Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most deadly types of cancer and the seventh leading cause of
cancer death in both sexes. The overall five-year survival rate is
about 6% (range from 2 to 9%) (1). It
is a highly malignant digestive system tumor with characteristic
features of late discovery, early metastasis, rapid progress, and
poor prognosis. Surgical resection is currently the only effective
treatment, but only 20% of PDAC patients are eligible for operation
when diagnosed (2). Earlier diagnosis
and earlier treatment of pancreatic cancer have thus become
particularly important. However, neither reliable screening tests
for early diagnosis nor useful biomarkers to predict treatment
efficacy is currently available (3,4). Although
research in this field has increased significantly over the past
decade, no consistent conclusion has been reached. Therefore, it is
necessary to find biomarkers that can help to diagnose pancreatic
cancer at early stage, predict prognosis, or predict response to
chemotherapeutic drugs.
Checkpoint with FHA domain and RING-finger (CHFR)
was initially identified in a screen to find novel mitotic
checkpoint proteins (5). The CHFR
protein is a 664 amino acid protein with forkhead-associated (FHA)
and RING-finger domains within its amino terminus and a
cysteine-rich region within its carboxy terminus, which is very
highly conserved between humans and mice (6–8). FHA
domain is a phosphothreonine-binding domain, which was frequently
found in DNA repair and checkpoint proteins (6,9). Together
previous studies indicate that, as a checkpoint protein, CHFR might
play different roles at different phases of cell cycle, although
there has been no unified conclusion (5,10,11). Yu et al (10) firstly indicated CHFR was a tumor
suppressor by the creation of CHFR knockout mouse. The study
demonstrated that CHFR was important for maintaining genomic
stability. However, another study in colon cancers was not arriving
at the same conclusion (12). Last
but not least, among the studies focused on the relationship
between CHFR expression and the sensitivity to chemotherapeutics,
inconsistent conclusions can also be found in different kinds of
cancers (13–15). Consequently, the role of CHFR in
different cancers seem to be different. So far, there is no
published investigation focused on the role of CHFR in pancreatic
cancer.
In the present study, the expression of CHFR was
examined in several pancreatic cancer cell lines. We explored the
function of CHFR in pancreatic cancer cells in vitro by
examining the effects of altered CHFR expression on the malignant
biological behaviors and chemotherapy sensitivity. Meanwhile, in
order to investigate the potential of targeting CHFR as an index to
predict prognosis, we also examined the associations among CHFR
expression level, clinicopathologic parameters, and survival rate
in PDAC patients.
Materials and methods
Cell culture and transfection
The pancreatic cancer cell lines CFPAC-1, BxPC-3,
Capan-1, PANC-1, and SW1990 were purchased from the Cell Bank of
the Chinese Science Academy (Shanghai, China) and cultured in the
appropriate media (CFPAC-1, BxPC-3 and Capan-1 cells in RPMI-1640,
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA; PANC-1
cells in DMEM; Thermo Fisher Scientific, Inc.; and SW1990 cells in
L-15; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (FBS; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Thermo Fisher Scientific, Inc.). To
generate a stable Capan-1 cell line with CHFR overexpression, the
complete human cDNA sequence of CHFR (NM_001161344) was cloned into
the GV358 vector backbone downstream from an Ubi promoter
(Ubi-CHFR-MCS-3FLAG-EGFP-IRES-piromycin) to produce GV358-CHFR
vector, which carries the enhanced green fluorescent protein (EGFP)
receptor gene. GV358 empty vectors were used to generate control
cells. The GV358-CHFR vector and control vector were designed and
synthesized by Genechem Co., Ltd. (Shanghai, China). Capan-1 cells
were transfected with the appropriate lentiviral vector at a
multiplicity of infection (MOI)=25 in medium containing 5 µg/ml
polybrene (Genechem Co., Ltd.) according to the manufacturer's
protocol. After 72 h of transfection, the medium was replaced with
2 ml complete culture medium. Cells were selected with 0.2 µg/ml
puromycin (Genechem Co., Ltd.) for 48 h and maintained until
experimental analyses. The stable cell lines were confirmed by
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) and western blot analysis.
RT-qPCR
Total RNA was extracted with RNAiso Plus (#9108;
Takara Bio, Inc., Kusatsu, Japan) according to the manufacturer's
recommended protocol and then reverse transcribed to cDNA using the
Prime Script™ RT reagent kit with gDNA Eraser (#RR047;
Takara Bio, Inc.). Real-time amplification was carried out with a
LightCycler® 480 system (Roche Molecular Diagnostics,
Pleasanton, CA, USA) and the product was quantified using an
intercalating dye (SYBR® Premix Ex Taq™ II;
#RR820A; Takara Bio, Inc.) that exhibits an increased fluorescence
upon binding double-stranded DNA. The housekeeping gene β-actin was
used as the internal control. All primers were purchased from
Takara Bio, Inc. cDNA was subjected to denaturing (95°C, 30 sec),
annealing (55°C, 15 sec), and extension (95°C 5 sec and 60°C 30
sec) for 40 cycles. The relative expression of CHFR mRNA was
quantified using the 2−ΔΔCq method (16). PCR amplification was performed using
the following primers: CHFR forward, 5′-CAGCTTCCGTGAGCTGACCTATC-3′
and reverse, 5′-GCGTGGTGAGCTTTCACCTG-3′; and β-actin forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′.
Western blot analysis
Cells were lysed on ice using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China). Cell lysates were quantified by a
BCA Protein Assay kit (#P0010S; Beyotime Institute of
Biotechnology) and equal amounts of cell protein were loaded into
each well of 10% SDS-PAGE gels. After electrophoresis, proteins on
gels were transferred to polyvinylidene difluoride membranes (Merck
Millipore, Darmstadt, Germany). Membranes were blocked with 5%
(w/v) non-fat milk and then incubated with primary antibodies
(rabbit anti-human CHFR polyclonal antibody, #4297S; dilution,
1:1,000; Cell Signaling Technology, Inc., Boston, MA, USA; and
mouse anti-human FLAG monoclonal antibody, #F1804; dilution,
1:3,000; Sigma-Aldrich Co. LLC., Santa Clara, CA, USA) overnight at
4°C. Membranes were washed four times, and then incubated with goat
anti-rabbit or anti-mouse secondary antibodies (goat anti-rabbit,
#sc-2004; and goat anti-mouse, #sc-2005; dilution, 1:5,000; both
from Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
membranes were the incubated in ECL western blot substrates
(#M3121/1859022; Thermo Fisher Scientific, Inc.) for 1 min, and
exposed to X-ray film. β-actin was used as a control.
