Introduction
Cutaneous melanoma is a heterogeneous disease
affecting the regulation of multiple genes and proteins that
contribute to the strong proliferation and invasion of degenerated
melanocytes into the dermis and subsequent metastatic dissemination
of melanoma cells and progression of the disease (1).
Ultraviolet (UV) light exposure is considered an
important predisposing factor that triggers continuous
proliferation of certain melanocytes, which do not undergo
senescence, and therefore support the development of melanoma
(2). In melanocytes, UV light usually
induces pigmentation. In this case, proliferation and pigment
production is stimulated by UV-induced DNA damage to keratinocytes,
which subsequently secrete α-melanocyte stimulating hormone (αMSH)
(3). αMSH binds to the melanocortin 1
receptor, which is expressed on melanocytes (4). Additional research demonstrated that
UVB-induced DNA lesions also cause genetic mutations directly in
melanocytes, with C→T transitions at dipyrimidine sites (5).
However, recent findings revealed that the
photo-carcinogenesis pathway is more complex, with consequences in
which each of these processes, mediated by various cellular,
biochemical and molecular changes, are closely associated with each
other (6).
Although UVA is the most prevalent component of
solar UV radiation reaching the surface of the Earth, it mainly
causes skin photo-aging (solar elastosis), but it is less
carcinogenic compared to UVB radiation (7). By contrast, although UVB radiation only
constitutes a minor part of solar radiation, it is carcinogenic at
significantly lower doses compared with UVA radiation. UVB has a
direct mutagenic effect on DNA as it is maximally absorbed by this
primary chromophore (8). UV photon
energy absorption by DNA decreases constantly at longer wavelengths
(in the UVA range); therefore, UVB radiation is considered the
major cause of skin cancer (9,10).
Notably, Noonan et al (11)
reported that only a single dose of burning UV radiation to neonate
hepatocyte growth factor/scatter factor-transgenic mice is
necessary and sufficient to induce melanoma with a high incidence.
Although supported by several studies, the molecular mechanism of
melanoma induction by UV irradiation is not fully understood
(12,13).
Cyld was first identified as a gene
associated with familial cylindromatosis, a disease showing
multiple benign skin tumors that result from Cyld germline
mutations (usually nonsense or missense mutations) associated with
somatic mutations in dermal cells (loss of heterozygosity)
(14). In previous studies, the
expression and function of CYLD in malignant melanoma and basal
cell carcinoma has been analyzed (15,16). These
uncovered a new mechanism, revealing that the zinc-finger
transcription factor SNAIL1 drives melanoma cells to a mitogenic
and metastatic phenotype via downregulating the expression of
CYLD. Loss of CYLD paves the way for activation of p50/p52
subunits of nuclear factor-κB (NF-κB), resulting in stimulation of
the expression of genes, including cyclin D1 and N-cadherin
expression. The induced target genes consequently lead to enhanced
proliferation, migration and invasiveness of melanoma cells in
vitro, as well as tumor growth and metastasis in vivo.
Notably, increased SNAIL1 expression and reduced CYLD levels are
inversely correlated with progression-free and overall survival of
melanoma patients (16). Thus, it was
shown that SNAIL1 has an important role in melanoma progression
(17) and that one of the molecular
mechanisms involved is the downregulation of CYLD. However, the
role of SNAIL1 and CYLD in melanocyte proliferation and migration,
as well as early malignant transformation, remains elusive.
In the present study, the effect of UVB radiation,
one of the major promoting factors for the development of skin
cancer, on SNAIL1 expression was investigated. Induced
signaling via the ERK-SNAIL1 axis in normal primary human
melanocytes, which reduces CYLD expression in UVB dependency, was
identified.
Materials and methods
Cells and cell culture
The melanoma cell line Mel Ei was derived from a
primary cutaneous melanoma, and the melanoma cell line Mel Im was
isolated from metastasis. These cell lines were provided by
Professor Judith P. Johnson (Cancer Immunology, Ludwig-Maximilians
University, Munich, Germany) (18).
The cell lines were cultured in Dulbecco's modified Eagle's medium,
supplemented with penicillin (400 U/ml), streptomycin (50 µg/ml)
and 10% fetal calf serum (all Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Primary normal human epidermal melanocytes (NHEMs) were
isolated and cultured as described in a previous study (18,19).
Melanocytes were cultivated in M2 medium (PromoCell GmbH,
Heidelberg, Germany). All cell lines were incubated at 37°C in an
8% CO2 humidified atmosphere.
