc | 1.46 | (1.25–1.68) |
<0.001b |
|
PIK3CGc | 2.22 | (1.61–2.82) |
<0.001b |
|
PLK3c | 1.56 | (1.23–1.88) |
<0.001b |
|
PTENc | 1.29 | (1.05–1.53) | 0.006 |
|
PTK2Bc | 1.68 | (1.44–1.91) | <0.001b |
|
RAC1c | 1.65 | (1.34–1.96) | <0.001b |
| RAF1 | 0.62 | (0.54–0.69) |
<0.001b |
|
RALAc | 1.43 | (1.27–1.59) |
<0.001b |
|
RALBP1c | 1.60 | (1.39–1.81) |
<0.001b |
|
RAP1Ac | 1.18 | (1.07–1.29) |
<0.001b |
|
RASA1c | 1.28 | (1.12–1.43) |
<0.001b |
|
RASA2c | 1.87 | (1.51–2.23) |
<0.001b |
|
RHOAc | 1.23 | (1.13–1.34) |
<0.001b |
|
RIN1c | 1.39 | (1.10–1.67) | 0.002 |
| RPS6KA2 | 0.65 | (0.49–0.81) | 0.001b |
|
RPS6KA4c | 1.76 | (1.45–2.08) |
<0.001b |
|
SHC1c | 1.24 | (1.09–1.38) | 0.001b |
|
SOS1c | 1.32 | (1.14–1.50) |
<0.001b |
| SOS2 | 0.68 | (0.59–0.77) | <0.001b |
|
SRCc | 1.43 | (1.16–1.71) | <0.001b |
However, no association between KRAS
downstream signaling pathway gene expression and tumor
characteristics, including tumor size, grade, angioinvasion, lymph
node metastasis or perineural invasion, passed the significance
threshold of the Bonferroni test.
Impact of KRAS mutation status on
transcript levels of target genes
From the 80% of tumor samples with KRAS
mutations, the most common mutation, KRASG12D,
was present in 33% (n=15) of the tumors. Only 1 tumor was found
with a mutation in codon 13, and 2 cases with a mutation in codon
61 (Table II).
Patients divided by the KRAS mutation status
significantly differed in terms of the gene expression of 5 of the
52 analyzed genes [BRAF, mitogen-activated protein kinase
kinase kinase 4, mitogen-activated protein kinase 8, MKNK1
and son of sevenless, Drosophila, homolog 2 (SOS2;
P<0.05; Table IV)], but none of
these associations passed the threshold for the multiple testing
correction. The expression profiles of the KRAS signaling pathway
as a whole also did not significantly differ between KRAS
wild-type and KRAS-mutated tumors (Fig. 3).
| Table IV.Downregulation of KRAS pathway genes
in PDAC KRAS-mutated tumors compared with cases with
wild-type KRAS. |
Table IV.
Downregulation of KRAS pathway genes
in PDAC KRAS-mutated tumors compared with cases with
wild-type KRAS.
| Gene |
Fold-changea (tumor vs. non-tumor) | 95% confidence
intervala |
P-valuea |
|---|
| BRAF | 0.84 | (0.72–0.95) | 0.021 |
| MAP3K4 | 0.79 | (0.67–0.91) | 0.035 |
| MAPK8 | 0.84 | (0.71–0.97) | 0.027 |
| MKNK1 | 0.72 | (0.45–0.99) | 0.033 |
| SOS2 | 0.77 | (0.59–0.94) | 0.003 |
KRAS mutation status had no significant
effect on the OS time of the PDAC patients. KRAS wild-type
patients experienced a median OS time of 22.3 months, and patients
with KRAS mutation experienced a median OS time of 21.0
months (P=0.182).
There was also no association between KRAS
mRNA transcript levels and OS time. In contrast to the rest of
pathway, RAF1 showed a significant association with the OS
time of the PDAC patients. Patients with RAF1 expression
levels lower than the median experienced longer OS times than
patients with higher RAF1 expression levels (P=0.030)
(Fig. 4). However, this association
did not pass Bonferroni correction for multiple testing.
Discussion
Mutation analysis of the present cohort of patients
with operable PDAC aligns with that of prior studies reporting the
presence of KRAS mutation in the majority of PDAC cases
(18,19). Additionally, the genes of four KRAS
downstream signaling pathways, including the PI3K/PDK1/AKT,
RAL guanine nucleotide exchange factor, RIN1/ABL and
RAF/MAPK pathways, exhibited differential expression in PDAC
compared with that of the adjacent normal tissues, although no
significant differences were observed in the expression of these
genes between patients with KRAS-mutated and wild-type
tumors. The expression profiles of KRAS downstream signaling
pathways were not associated with pathological characteristics that
reflect tumor biology, including angioinvasion, perineural
invasion, grade or presence of lymph node metastasis.