Cell proliferation, migration and
invasion assays
For cell proliferation assay, Capan-1_CHFR or
Capan-1_Ctrl cells (6×103/100 µl) were seeded in 96-well
plates and cultured for 0, 24, 48, 72 and 96 h at 37°C. Next, 100
µl serum-free RPMI-1640 containing 10% Cell Counting Kit-8 (CCK-8)
reagent (Dojindo, Tokyo, Japan) was added in each well, and cells
were incubated for 4 h at 37°C. Optical density values (OD) were
read at 450 nm using a 96-well plate reader (Thermo Fisher
Scientific, Inc.). Transwell assays were performed to assess cell
migration and invasion. For migration assays, Capan-1_CHFR or
Capan-1_Ctrl cells (4×104 cells) were seeded into the
upper chambers of the inserts with non-coated membrane (24-well
insert; 8-µm pore size; Corning Inc., Corning, NY, USA). For
invasion assays, 4×104 cells were plated in the upper
chambers with Matrigel-coated membrane (Corning Inc.). 100 µl
serum-free RPMI-1640 was added to the upper chamber and 600 µl
RPMI-1640 medium containing 10% FBS was added to the lower chamber.
Cells were incubated for 48 h at 37°C in a 5% CO2
atmosphere. The cells were then fixed in 4% paraformaldehyde
solution for 30 min and stained with 5% crystal violet solution for
5 min. Cells that invaded through the pores to the lower surface of
the inserts were photographed and counted under an inverted
microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometric analysis
For cell cycle analysis, cells were cultured to
logarithmic phase, harvested and fixed in ice-cold 70% ethanol
overnight at 4°C. Cells were then incubated with RNase A at 37°C
for 30 min and stained with propidium iodide (#KGA512; Nanjing
KeyGen Biotech., Co., Ltd., Nanjing, China) for 30 min in the dark.
The cellular DNA content and cell cycle phase distribution were
analyzed with a FACS Caliber instrument (BD Biosciences, Franklin
Lakes, NJ, USA). Apoptotic cells were detected by flow cytometry
using the Annexin V-APC/7-AAD Apoptosis Detection kit (#KGA1026;
Nanjing KeyGen Biotech., Co., Ltd.). Capan-1_CHFR or Capan-1_Ctrl
cells were inoculated into 25 cm2 culture flasks
(8×105 cells/flask). After cells adhered, they were
treated with 100, 200, or 400 nM gemcitabine or 2, 4, or 8 nM
docetaxel (Sigma-Aldrich Co. LLC.) for 24 h and then harvested. The
staining was performed according to the manufacturer's
instructions. Flow cytometry evaluation of the apoptotic rate was
performed using a FACS Caliber instrument (BD Biosciences). The
percentage of specific apoptosis was calculated using the following
equation: % specific apoptosis % = {1 - [100% - % (Annexin
V+ + Annexin V 7-AAD+)] - [% spontaneous
(Annexin V+ + Annexin V 7-AAD+)]}. Three
wells were assessed at each condition and the experiment was
repeated three times.
Patients and tissue specimens
The study was approved by the Ethics Committee of
Xuanwu Hospital, Capital Medical University (Beijing, China) in
accordance with the guidelines for the protection of human subjects
(17). Patients with PDAC who had
undergone radical resection in General Surgery Department at
Beijing Xuanwu Hospital between March 2008 and October 2015 were
retrospectively identified via medical records and pathology
reports. None of the patients received chemotherapy or radiation
therapy prior to cancer resection. All tissue samples were obtained
following informed consent. Selected hematoxylin and eosin-stained
slides were reviewed by an experienced pathologist at Xuanwu
Hospital to confirm the original pathological diagnoses and to
choose representative areas. The pathologic specimens should
contain both tumor tissues and adjacent non-tumor tissues. All
tumors were staged according to the pathological
tumor-node-metastasis (TNM) staging system of the American Joint
Committee on Cancer.
Immunohistochemistry
Twenty-seven PDAC samples and paired non-tumor
tissue samples were immunohistochemically stained by the Envision
method. The sections (5 µm thick) were deparaffinized, rehydrated
with graded concentrations of ethanol, and incubated with 3%
H2O2 for 15 min. The sections were then
incubated with the primary antibody (mouse anti-human CHFR
monoclonal antibodies, #H00055743-M01; dilution, 1:100; Abnova,
Taibei, Taiwan) in a moist chamber (dark, 4°C, overnight), followed
by incubation with secondary antibodies (anti-mouse/rabbit,
#KIT-9922; Fuzhou Maixin Biotech Co., Ltd., Fuzhou, China) for 15
min at room temperature and DAB reagent (Fuzhou Maixin Biotech Co.,
Ltd.). The sections were then counterstained with hematoxylin and
dehydrated. CHFR staining was scored both for nuclear and
cytoplasmic staining based on intensity (0, no staining; 1, weak
staining; 2, strong staining) and percentage of cells stained (0,
<10%; 1, 10–50%; 2, >50%) (15,18).
Scores for intensity and percentage of stained cells were added for
a maximum score of 4. Scores of 4 were considered ‘high’
expression, while all others were ‘low’ expression.
Immunohistochemistry was reviewed for accuracy of diagnosis and for
scoring by an experienced pathologist who was blinded to the
clinical outcomes of the patients.
Statistical analysis
Statistical analysis was performed using SPSS
version 20.0 (IBM Corp., Armonk, NY, USA). A Mann-Whitney U test
(comparisons between two groups) was used for analysis and P≤0.05
was considered to indicate a statistically significant difference.
The association of CHFR expression with histological or clinical
factors was analyzed using Chi-square test or Fisher's exact tests.
Kaplan-Meier and time series tests (log-rank test) were used for
univariate survival analysis.
Results
CHFR expression in pancreatic cancer
cells
We first examined endogenous CHFR mRNA and protein
levels in five human pancreatic cancer cell lines, CFPAC-1, BxPC-3,
Capan-1, PANC-1, and SW1990, using RT-qPCR and western blot
analysis. The results showed that CHFR levels varied across all
cell lines (Fig. 1A and B). Capan-1
cells expressed the lowest level of CHFR, and thus we selected
these cells for further analyses. We next established Capan-1 cells
with stable overexpression of CHFR by lentiviral infection;
Capan-1_Ctrl cells were established as controls (Fig. 1E). We confirmed that CHFR mRNA levels
were significantly upregulated in the Capan-1_CHFR cells compared
to the Capan-1_Ctrl cells (P<0.01; Fig. 1D). Exogenous CHFR protein expression
was also confirmed in Capan-1_CHFR cells, but not in control cells
(Fig. 1C).