NHEMs were treated with the chemical
mitogen-activated protein (MAP) kinase inhibitors PD98059 and UO126
[specific for MAP kinase kinase (MEK) 1 and MEK2; both Calbiochem;
Merck KGaA, Darmstadt, Germany] for 6 h. Control cells were
incubated with the vehicle dimethyl sulfoxide alone.
UVB radiation
UVB radiation of NHEMs seeded in M2 medium
(PromoCell, Heidelberg, Germany) without PMA was performed with
defined UVB doses (Whatman Biometra Transilluminator; Biometra
GmbH, Göttingen, Germany). Kinase inhibitors were added before UVB
radiation at a concentration of 20 µM, and due to their light
sensitivity, renewed immediately after radiation in fresh medium at
a concentration of 10 µM. For quenching of singlet oxygen, cells
were treated with histidine (50 mM in PBS) 1 h prior to and during
UVB administration (80 mJ/cm2). The irradiated cells
were maintained at 37°C in a 5% CO2 atmosphere for 3 h
(Mel Ei cells) and 5 h (NHEMs).
Expression analysis
Isolation of total cellular RNA from the Mel Ei, Mel
Im cell lines and primary NHEM was performed using the E.Z.N.A.
MicroElute Total RNA kit (Omega Bio-Tek, VWR Darmstadt, Germany)
according to the manufacturer's protocol. RNA concentration was
measured with a NanoDrop spectrophotometer (NanoDrop Technologies;
Thermo Fisher Scientific, Inc., Wilmington, DE, USA) and cDNA was
generated by reverse transcription using the Super Script II
Reverse Transcriptase kit (Thermo Fisher Scientific. Inc., Waltham,
MA, USA), with each reaction containing 500 ng of total RNA
according to the protocol of the manufacturer. Analysis of mRNA
expression was performed using quantitative Real-Time PCR on the
LightCycler 480 system (Roche Diagnostics GmbH, Mannheim, Germany).
A volume of 1 µl cDNA template, 0.5 µl of forward and reverse
primers (each 20 µM) and 10 µl of SYBR Green I (Roche Diagnostics
GmbH) were combined to a total volume of 20 µl. Specific primers
for CYLD, cyclin D1, N-cadherin and SNAIL1 expression analysis are
summarized in Table I. The
housekeeping gene was β-actin (Table
I).
 | Table I.Primer sequences for expression
analysis of cyclin D1, CYLD, N-cadherin, SNAIL and β-actin. |
Table I.
Primer sequences for expression
analysis of cyclin D1, CYLD, N-cadherin, SNAIL and β-actin.
| Gene | Primer
sequence |
|---|
| Cyclin D1 | F:
5′-GCCTGTGATGCTGGGCACTTCATC-3′ |
|
| R:
5′-TTTGGTTCGGCAGCTTGCTAGGTG-3′ |
| CYLD | F:
5′-TGCCTTCCAACTCTCGTCTTG-3′ |
|
| R:
5′-AATCCGCTCTTCCCAGTAGG-3′ |
| N-cadherin | F:
5′-TGGATGAAGATGGCATGG-3′ |
|
| R:
5′-AGGTGGCCACTGTGCTTAC-3′ |
| SNAIL | F:
5′-AGGCCCTGGCTGCTACAAG-3′ |
|
| R:
5′-ACATCTGAGTGGGTCTGGAG-3′ |
| β-actin | F:
5′-CTACGTCGCCCTGGACTTCGAGC-3′ |
|
| R:
5′-GATGGAGCCGCCGATCCACACGG-3′ |
Protein analysis
Protein extraction, analysis and western blotting
were performed as previously described (20), applying the following primary
antibodies: Polyclonal anti-CYLD [cat. no. 4495; dilution, 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA; (21)], anti-p44/42 MAP kinase (cat. no. 9102;
dilution, 1:1,000) or anti-phospho-p44/42 MAP kinase (cat. no.
4370; dilution, 1:1,000; both Cell Signaling Technology, Inc.) and
anti- β-actin (cat. no. A5441; dilution, 1:5,000; Sigma-Aldrich;
Merck KGaA).
Transfection/transduction of cell
lines
To express p65 and an adenoviral negative form of
IκB (AdVdnIκB) (22), melanoma cells
were transiently transfected or transduced with the expression
plasmid or adenovirus, respectively as previously described
(23).
Statistical analysis
All experiments were performed on at least 3
independent occasions. Results are presented as the mean ± standard
error of the mean (SEM). Comparison between groups was made using a
one-way analysis of variance followed by a Kruskal-Wallis test, and
comparisons between CYLD expression in NHEM cells with and without
UVB exposure were calculated by unpaired Student's t-test. All
calculations were performed using the GraphPad Prism 7 software
(GraphPad Software Inc., San Diego, CA, USA).