Similar to earlier studies (20–22), the
present data indicated that in this cohort of patients (with
early-stage disease and following radical surgery) the presence of
a KRAS mutation had no effect on the OS time of the
patients, although there was limited power to determine
associations indicating more minor effects due to the limited size
of the patient cohort. Moreover, with the exception of RAF1, no
impact was observed of the expression profile of the KRAS
downstream major effective signaling pathways on OS. These findings
may explain why all previous efforts targeting KRAS failed to
improve the patient outcome.
Despite sustained efforts in preclinical and
clinical research, PDAC remains a malignancy with an almost
uniformly fatal prognosis (23). In
contrast to other solid tumors, there has been no major progress in
the systemic therapy of PDAC during the last decade. In particular,
there is currently no targeted agent with clinically significant
activity against this tumor.
Although molecular biomarkers play a crucial role in
the management of numerous solid tumors (24), there are currently no useful
biomarkers for treatment selection in PDAC. In recent years, a
number of negative trials of targeted therapy have been conducted
in PDAC (25,26). Consequently, there is an urgent
requirement to improve the understanding of PDAC pathogenesis and
biology in order to identify novel therapeutic approaches and to
define subgroups of patients for tailored therapies. It has been
demonstrated that KRAS mutations represent the driver
mutations in the majority of PDAC cases. KRAS-targeted agents can
be classified into several categories according to the mechanism of
action, namely small-molecule RAS-binding ligands, inhibitors of
KRAS membrane anchorage, inhibitors that bind to RAS-binding
domains of RAS-effector proteins and inhibitors of KRAS expression
(27). However, attempts to
therapeutically target KRAS or the downstream pathways have all
thus far failed in clinical trials (28–32).
In conclusion, as expected, KRAS was mutated
in the majority of PDAC cases. The genes of the KRAS downstream
signaling pathways, including the PI3K/PDK1/AKT, RAL
guanine nucleotide exchange factor, RIN1/ABL and
RAF/MAPK pathways, were differentially expressed in PDAC
compared with those in adjacent non-neoplastic tissues. However,
neither the presence of KRAS mutation nor the extent of KRAS
signaling dysregulation was associated with OS time. Among the KRAS
downstream signaling pathway genes investigated, only RAF1
expression was predictive of outcome. It is possible that the
analysis of post-transcriptional and epigenetic factors associated
with KRAS signaling may shed more light onto the molecular biology
of PDAC.
Acknowledgements
This study was supported by projects of the Czech
Science Foundation (grant no. P301/12/1734), the Ministry of Health
of the Czech Republic (grant no. 16-28375A), the Ministry of
Education Youth and Sports of the Czech Republic (no. LO1503) and
the Czech Ministry of Education (nos. NPU I LO1304 and RVO
61989592).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morris JP IV, Wang SC and Hebrok M: KRAS,
hedgehog, Wnt and the twisted developmental biology of pancreatic
ductal adenocarcinoma. Nat Rev Cancer. 10:683–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eser S, Schnieke A, Schneider G and Saur
D: Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer.
111:817–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eser S, Reiff N, Messer M, Seidler B,
Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et
al: Selective requirement of PI3K/PDK1 signaling for Kras
oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell.
23:406–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Collisson EA, Trejo CL, Silva JM, Gu S,
Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort
D, et al: A central role for RAF→MEK→ERK signaling in the genesis
of pancreatic ductal adenocarcinoma. Cancer Discov. 2:685–693.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim KH, Baines AT, Fiordalisi JJ,
Shipitsin M, Feig LA, Cox AD, Der CJ and Counter CM: Activation of
RalA is critical for Ras-induced tumorigenesis of human cells.
Cancer Cell. 7:533–545. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Feldmann G, Mishra A, Hong SM, Bisht S,
Strock CJ, Ball DW, Goggins M, Maitra A and Nelkin BD: Inhibiting
the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation
and progression through the suppression of Ras-Ral signaling.
Cancer Res. 70:4460–4469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dhaka A, Costa RM, Hu H, Irvin DK, Patel
A, Kornblum HI, Silva AJ, O'Dell TJ and Colicelli J: The RAS
effector RIN1 modulates the formation of aversive memories. J
Neurosci. 23:748–757. 2003.PubMed/NCBI
|
|
11
|
Mohelnikova-Duchonova B, Kocik M,
Duchonova B, Brynychova V, Oliverius M, Hlavsa J, Honsova E,
Mazanec J, Kala Z, Ojima I, et al: Hedgehog pathway overexpression
in pancreatic cancer is abrogated by new-generation taxoid
SB-T-1216. Pharmacogenomics J. Aug 30–2016.(Epub ahead of print).