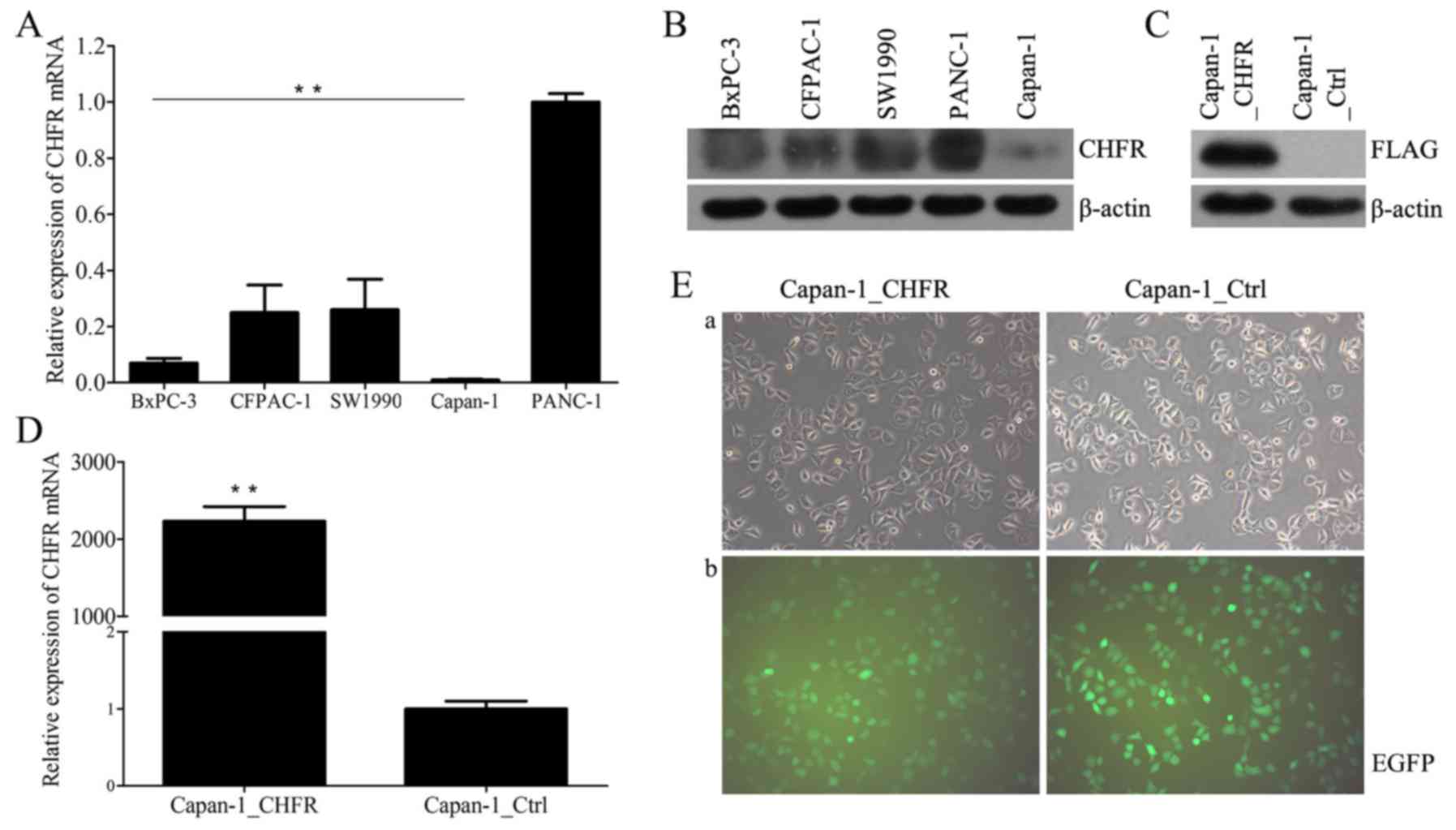 | Figure 1.Expression of CHFR in pancreatic
cancer cell lines. (A) Endogenous CHFR mRNA expressions in five
human pancreatic cancer cell lines, BxPC-3, CFPAC-1, SW1990,
Capan-1, and PANC-1 were determined by RT-qPCR. **P<0.01. (B)
CHFR protein expression in five human pancreatic cancer cell lines,
BxPC-3, CFPAC-1, SW1990, PANC-1 and Capan-1 were determined by
western blotting. (C) Exogenous CHFR protein expression in
Capan-1_CHFR and Capan-1_Ctrl cells were determined by western
blotting. (D) CHFR mRNA expressions in Capan-1_CHFR and
Capan-1_Ctrl cells were determined by RT-qPCR. **P<0.01. (E)
After transfection, Capan-1_CHFR and Capan-1_Ctrl cells were
observed by (a) inverted optical microscopy and (b) inverted
fluorescence microscopy. Note that EGFP fluorescence could be
observed in most of the cells, which is indicative of successful
transfection. Original magnification, ×200. CHFR, checkpoint with
forkhead and ring-finger; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; EGFP,
enhanced green fluorescent protein. |
Effects of CHFR overexpression on cell
proliferation and cell cycle
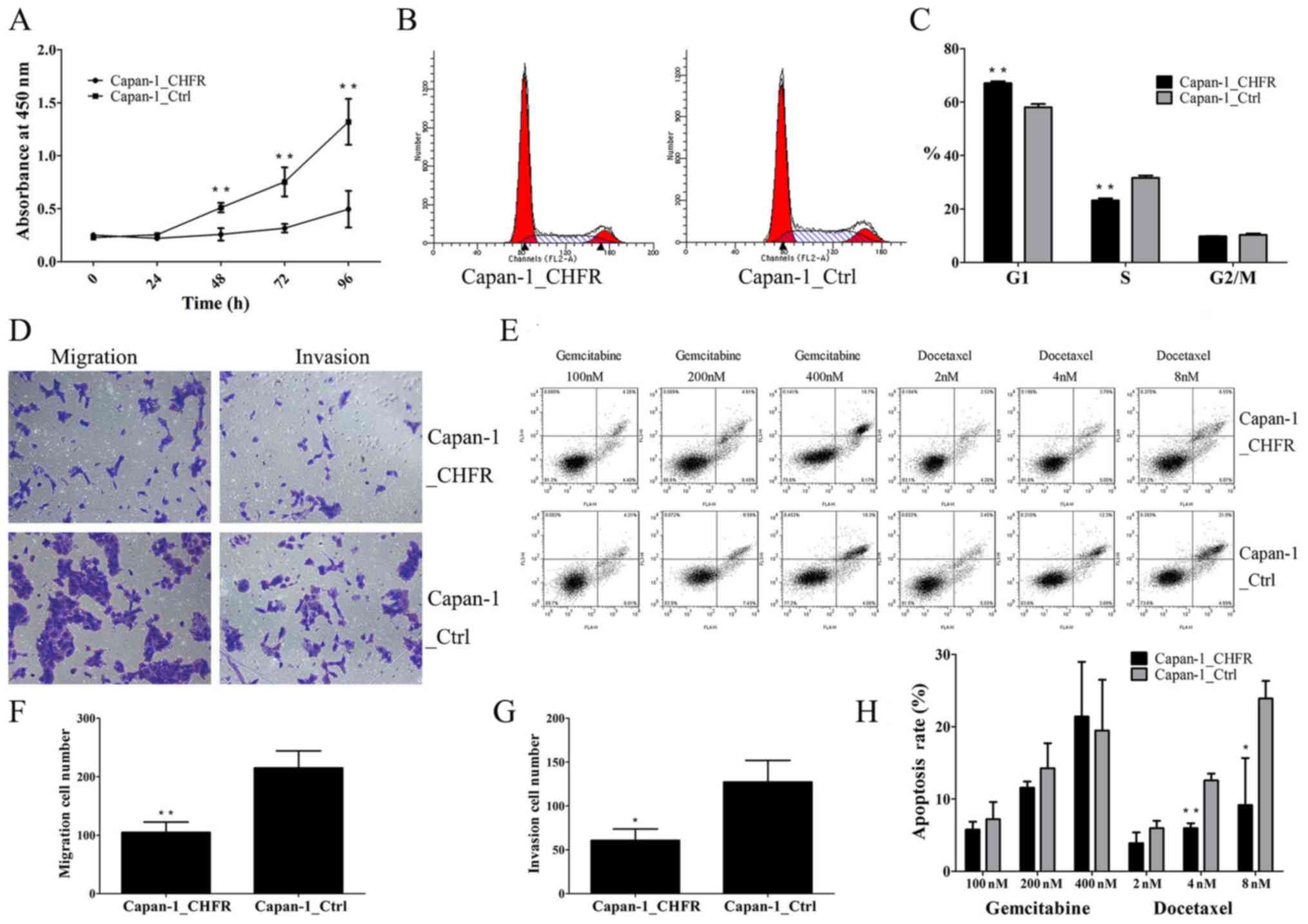
As shown in Fig. 2A,
the proliferation of Capan-1_CHFR cells was significantly inhibited
at 48, 72 and 96 h compared to Capan-1_Ctrl cells (P<0.01). To
evaluate the mechanism underlying the inhibitory effect of CHFR in
more detail, we examined the cell cycle phase distribution of
Capan-1 stable cell lines using flow cytometry after staining with
PI. Interestingly, the results showed that the percentage of cells
in the G1 phase was significantly increased in Capan-1_CHFR cells
compared to Capan-1_Ctrl cells (P<0.01; Fig. 2B and C). These results indicated that
CHFR inhibited the proliferation of Capan-1 cells by arresting
cells at G1 phase.
Effects of CHFR overexpression on cell
migration and invasion
Cell migration and invasion abilities were next
determined by Transwell assays. We found that, compared with
Capan-1_Ctrl cells, Capan-1_CHFR cell migration was significantly
reduced (P<0.01) (Fig. 2D and F).
The Transwell invasion assays also showed that cell invasion was
inhibited in Capan-1_CHFR cells compared to control cells
(P<0.05) (Fig. 2D and G).
Effect of CHFR overexpression on
apoptosis of Capan-1 cells
We next evaluated the apoptotic response of
Capan-1_CHFR and Capan-1_Ctrl cells by treating cells with
different concentrations of gemcitabine or docetaxel for 24 h
(Fig. 2E and H). Before formal
experiments, we have tried different action times of the drugs.
When the action time was prolonged to 48 h, in the both groups