Results
ERK and SNAIL regulation by UVB
irradiation in melanocytes
As the processes of proliferation and migration in
melanocytes have a physiological as well as pathophysiological
role, the present study determined whether there is an association
between UVB radiation exposure and expression of ERK and SNAIL1 in
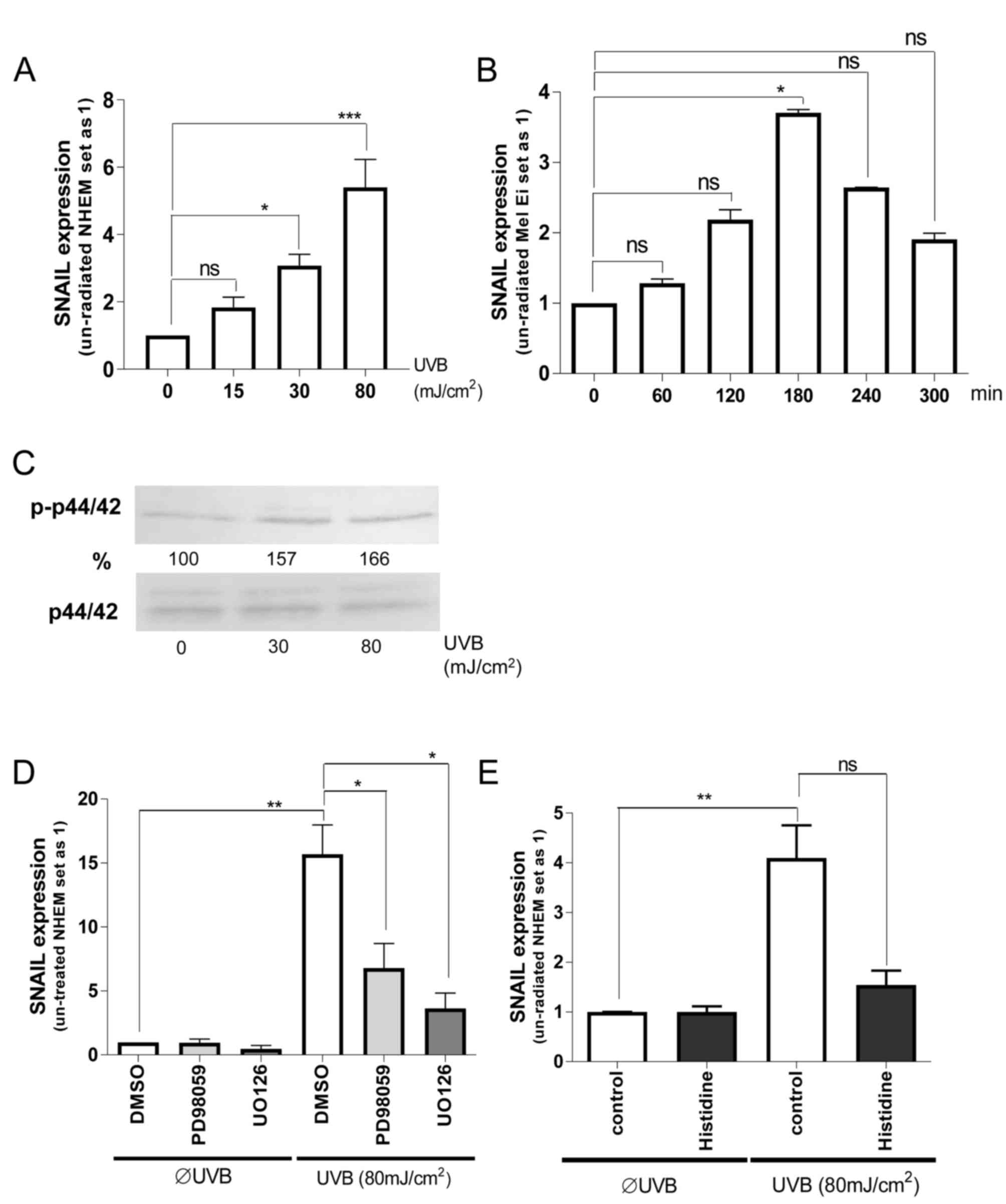
melanocytes. It was identified that UVB-radiation induced
SNAIL1 mRNA expression in a dose-dependent manner in NHEMs;
the most effective dose was 80 mJ/cm2 (Fig. 1A). This induction was also observed in
the primary melanoma Mel Ei cells using the most effective UVB dose
of 80 mJ/cm2 (Fig. 1B). In
the present study, it was further revealed that the maximum SNAIL1
mRNA level was reached after 180 min of UVB radiation,
demonstrating a fast regulation of transcription.