PubMed/NCBI
|
|
12
|
Mohelnikova-Duchonova B, Brynychova V,
Hlavac V, Kocik M, Oliverius M, Hlavsa J, Honsova E, Mazanec J,
Kala Z, Melichar B and Soucek P: The association between the
expression of solute carrier transporters and the prognosis of
pancreatic cancer. Cancer Chemother Pharmacol. 72:669–682. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohelnikova-Duchonova B, Brynychova V,
Oliverius M, Honsova E, Kala Z, Muckova K and Soucek P: Differences
in transcript levels of ABC transporters between pancreatic
adenocarcinoma and nonneoplastic tissues. Pancreas. 42:707–716.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soucek P, Anzenbacher P, Skoumalova I and
Dvorak M: Expression of cytochrome P450 genes in CD34+
hematopoietic stem and progenitor cells. Stem Cells. 23:1417–1422.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mohelnikova-Duchonova B, Oliverius M,
Honsova E and Soucek P: Evaluation of reference genes and
normalization strategy for quantitative real-time PCR in human
pancreatic carcinoma. Dis Markers. 32:203–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 55:611–622.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Caldas C, Hahn SA, Hruban RH, Redston MS,
Yeo CJ and Kern SE: Detection of K-ras mutations inthe stool of
patients with pancreatic adenocarcinoma and pancreatic ductal
hyperplasia. Cancer Res. 54:3568–3573. 1994.PubMed/NCBI
|
|
19
|
Bournet B, Buscail C, Muscari F, Cordelier
P and Buscail L: Targeting KRAS for diagnosis, prognosis, and
treatment of pancreatic cancer: Hopes and realities. Eur J Cancer.
54:75–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou L, Baba Y, Kitano Y, Miyake K, Zhang
X, Yamamura K, Kosumi K, Kaida T, Arima K, Taki K, et al: KRAS,
BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic
cancers: Pyrosequencing technology and literature review. Med
Oncol. 33:322016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oliveira-Cunha M, Hadfield KD, Siriwardena
AK and Newman W: EGFR and KRAS mutational analysis and their
correlation to survival in pancreatic and periampullary cancer.
Pancreas. 41:428–434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schultz NA, Roslind A, Christensen IJ,
Horn T, Høgdall E, Pedersen LN, Kruhøffer M, Burcharth F, Wøjdemann
M and Johansen JS: Frequencies and prognostic role of KRAS and BRAF
mutations in patients with localized pancreatic and ampullary
adenocarcinomas. Pancreas. 41:759–766. 2012.PubMed/NCBI
|
|
23
|
Lemstrova R, Melichar B and
Mohelnikova-Duchonova B: Therapeutic potential of taxanes in the
treatment of metastatic pancreatic cancer. Cancer Chemother
Pharmacol. 78:1101–1111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Melichar B: Laboratory medicine and
medical oncology: The tale of two Cinderellas. Clin Chem Lab Med.
51:99–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fuchs CS, Azevedo S, Okusaka T, Van
Laethem JL, Lipton LR, Riess H, Szczylik C, Moore MJ, Peeters M and
Bodoky G: A phase 3 randomized, double-blind, placebo-controlled
trial of ganitumab or placebo in combination with gemcitabine as
first-line therapy for metastatic adenocarcinoma of the pancreas:
The GAMMA trial. Ann Oncol. 26:921–927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deplanque G, Demarchi M, Hebbar M, Flynn
P, Melichar B, Atkins J, Nowara E, Moyé L, Piquemal D, Ritter D, et
al: A randomized, placebo-controlled phase III trial of masitinib
plus gemcitabine in the treatment of advanced pancreatic cancer.
Ann Oncol. 26:1194–1200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chuang HC, Huang PH, Kulp SK and Chen CS:
Pharmacological strategies to target oncogenic KRAS signaling in
pancreatic cancer. Pharmacol Res. 117:370–376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chung V, McDonough S, Philip PA, Cardin D,
Wang-Gillam A, Hui L, Tejani MA, Seery TE, Dy IA, Al Baghdadi T, et
al: Effect of Selumetinib and MK-2206 vs oxaliplatin and
fluorouracil in patients with metastatic pancreatic cancer after
prior therapy: SWOG S1115 study randomized clinical trial. JAMA
Oncol. 3:516–522. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Riely GJ, Johnson ML, Medina C, Rizvi NA,
Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W and
Ginsberg MS: A phase II trial of Salirasib in patients with lung
adenocarcinomas with KRAS mutations. J Thoracic Oncol. 6:1435–1437.
2011. View Article : Google Scholar
|
|
30
|
Karp JE, Vener TI, Raponi M, Ritchie EK,
Smith BD, Gore SD, Morris LE, Feldman EJ, Greer JM, Malek S, et al:
Multi-institutional phase 2 clinical and pharmacogenomic trial of
tipifarnib plus etoposide for elderly adults with newly diagnosed
acute myelogenous leukemia. Blood. 119:55–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rao S, Cunningham D, de Gramont A,
Scheithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T,
Dostalova J, et al: Phase III double-blind placebo-controlled study
of farnesyl transferase inhibitor R115777 in patients with
refractory advanced colorectal cancer. J Clin Oncol. 22:3950–3957.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Van CE, van de Velde H, Karasek P, Oettle
H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C,
Neumann H, et al: Phase III trial of gemcitabine plus tipifarnib
compared with gemcitabine plus placebo in advanced pancreatic
cancer. J Clin Oncol. 22:1430–1438. 2004. View Article : Google Scholar : PubMed/NCBI
|