treated with gemcitabine at the concentrations of 400 nM, there
were more than 70% cells in the right upper quadrant (Annexin V
7-AAD+), which contained both necrosis and the late stage apoptotic
cells (data not shown). When the action time was prolonged to 72 h,
after two times of washing with PBS, the rest cells in the tubes
were even not enough to be analyzed with the instrument. Besides,
we also tried the time point shorter than 24 h. When the action
time was 12 h, the apoptosis rates of the both groups were too
close to the basal apoptotic level at the concentration of
docetaxel 4 nM (data not shown). Therefore, we chose the time point
of 24 h for our formal experiments. The results showed that
calculated with the equation mentioned in Materials and methods, a
decreased apoptotic rate was detected in Capan-1_CHFR cells treated
with docetaxel at the concentrations of 4 and 8 nM compared with
Capan-1_Ctrl cells. However, no statistically significant
differences in apoptosis were observed in response to gemcitabine
treatment (P<0.05; P<0.01) (Fig.
2H).
Correlation between CHFR
immunohistochemical expression and clinicopathological
parameters
Considering the lasting time span of the PDAC cases,
some of the pathological data were not intact, while some patients
lost to follow-up. Only 27 patients were finally included in our
research. The patients' ages ranged from 48 to 79 years (median, 64
years). The median follow-up period was 16.9 months (range, 0–59
months). The patients' clinicopathological data are summarized in
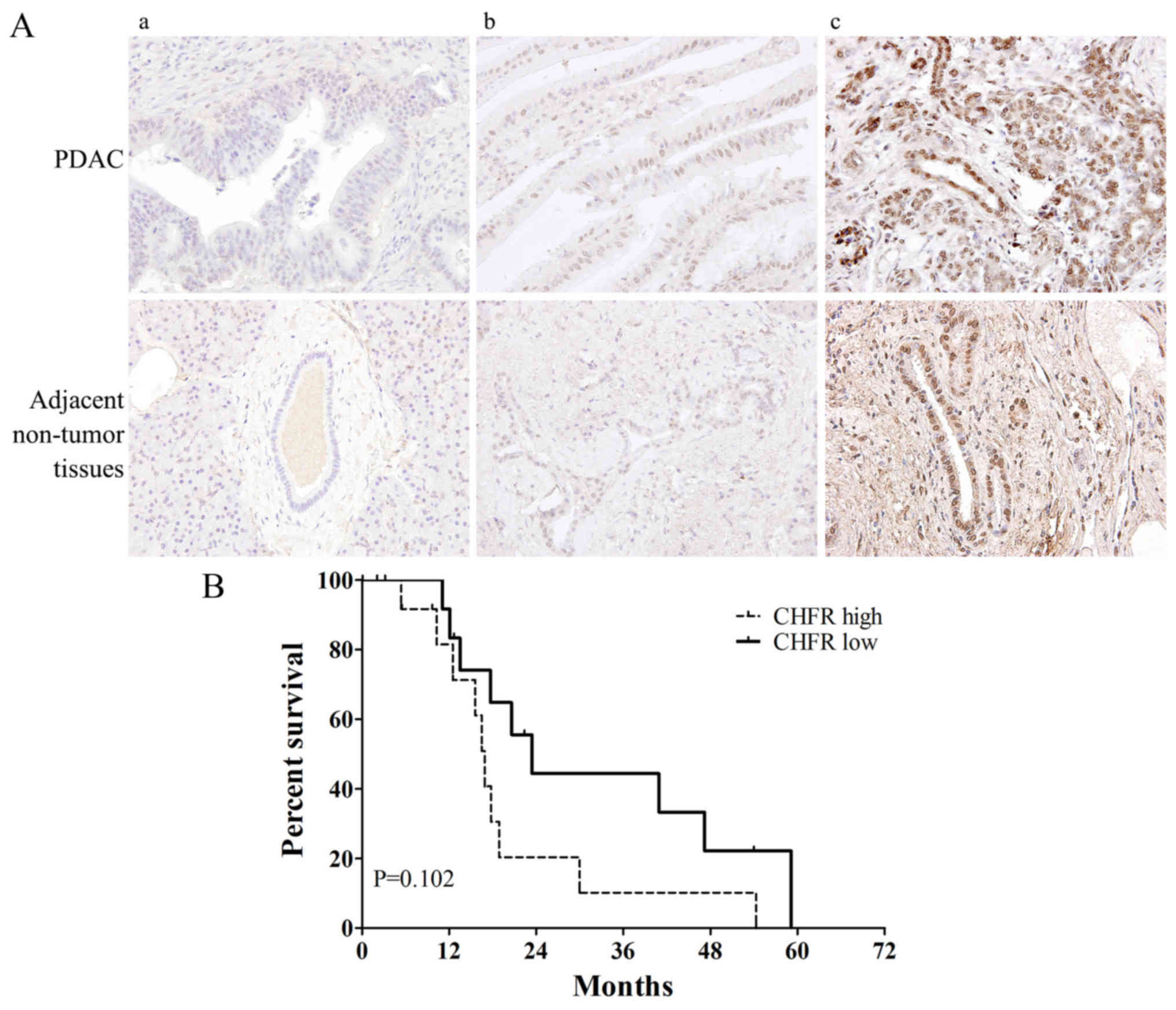
Table I. Fig. 3A shows the representative samples of
(panels a and b) CHFR ‘low’ and (panel c) ‘high’ expression in
tumor and adjacent non-tumor tissues. Among the total 27 PDAC tumor
samples, CHFR low expression was observed in 14 samples (51.9%) and
CHFR high expression was observed in 13 samples (48.1%). Compared
with adjacent non-tumor tissues, CHFR expression level was
significantly increased in tumor tissue samples (P<0.05). We
next analyzed the relationship between CHFR expression level and
clinicopathologic factors. Interestingly, there was a striking
significant correlation between high CHFR expression and earlier
T-stage (P=0.016) (Table I). Then we
performed Kaplan-Meier survival curve analysis for the patients
according to CHFR expression level. However, the log-rank test
demonstrated no statistically significant differences between
overall survival and CHFR expression levels (P=0.102; Fig. 3B). The 5-year survival rate was
0%.
 | Table I.Clinicopathological characteristics
of pancreatic ductal adenocarcinoma cases by CHFR
immunohistochemical expression. |
Table I.