To determine the mechanism of this UVB mediated
SNAIL expression, ERK signaling was first concentrated on.
Dose-dependent ERK activation by UV radiation in melanocytes was
identified (Fig. 1C). Pretreatment of
NHEMs with ERK-inhibitors (UO126 or PD98059) (Fig. 1D) significantly inhibited UVB
induction of SNAIL1 expression, although the base level was
not completely reached.
To determine the role of UV-induced reactive oxygen
species (ROS), NHEMs were pretreated with histidine (quencher to
inhibit the formation of free radicals) prior to UV irradiation.
Notably, histidine treatment almost completely inhibited UVB
effects on SNAIL1 expression (Fig.
1E). This indicates that UVB can induce SNAIL1
expression in NHEMs via free radical-mediated ERK activation.
CYLD regulation by UVB irradiation in
melanocytes
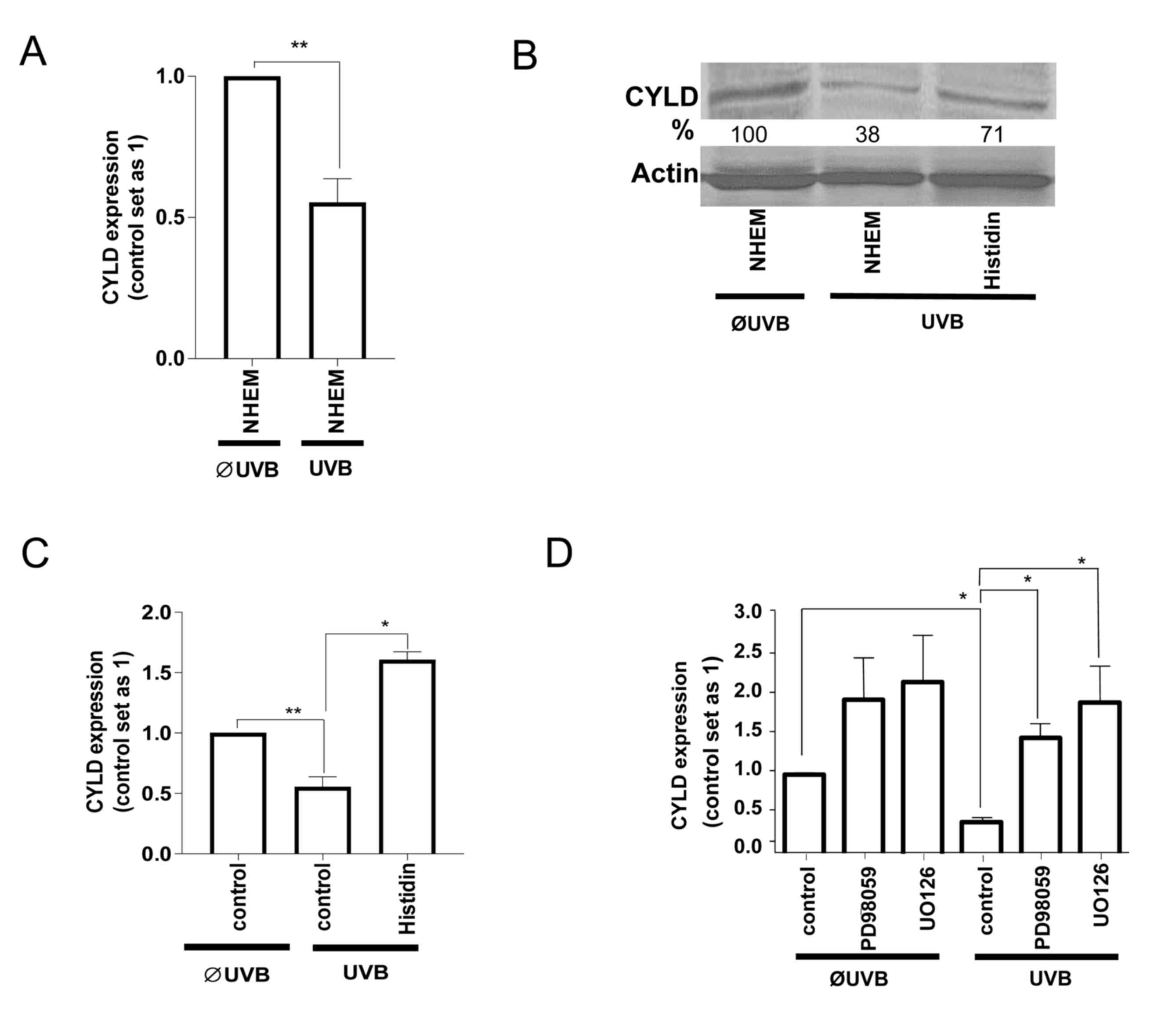
Since SNAIL1 inhibited CYLD expression in
melanoma cells (16), it was
investigated whether the UVB-induced increase in SNAIL1 also
affects CYLD levels in NHEMs or whether this is a cancer-specific
regulation. Notably, UVB-radiation also led to the downregulation
of CYLD mRNA (Fig. 2A) and protein
(Fig. 2B) levels in NHEMs. Treatment
with histidine (Fig. 2B and C) as
well as with ERK-inhibitors (Fig. 2D)
inhibited the UVB-induced downregulation of CYLD. These data
indicate that UVB-induces ERK activation, and as a consequence,
SNAIL1 expression led to the downregulation of CYLD in non-tumorous
melanocytes similar, to the regulation found in malignant melanoma
cells.
NF-κB involvement in the UVB dependent
signaling cascade in melanocytes
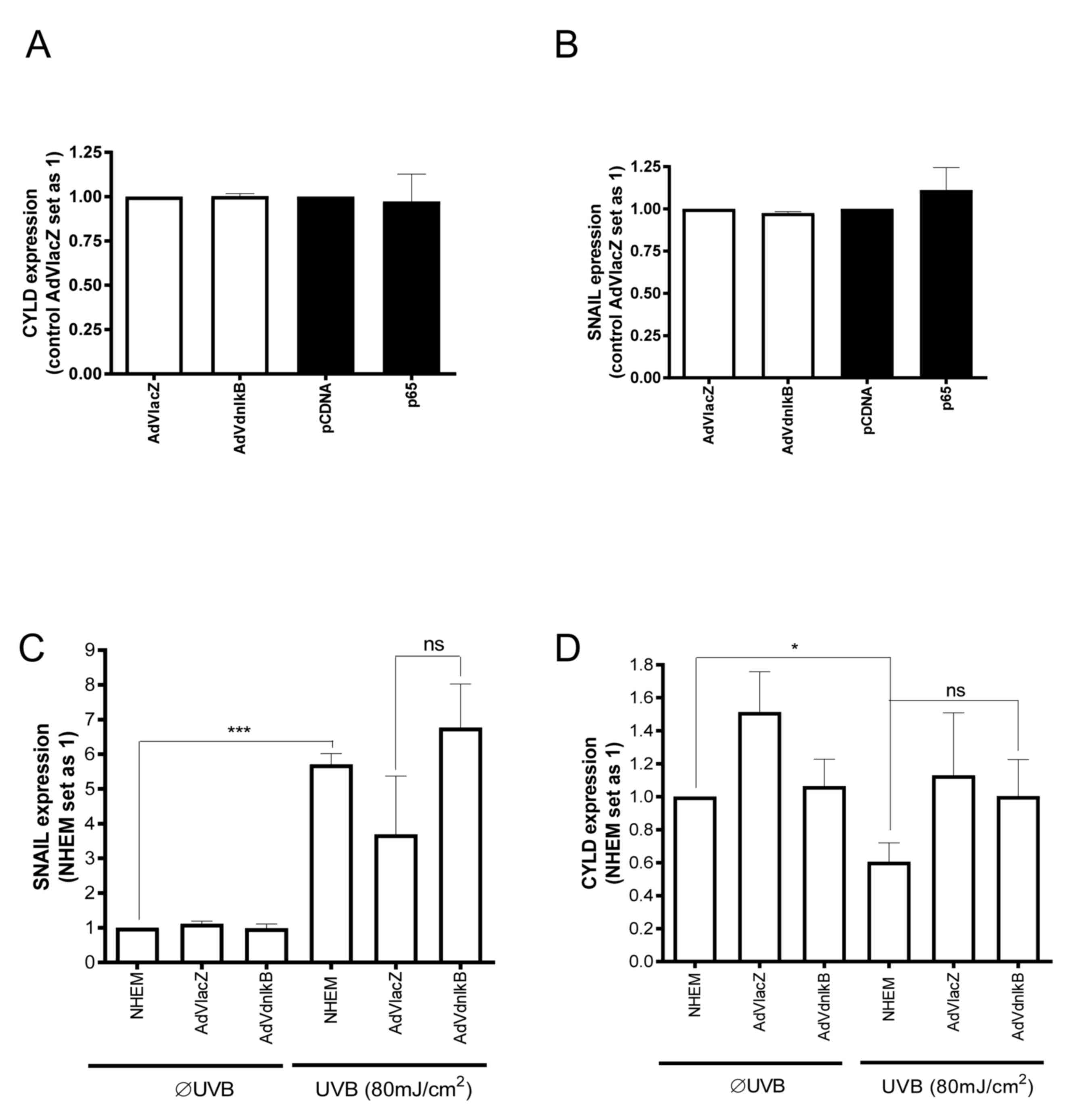
As previous studies have suggested that SNAIL1 is a
possible downstream target of NF-κB (24), the present study further explored the
role of this signaling pathway on SNAIL1 and CYLD expression in
NHEMs and melanoma cells. Adenoviral transduction of a dominant
negative form of IκB (AdVdnIκB) (25)
into Mel Im melanoma cells was performed, and the Mel Im cells were
transiently transfected with a p65 (NF-κB subunit) expression
plasmid. Neither SNAIL1 nor CYLD mRNA expression were
significantly affected by p65 overexpression or AdVdnIκB (Fig. 3A and B). Expression of a
dominant-negative form of IκB also failed to inhibit UVB-induced
SNAIL1 expression in NHEMs (Fig.