Clinicopathological characteristics
of pancreatic ductal adenocarcinoma cases by CHFR
immunohistochemical expression.
| Clinicopathological
characteristics | Total N=27 (%) | CHFR low N=14
(%) | CHFR high N=13
(%) | P-value |
|---|
| Age (years) |
|
|
| 0.706 |
|
≥65 | 13 (48.1) | 6 (42.9) | 7 (53.8) |
|
|
<65 | 14 (51.9) | 8 (57.1) | 6 (46.2) |
|
| Sex |
|
|
| 1.00 |
|
Male | 13 (48.1) | 7 (50) | 6 (46.2) |
|
|
Female | 14 (51.9) | 7 (50) | 7 (53.8) |
|
| Site |
|
|
| 0.440 |
|
Head | 17 (63) | 10 (71.4) | 7 (53.8) |
|
| Body
and tail | 10 (37) | 4 (28.6) | 6 (46.2) |
|
| Maximum diameter of
tumor, cm |
|
|
| 1.00 |
|
>4 | 6 (22.2) | 3 (21.4) | 3 (23.1) |
|
| ≤4 | 21 (77.8) | 11 (78.6) | 10 (76.9) |
|
|
Differentiation |
|
|
| 0.098 |
|
Well | 2 (7.4) | 2 (14.3) | 0 (0) |
|
|
Moderately | 23 (85.2) | 10 (71.4) | 13 (100) |
|
|
Poorly | 2 (7.4) | 2 (14.3) | 0 (0) |
|
| T-stage |
|
|
| 0.016 |
|
T1-2 | 5 (18.5) | 0 (0) | 5 (38.5) |
|
|
T3-4 | 22 (81.5) | 14 (100) | 8 (61.5) |
|
| N-stage |
|
|
| 0.449 |
| N0 | 14 (51.9) | 6 (42.9) | 8 (61.5) |
|
| N1 | 13 (48.1) | 8 (57.1) | 5 (38.5) |
|
Discussion
CHFR was initially identified as a member of a small
family of proteins that contain the FHA and RING-finger domains.
This protein functions as a cell cycle checkpoint regulator, and
thus we named it CHFR. The FHA domain of CHFR was confirmed to be
responsible for its anti-proliferative effects (5), while the RING-finger domain has proven
to be related to the mitotic checkpoint function (19). E3 ubiquitin ligase activity of CHFR
and its autoubiquitination process also requires the RING-finger
domain (20,21). In addition, CHFR contains a unique PBZ
motif in the C-terminal cysteine-rich region, which can not be
found among other members of the FHA-RING protein family (22). However, the function of this motif
remains largely unknown.
It was the first time to discuss the relationship
between CHFR and pancreatic cancers. Our CCK-8 proliferation assays
showed that the proliferation of Capan-1_CHFR cells was
significantly inhibited compared to control cells. Flow cytometric
analysis revealed Capan-1 cells stably overexpressing CHFR showed a
significant increase in the G1 cell population, which may indicate
that CHFR inhibited cell proliferation by arresting cell cycle at
G1 phase. This is quite different from several studies that
reported the mitotic checkpoint function of CHFR (5,23,24). In these studies, in the presence of
mitotic stress, which was induced by adding microtubule poisons
such as nocodazole, colcemid, and taxanes, CHFR played a role as an
early mitotic checkpoint in antephase and in the arrest of cells at
the G2-to-M transition (22).