3C). Furthermore, inhibition of the NF-κB pathway did not
significantly affect UV-mediated CYLD expression in NHEMs (Fig. 3D). Based on these data, it was
concluded that UV-dependent downregulation of CYLD expression in
NHEMs is not mediated by the NF-κB pathway, but rather by the
ROS/ERK/SNAIL1 axis.
Discussion
UV-radiation is as an initiating factor in nevus
formation. On the basis of the anatomical distribution of acquired
nevi and a UV-associated mutation signature in the majority of the
somatic mutations of nevi, it is hypothesized that UV radiation
also contributes to melanoma initiation (4,9).
When pigment cells undergo neoplastic
transformation, these pathological derivatives are usually termed
according to the pigment that characterized the original cell
lineage, such as melanoma cells from melanocytes. Melanoma can
arise spontaneously in a variety of animals, including dogs,
horses, pigs and several fish. However, melanoma can also be
induced by exposure to UV irradiation and carcinogens, or by the
presence of relevant genetic changes in melanocytes (26). In previous years, varieties of genetic
changes have been characterized in human melanocytic neoplasms and
often correlate strongly with specific morphological
characteristics (4). While progress
has been made on defining the genetic changes present in melanoma,
much remains to be learned about the specific characteristics of
early malignant transformation of the melanocytes. As an example,
approximately one-half of all melanomas in humans harbor oncogenic
mutations in the BRAF gene (e.g., V600E), which leads to
constitutive activation of the RAS-RAF-MEK-ERK (p44/p42; MAPK)
signaling pathway (4,27). In contrast to melanoma cells,
melanocytes exhibit low ERK-activity and the question of which
early processes provoke pathological processes in melanocytes
remains.
Epidemiological studies indicate the importance of
UV radiation in the etiology of melanoma, and since UV-associated
mutations are relatively rare in melanoma, it was speculated that
UV radiation may support melanoma development by indirect effects
(28,29). As a physiological response,
melanocytes migrate to skin areas exposed to solar radiation, and
start to proliferate and produce melanin, thereby exhibiting their
protective effect against UV radiation (30). The signaling molecules involved in the
stimulation of migration and proliferation of melanocytes following
UV exposure remain unclear. However, the present study demonstrated
that UVB radiation induces ERK-activity in melanocytes (NHEMs),
similarly as previously shown in keratinocytes and melanoma cells
(31,32). Additionally, UVB radiation triggered
ERK-mediated SNAIL1 induction and downregulation of CYLD
expression in NHEMs. Following malignant transformation,
constitutive high ERK activity and SNAIL1 expression allow melanoma
cells permanently to exploit this mechanism to obtain a more
aggressive phenotype (16,17,23).
Previously, UVB light was identified as a trigger
for direct association of CYLD with B-cell lymphoma-3 (BCL-3)
(21). Following this association,
CYLD removes lysine 63-linked polyubiquitin chains from BCL-3,
which in turn prevents BCL-3 from translocating into the nucleus
and further expressing different genes in keratinocytes (21). It was observed in malignant melanoma
that loss of CYLD also induces nuclear accumulation of BCL-3 and
NF-κB activation, with the consequence of induced N-cadherin
(migration) and cyclin D1 (proliferation) activity (16). Data from embryogenesis and studies in
Drosophila (24,33–35),
suggesting that SNAIL1 is regulated by Dorsal (NF-κB), were
reassessed in the present study. However, it was found that in
melanoma ERK signaling appears to be the major pathway responsible
for the high constitutive activation of SNAIL1. While the migration
and proliferation of melanocytes in response to UV are
physiological with regards to skin protection by melanin,
long-lasting UV radiation, which is considered as one of the main
risk factors for the development of melanoma, may shift these cells
towards malignant transformation. In normal epidermal melanocytes,
UVB radiation induces ERK-activation and subsequently upregulates
SNAIL1 expression. The repression of CYLD by SNAIL1 allows
degeneration of melanocytes.
In melanoma cells, constitutively high ERK-activity
(potentially caused by B-RAF mutations) leads to high SNAIL1
expression, which in turn causes a high and strong suppression of
CYLD. Activation of cyclinD1 and N-cadherin leads to an increased
proliferation rate of melanoma cells and contributes to the
progression and metastasis of tumors.
Acknowledgements
The authors would like to thank the technicians Mrs.
Lisa Ellmann (University Hospital, Regensburg, Germany) and Mrs.
Nadja Schneider (Friedrich-Alexander University, Erlangen, Germany)
for technical assistance. This study was supported by grants from
the German Research Foundation to S.K., C.H. and A.B. within the
research consortium (grant no. FOR2172).
Glossary
Abbreviations
Abbreviations:
|
UVB
|
ultraviolet B
|
|
SNAIL1
|
snail family transcriptional repressor
1
|
|
NHEMs
|
normal human epidermal melanocytes
|
|
ERK
|
extracellular signal-regulated
kinase
|
References
|
1
|
Rowe CJ and Khosrotehrani K: Clinical and
biological determinants of melanoma progression: Should all be
considered for clinical management? Australas J Dermatol.
57:175–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qadir MI: Skin cancer: Etiology and
management. Pak J Pharm Sci. 29:999–1003. 2016.PubMed/NCBI
|
|
3
|
Cui R, Widlund HR, Feige E, Lin JY,
Wilensky DL, Igras VE, D'Orazio J, Fung CY, Schanbacher CF, Granter
SR and Fisher DE: Central role of p53 in the suntan response and
pathologic hyperpigmentation. Cell. 128:853–864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bastian BC: The molecular pathology of
melanoma: An integrated taxonomy of melanocytic neoplasia. Annu Rev
Pathol. 9:239–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Premi S, Wallisch S, Mano CM, Weiner AB,
Bacchiocchi A, Wakamatsu K, Bechara EJ, Halaban R, Douki T and
Brash DE: Photochemistry. Chemiexcitation of melanin derivatives
induces DNA photoproducts long after UV exposure. Science.
347:842–847. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishisgori C: Current concept of
photocarcinogenesis. Photochem Photobiol Sci. 14:1713–1721. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Gruijl FR: Photocarcinogenesis: UVA vs
UVB. Methods Enzymol. 319:359–366. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tornaletti S and Pfeifer GP: UV damage and
repair mechanisms in mammalian cells. Bioessays. 18:221–228. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seebode C, Lehmann J and Emmert S:
Photocarcinogenesis and Skin Cancer Prevention Strategies.
Anticancer Res. 36:1371–1378. 2016.PubMed/NCBI
|
|
10
|
De Fabo EC, Noonan FP, Fears T and Merlino
G: Ultraviolet B but not ultraviolet A radiation initiates
melanoma. Cancer Res. 64:6372–6376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Noonan FP, Recio JA, Takayama H, Duray P,
Anver MR, Rush WL, De Fabo EC and Merlino G: Neonatal sunburn and
melanoma in mice. Nature. 413:271–272. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Halliday GM, Agar NS, Barnetson RS,
Ananthaswamy HN and Jones AM: UV-A fingerprint mutations in human
skin cancer. Photochem Photobiol. 81:3–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gandini S, Sera F, Cattaruzza MS, Pasquini
P, Picconi O, Boyle P and Melchi CF: Meta-analysis of risk factors
for cutaneous melanoma: II. Sun exposure. Eur J Cancer. 41:45–60.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bignell GR, Warren W, Seal S, Takahashi M,
Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al:
Identification of the familial cylindromatosis tumour-suppressor
gene. Nat Genet. 25:160–165. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuphal S, Shaw-Hallgren G, Eberl M, Karrer
S, Aberger F, Bosserhoff AK and Massoumi R: GLI1-dependent
transcriptional repression of CYLD in basal cell carcinoma.