However, some studies presented other data. Oh YM (11) used Hela CHFR-overexpression cells to
show that CHFR may play a role in checkpoint function by cell cycle
arrest in G1 phase. In the study of Chf1 and Chf2 in S.
cerevisiae (25), the members of
FHA-RING protein family, which were considered to be orthologous to
CHFR protein, overexpression of either Chf protein led to growth
retardation and a large increase in G1 cells. Our results are in
agreement with the findings. This observation may mean that in the
absence of mitotic stress, in pancreatic cancer cells, CHFR
contributes to checkpoint function during interphase of the cell
cycle rather than mitosis phase. A few reports have also indicated
that CHFR may also control the cell cycle at anaphase of mitosis
(10). In our Transwell assays,
Capan-1_CHFR cells showed reduced migration and invasion abilities
compared to Capan-1_Ctrl cells. Although the cell numbers had
certain growth after cultured for 48 h, proliferation of the cells
was restricted by the limited bottom areas of the inserts.
Therefore, we believed that the exogenous CHFR should be the major
factor to reduce the migration and invasion abilities of
Capan-1_CHFR cells compared to control cells. Researches on the
mechanism of this field indicated that, CHFR is able to inhibit the
NF-κB signaling pathway and IL-8, which subsequently resulted in
decreased angiogenesis and cell migration (26,27). Thus,
we suggested that the inhibitory effects of CHFR on proliferative
and metastatic activities in pancreatic cancer cells were
consistent with previous findings in other kinds of cancers.
However, as the specific checkpoint function of CHFR has not been
clearly illustrated, it is possible it depends on cell specificity,
and/or the expression level of relevant factors, such as Plk1
(28), Cyclin B1 (29), and Aurora A (30). The purpose of our study is to reveal
the function of CHFR in pancreatic cancers. Our CCK-8 assay and
flow cytometric analysis indicated that CHFR inhibited cell
proliferation by arresting cell cycle at G1 phase. But the
mechanism of this phenomenon has not been clearly illustrated.
Inactive Cdk1/Cyclin B1 is a feature of the CHFR-dependent arrest
state (28,31,32). The
major problem is identifying the direct protein targets of CHFR.
Besides, a key protein that regulates the activity and
translocation of cyclin B1 to the nucleus to initiate mitosis is
Aurora A kinase (30). Therefore,
Aurora A has also been speculated to be a target for ubiquitination
by CHFR. The interaction of these factors and CHFR will be pursued
in our further studies.
Some tumors with CHFR deficiency show a better
chemotherapeutic response to taxanes, such as paclitaxel and
docetaxel (14,15), which may indicate an aberrant mitosis
checkpoint among these cancers. One probable cause of the
phenomenon might be that cancer cells with intact CHFR expression
possess an early mitotic checkpoint with normal function that
delays their entry into mitosis, and microtubular damage repair was
performed. In contrast, CHFR downregulation or deficient cells
would enter mitosis with a non-functional mitotic checkpoint and
thus undergo mitotic catastrophy, eventually resulting in
apoptosis.
In clinical practice, gemcitabine is currently the
first line chemotherapy for pancreatic adenocarcinoma, and taxanes,
such as docetaxel and paclitaxel, have not shown superior clinical
benefits compared with gemcitabine (33). For this reason, in addition to
docetaxel, we also included gemcitabine in our study. We did not
detect any difference in the basal apoptotic level of Capan-1_CHFR
and Capan-1_Ctrl cells. However, Capan-1_CHFR cells showed a lower
apoptosis rate upon treatment with the anti-microtubule agent
docetaxel compared with the control cells, which was not observed
with gemcitabine treatment. Therefore, we speculate that if the
patients of pancreatic cancer were divided into different groups
based on CHFR expression level, the CHFR low group might show a
higher sensitivity to taxanes, and thus benefit more. Combined with
the experimental results of proliferation, migration and invasion
assays, we find an interesting dual character of CHFR. On one hand,
overexpression of CHFR results in an inhibitory effect on multiple
tumor malignant biological behaviors; on the other hand, CHFR
downregulation might indicate a higher effective rate in response
to anti-microtubule agents. This phenomenon fundamentally related
to the cell cycle checkpoint function of CHFR. After radical
resections, some of the patients may receive chemotherapy. When the
formation of normal mitotic spindles was inhibited by docetaxel,
CHFR downregulation cells would enter mitosis with a non-functional
checkpoint, undergo mitotic catastrophy, and eventually resulting
in apoptosis. Thus, treatment with docetaxel may benefit the
patients with CHFR low expression. In vitro experiments, the
use of only one cell line was the limitation of our present study.
However, according to the finding of our research, as well as other
studies reported previously (11,25–27), we
firmly believed that our conclusion was reliable. In our further
study, we will try our best to improve the methods and break this
limitation.
On the basis of published investigations, promoter
CpG island methylation is the most common reason leading to CHFR
inactivation (34). Besides,
diminished CHFR expression also needed to be drew attention
(15). So far, earlier diagnosis and
treatment of PDAC is now still a grate problem for clinical
practice. In the fact, most of the patients who came to see a
doctor have already developed symptoms, which means the TNM stage
was often not too early. Since all the cases involved in our study
had undergone radical resection, most of them were at II or III
stage when received operations. Patients at stage IV have lost the
opportunities of radical surgery, while the detection of patients
at I stage is really difficult. For these reasons, T1-2 case number
was much less than T3-4. And it is the same case with the tumor
size and differentiation. Smaller tumor size often means higher
resection rate. Tumors with well differentiation and slow progress
usually difficult to be discovered at an early stage. Patients with
poorly differentiation tumor mostly lost the opportunity of
surgical therapy because of early metastasis and rapid progress.
Therefore, in our study, uneven distribution of the
clinicopathological data might be inevitable. In the report of CHFR
expression in malignant peripheral nerve sheath tumors in 2006
(18), the correlation between the
immunohistochemical expression of CHFR and the clinicopathologic
parameters was assessed by t-test, Chi-square test, and Fisher's
exact test. In the data of tumor depth, tumor size and AJCC stage,
there were big differences of case numbers in groups. In 2013,
Pillai et al (15) reported
CHFR protein expression in metastatic NSCLC. In the text,
differences between CHFR high vs. low expression were assessed
using ANOVA for numerical covariates and Chi-square test or
Fisher's exact test for categorical variables, where appropriate.
In the text, the sex distribution of cases was also very uneven (40
vs. 1). And it is the same case with the data of response. In 2014,
Gebauer et al (35) studied
the relationship between carcinoembryonic antigen-related cell
adhesion molecules (CEACAM) and pancreatic cancer. For explorative
statistical analysis of the individual patient groups, either a
two-sided Chi-square test or a Fisher's exact test was used. There
were also too much differences of case numbers between the groups
of N0 and N1 (119 vs. 18). All the researches above used Chi-square
test or Fisher's exact test for categorical variables. However, in
the parts of discussion of these texts, we have not found any
comment on the impact of uneven distribution of the data. In
addition, we thought when we analyzed the correlation between CHFR
expression and clinicopathologic features, what we really comparing
was the groups of CHFR high and low expression, but not the groups
of T1-2 and T3-4, or the tumor sizes. For these reasons, we
believed that uneven distribution of clinicopathological data was
ubiquitous in clinical researches, especially for malignant tumor,
and the methods of statistical analysis we chose was appropriate.
In PDAC patients, approximately half (51.9%) of PDAC samples showed
low expression level of CHFR in our study. Furthermore, we found a
correlation between CHFR expression and T-stage, suggesting a
certain connection between CHFR gene expression level and invasion
ability of the tumors. This is consistent with our in vitro
observations in invasion assays. Nevertheless, when compared with
adjacent non-tumor tissues, we were surprised to find CHFR
expression level dramatically increased. This result is very
different from some previous reports in other tumors (18,36,37). In
considering possible reasons, we speculate that single-nucleotide
polymorphisms (SNPs) of CHFR may play a role. In the initial report
of CHFR in 2000 (5), the authors
examined five cancer cell lines and found that the U2OS cell line
contains a variation in the CHFR gene in the non-coding strand.
This variation could be a mutation, because it occurred within a CG
dinucleotide, which is a mutagenesis hot-spot. Further experiments
confirmed that this variation led to the loss of partial functions
of CHFR, but had no effect on its protein expression. Although few
mutations of the CHFR gene have been detected in human tumor cells,
several SNPs have been reported (12,31,38).
Therefore, we speculate in pancreatic cancer tissues, there might
exist some SNPs, which affect partial function of CHFR, but do not
affect the expression level of CHFR protein. Finally, although our
survival analysis demonstrated no relationship between CHFR
expression level and overall survival, as shown in Fig. 3B, patients with CHFR high expression
tended to show a poorer survival rate compared to CHFR-low
expression patients (although P=0.102). We think there may be two
possibilities for this phenomenon. Firstly, after radical
resections, some of the patients might receive chemotherapy. In the
chemotherapy regimens which included docetaxel, CHFR expression
level might be able to influence the treatment effects, and affect
the patients' prognosis. Secondly, as mentioned previously, SNPs or
mutations of CHFR may exist in pancreatic cancers, which caused the
loss of partial functions of the gene, and even became an
unbeneficial factor of the patients' outcome. However, there has
not been a consistent conclusion in this field. In metastatic NSCLC
(16) diminished CHFR expression
predicted a better prognosis, but CHFR inactivation in stage II
colorectal cancer (39) and acute
myeloid leukemia (40) were
associated with an adverse outcome. In our study, although a
significant association between CHFR expression and prognosis of
PDAC patients was not observed, further studies with a larger
sample size and more clinicopathological data are required to
reveal deeper relationship between CHFR and PDAC.
In conclusion, our data show that CHFR regulates the
cell cycle of pancreatic cancer cells by G1 phase arrest and it
also functions as an important tumor suppressor in vitro.
Nevertheless, CHFR overexpression is not a favorable factor in
apoptosis induced by microtubule inhibitors. In PDAC patients,
aberrant high expression of CHFR in tumor tissues compared with
adjacent non-tumor tissues may not play its role effectively. CHFR
expression level was significantly associated with the pathological
stage; however, it is insufficient to function as a biomarker in
prognosis. In our follow-up study, we will do more research on the
mechanisms of CHFR in pancreatic cancer and the interaction of
relevant factors. In addition, in vivo experiments are
necessary to expand our current findings.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81272756) and Beijing Natural
Science Foundation (no. 7162076). The authors would like to
acknowledge L.H. Teng for writing assistance, and Y.P. Zhang for
proof reading the article.
References
|
1
|
Ilic M and Ilic I: Epidemiology of
pancreatic cancer: World. J Gastroenterol. 22:9694–9705. 2016.
|
|
2
|
Pannala R, Leirness JB, Bamlet WR, Basu A,
Petersen GM and Chari ST: Prevalence and clinical profile of
pancreatic cancer-associated diabetes mellitus. Gastroenterology.
134:981–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenhalf W, Grocock C, Harcus M and
Neoptolemos J: Screening of high-risk families for pancreatic
cancer. Pancreatology. 9:215–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shin EJ and Canto MI: Pancreatic cancer
screening. Gastroenterol Clin North Am. 41:143–157. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scolnick DM and Halazonetis TD: Chfr
defines a mitotic stress checkpoint that delays entry into
metaphase. Nature. 406:430–435. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hofmann K and Bucher P: The FHA domain: A
putative nuclear signalling domain found in protein kinases and
transcription factors. Trends Biochem Sci. 20:347–349. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lovering R, Hanson IM, Borden KL, Martin
S, O'Reilly NJ, Evan GI, Rahman D, Pappin DJ, Trowsdale J and
Freemont PS: Identification and preliminary characterization of a
protein motif related to the zinc finger. Proc Natl Acad Sci USA.
90:pp. 2112–2116. 1993, View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Borden KL, Boddy MN, Lally J, O'Reilly NJ,
Martin S, Howe K, Solomon E and Freemont PS: The solution structure
of the RING finger domain from the acute promyelocytic leukaemia
proto-oncoprotein PML. EMBO J. 14:1532–1541. 1995.PubMed/NCBI
|
|
9
|
Durocher D and Jackson SP: The FHA domain.
FEBS Lett. 513:58–66. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu X, Minter-Dykhouse K, Malureanu L, Zhao
WM, Zhang D, Merkle CJ, Ward IM, Saya H, Fang G, van Deursen J and
Chen J: Chfr is required for tumor suppression and aurora a
regulation. Nat Genet. 37:401–406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oh YM, Kwon YE, Kim JM, Bae SJ, Lee BK,
Yoo SJ, Chung CH, Deshaies RJ and Seol JH: Chfr is linked to tumour
metastasis through the downregulation of HDAC1. Nat Cell Biol.