Oncogene. 30:4523–4530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Massoumi R, Kuphal S, Hellerbrand C, Haas
B, Wild P, Spruss T, Pfeifer A, Fässler R and Bosserhoff AK:
Down-regulation of CYLD expression by Snail promotes tumor
progression in malignant melanoma. J Exp Med. 206:221–232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuphal S, Palm HG, Poser I and Bosserhoff
AK: Snail-regulated genes in malignant melanoma. Melanoma Res.
15:305–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ziegler-Heitbrock HW, Munker R, Johnson J,
Petersmann I, Schmoeckel C and Riethmuller G: In vitro
differentiation of human melanoma cells analyzed with monoclonal
antibodies. Cancer Res. 45:1344–1350. 1985.PubMed/NCBI
|
|
19
|
Tsuji T and Karasek M: A procedure for the
isolation of primary cultures of melanocytes from newborn and adult
human skin. J Invest Dermatol. 81:179–180. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spangler B, Kappelmann M, Schittek B,
Meierjohann S, Vardimon L, Bosserhoff AK and Kuphal S: ETS-1/RhoC
signaling regulates the transcription factor c-Jun in melanoma. Int
J Cancer. 130:2801–2811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Massoumi R, Chmielarska K, Hennecke K,
Pfeifer A and Fässler R: Cyld inhibits tumor cell proliferation by
blocking Bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jobin C, Hellerbrand C, Licato LL, Brenner
DA and Sartor RB: Mediation by NF-kappa B of cytokine induced
expression of intercellular adhesion molecule 1 (ICAM-1) in an
intestinal epithelial cell line, a process blocked by proteasome
inhibitors. Gut. 42:779–787. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kuphal S, Poser I, Jobin C, Hellerbrand C
and Bosserhoff AK: Loss of E-cadherin leads to upregulation of
NFkappaB activity in malignant melanoma. Oncogene. 23:8509–8519.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barberà MJ, Puig I, Domínguez D,
Julien-Grille S, Guaita-Esteruelas S, Peiró S, Baulida J, Francí C,
Dedhar S, Larue L, et al: Regulation of Snail transcription during
epithelial to mesenchymal transition of tumor cells. Oncogene.
23:7345–7354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hellerbrand C, Jobin C, Iimuro Y, Licato
L, Sartor RB and Brenner DA: Inhibition of NFkappaB in activated
rat hepatic stellate cells by proteasome inhibitors and an IkappaB
super-repressor. Hepatology. 27:1285–1295. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schartl M, Larue L, Goda M, Bosenberg MW,
Hashimoto H and Kelsh RN: What is a vertebrate pigment cell?
Pigment Cell Melanoma Res. 29:8–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guadarrama-Orozco JA, Ortega-Gómez A,
Ruiz-García EB, Astudillo-de la Vega H, Meneses-García A and
Lopez-Camarillo C: Braf V600E mutation in melanoma: Translational
current scenario. Clin Transl Oncol. 18:863–871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brenner M, Degitz K, Besch R and Berking
C: Differential expression of melanoma-associated growth factors in
keratinocytes and fibroblasts by ultraviolet A and ultraviolet B
radiation. Br J Dermatol. 153:733–739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jhappan C, Noonan FP and Merlino G:
Ultraviolet radiation and cutaneous malignant melanoma. Oncogene.
22:3099–3112. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Esparza-Soto M, Fox P and Westerhoff P:
Transformation of molecular weight distributions of dissolved
organic carbon and UV-absorbing compounds at full-scale
wastewater-treatment plants. Water Environ Res. 78:253–262. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pak BJ, Lee J, Thai BL, Fuchs SY, Shaked
Y, Ronai Z, Kerbel RS and Ben-David Y: Radiation resistance of
human melanoma analysed by retroviral insertional mutagenesis
reveals a possible role for dopachrome tautomerase. Oncogene.
23:30–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peus D, Meves A, Vasa RA, Beyerle A,
O'Brien T and Pittelkow MR: H2O2 is required for UVB-induced EGF
receptor and downstream signaling pathway activation. Free Radic
Biol Med. 27:1197–1202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ip YT: Transcriptional regulation.
Converting an activator into a repressor. Curr Biol. 5:1–3. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ip YT, Park RE, Kosman D, Yazdanbakhsh K
and Levine M: dorsal-twist interactions establish snail expression
in the presumptive mesoderm of the Drosophila embryo. Genes Dev.
6:1518–1530. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Marchal L, Foucaul A, Patissier G, Rosant
JM and Legrand J: Influence of flow patterns on chromatographic
efficiency in centrifugal partition chromatography. J Chromatogr A.
869:339–352. 2000. View Article : Google Scholar : PubMed/NCBI
|