11:295–302. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bertholon J, Wang Q, Falette N, Verny C,
Auclair J, Chassot C, Navarro C, Saurin JC and Puisieux A: Chfr
inactivation is not associated to chromosomal instability in colon
cancers. Oncogene. 22:8956–8960. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida K, Hamai Y, Suzuki T, Sanada Y,
Oue N and Yasui W: DNA methylation of CHFR is not a predictor of
the response to docetaxel and paclitaxel in advanced and recurrent
gastric cancer. Anticancer Res. 26:49–54. 2006.PubMed/NCBI
|
|
14
|
Yanokura M, Banno K, Kawaguchi M, Hirao N,
Hirasawa A, Susumu N, Tsukazaki K and Aoki D: Relationship of
aberrant DNA hypermethylation of CHFR with sensitivity to taxanes
in endometrial cancer. Oncol Rep. 17:41–48. 2007.PubMed/NCBI
|
|
15
|
Pillai RN, Brodie SA, Sica GL, Shaojin Y,
Li G, Nickleach DC, Yuan L, Varma VA, Bonta D, Herman JG, et al:
CHFR protein expression predicts outcomes to taxane-based first
line therapy in metastatic NSCLC. Clin Cancer Res. 19:1603–1611.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ketefian S: Ethical considerations in
research. Focus on vulnerable groups. Invest Educ Enferm.
33:164–172. 2015.PubMed/NCBI
|
|
18
|
Kobayashi C, Oda Y, Takahira T, Izumi T,
Kawaguchi K, Yamamoto H, Tamiya S, Yamada T, Iwamoto Y and
Tsuneyoshi M: Aberrant expression of CHFR in malignant peripheral
nerve sheath tumors. Mod Pathol. 19:524–532. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chin CF and Yeong FM: Safeguarding entry
into mitosis: The antephase checkpoint. Mol Cell Biol. 30:22–32.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bothos J, Summers MK, Venere M, Scolnick
DM and Halazonetis TD: The Chfr mitotic checkpoint protein
functions with Ubc13-Mms2 to form Lys63-linked polyubiquitin
chains. Oncogene. 22:7101–7107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chaturvedi P, Sudakin V, Bobiak ML, Fisher
PW, Mattern MR, Jablonski SA, Hurle MR, Zhu Y, Yen TJ and Zhou BB:
Chfr regulates a mitotic stress pathway through its RING-finger
domain with ubiquitin ligase activity. Cancer Res. 62:1797–1801.
2002.PubMed/NCBI
|
|
22
|
Brooks L III, Heimsath EG Jr, Loring GL
and Brenner C: FHA-RING ubiquitin ligases in cell division cycle
control. Cell Mol Life Sci. 65:3458–3466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Summers MK, Bothos J and Halazonetis TD:
The CHFR mitotic checkpoint protein delays cell cycle progression
by excluding Cyclin B1 from the nucleus. Oncogene. 24:2589–2598.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ogi K, Toyota M, Mita H, Satoh A, Kashima
L, Sasaki Y, Suzuki H, Akino K, Nishikawa N, Noguchi M, et al:
Small interfering RNA-induced CHFR silencing sensitizes oral
squamous cell cancer cells to microtubule inhibitors. Cancer Biol
Ther. 4:773–780. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bieganowski P, Shilinski K, Tsichlis PN
and Brenner C: Cdc123 and checkpoint forkhead associated with RING
proteins control the cell cycle by controlling eIF2gamma abundance.
J Biol Chem. 279:44656–44666. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kashima L, Toyota M, Mita H, Suzuki H,
Idogawa M, Ogi K, Sasaki Y and Tokino T: CHFR, a potential tumor
suppressor, downregulates interleukin-8 through the inhibition of
NF-kappaB. Oncogene. 28:2643–2653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee KH, Bae SH, Lee JL, Hyun MS, Kim SH,
Song SK and Kim HS: Relationship between urokinase-type plasminogen
receptor, interleukin-8 gene expression and clinicopathological
features in gastric cancer. Oncology. 66:210–217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang D, Chen J, Wong J and Fang G: The
checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and
inhibits Cdc2 at the G2 to M transition. J Cell Biol. 156:249–259.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hagting A, Jackman M, Simpson K and Pines
J: Translocation of cyclin B1 to the nucleus at prophase requires a
phosphorylation-dependent nuclear import signal. Curr Biol.
9:680–689. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hirota T, Kunitoku N, Sasayama T, Marumoto
T, Zhang D, Nitta M, Hatakeyama K and Saya H: Aurora A and an
interacting activator, the LIM protein Ajuba, are required for
mitotic commitment in human cells. Cell. 114:585–598. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsusaka T and Pines J: Chfr acts with
the p38 stress kinases to block entry to mitosis in mammalian
cells. J Cell Biol. 166:507–516. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Erson AE and Petty EM: CHFR-associated
early G2/M checkpoint defects in breast cancer cells. Mol Carcinog.
39:26–33. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Awasthi N, Zhang C, Schwarz AM, Hinz S,
Schwarz MA and Schwarz RE: Enhancement of nab-paclitaxel antitumor
activity through addition of multitargeting antiangiogenic agents
in experimental pancreatic cancer. Mol Cancer Ther. 13:1032–1043.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toyota M, Sasaki Y, Satoh A, Ogi K,
Kikuchi T, Suzuki H, Mita H, Tanaka N, Itoh F, Issa JP, et al:
Epigenetic inactivation of CHFR in human tumors. Proc Natl Acad Sci
USA. 100:pp. 7818–7823. 2003, View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gebauer F, Wicklein D, Horst J, Sundermann
P, Maar H, Streichert T, Tachezy M, Izbicki JR, Bockhorn M and
Schumacher U: Carcinoembryonic antigen-related cell adhesion
molecules (CEACAM) 1, 5 and 6 as biomarkers in pancreatic cancer.
PLoS One. 9:e1130232014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Privette LM, González ME, Ding L, Kleer CG
and Petty EM: Altered expression of the early mitotic checkpoint
protein, CHFR, in breast cancers: Implications for tumor
suppression. Cancer Res. 67:6064–6074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Soutto M, Peng D, Razvi M, Ruemmele P,
Hartmann A, Roessner A, Schneider-Stock R and El-Rifai W:
Epigenetic and genetic silencing of CHFR in esophageal
adenocarcinomas. Cancer. 116:4033–4042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mariatos G, Bothos J, Zacharatos P,
Summers MK, Scolnick DM, Kittas C, Halazonetis TD and Gorgoulis VG:
Inactivating mutations targeting the chfr mitotic checkpoint gene
in human lung cancer. Cancer Res. 63:7185–7189. 2003.PubMed/NCBI
|
|
39
|
Cleven AH, Derks S, Draht MX, Smits KM,
Melotte V, Van Neste L, Tournier B, Jooste V, Chapusot C,
Weijenberg MP, et al: CHFR promoter methylation indicates poor
prognosis in stage II microsatellite stable colorectal cancer. Clin
Cancer Res. 20:3261–3271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao L, Liu F, Zhang H, Sun J and Ma Y:
CHFR hypermethylation, a frequent event in acute myeloid leukemia,
is independently associated with an adverse outcome. Genes
Chromosomes Cancer. 55:158–168. 2016. View Article : Google Scholar : PubMed/NCBI
|