Introduction
Chloroquine (CQ) is an effective and well-tolerated
drug for the treatment of malaria (1), and is also a useful agent for the
treatment of systemic lupus erythematosus and rheumatoid arthritis
due to its anti-inflammatory properties (2,3). Recently,
a number of studies demonstrated that CQ exhibits antitumor
activity in different types of cancer cells, including human liver
cancer, gallbladder carcinoma, breast cancer and colon cancer cells
(4–6),
and that it is less toxic to non-tumor cells than standard
chemotherapy agents (7). However,
whether or not CQ exerts the same effect on acute promyelocytic
leukemia (APL) cells remains unknown.
Autophagy is an intracellular degradation process
that eliminates damaged organelles or proteins through lysosomal
activity (8). Autophagy serves an
important role in homeostasis under conditions of cellular stress,
including nutrient limitation, hypoxia and chemotherapy (9). A number of diseases, including cancer,
are associated with dysregulated autophagy processes (10–12).
However, the role of autophagy in tumors is very complex and is
suggested to have both pro-death and pro-survival effects in
different types of cancer (13). As
tumor cells proliferate rapidly, the relative deficiency of
nutrients and oxygen shifts the metabolic pathway of cancer cells
from aerobic oxidation towards glycolysis (14), and autophagy may serve as an
alternative source of energy for cancer cell survival (15). However, autophagy also leads to
programmed cell death, which may induce anticancer effects
(16). A previous study demonstrated
that high basal autophagy levels were a potential mechanism of
resistance to cell death in APL cell lines (17), indicating that autophagy promotes
survival and that autophagy inhibition may serve an antitumor role
in APL cells.
CQ is an inhibitor of late-stage autophagy, which
takes effect after autophagosomes have already formed (18,19). Based
on the results of the aforementioned studies, we hypothesized that
CQ may inhibit autophagy and exert anticancer effects on APL NB4
cells.
Arsenic trioxide (ATO) is an anticancer drug used
for the treatment of APL. However, ATO also induces autophagy in
APL cells, which may weaken its antitumor activity (20). The addition of CQ may compensate for
this defect to enhance the effect of ATO. In the present study, the
effect of CQ on the growth, apoptosis and cell cycle distribution
of NB4 cells was investigated. In addition, the combined effect of
CQ and ATO on the growth of NB4 cells was also determined.
Materials and methods
Cell lines, cell culture and
reagents
NB4 cells were obtained from the Third Affiliated
Hospital of Sun Yat-sen University (Guangzhou, China). The cells
were maintained in RPMI-1640 medium containing 10% fetal bovine
serum (both from Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) in a 37°C incubator containing 5% CO2. CQ and
ATO were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany). CQ was prepared as a 0.1 M stock solution in dimethyl
sulfoxide. ATO stock solution was made at a concentration of 1 mM
with normal saline. Stock solutions were diluted to working
concentrations for use.
Cell proliferation assay
The cytotoxicity of CQ and ATO on NB4 cells was
determined using an MTS assay (Promega Corporation, Madison, WI,
USA), as previously described (21).
A total of 104 NB4 cells were cultured in each well of a
96-well plate and were treated with various concentrations of CQ
for 48 or 72 h. Than 20 µl MTS was added and the absorbance at 490
nm was measured using a microplate reader (Thermo Fisher
Scientific, Inc.) after a 3-h incubation. The inhibition rate of
cell proliferation was calculated using the following formula: [1 -
(OD of experimental well/OD of control)] × 100%.
The potential synergy between CQ and ATO was
evaluated according to the following formula (21):
Q=E(CQ+ATO)E(CQ)+E(ATO)–E(CQ)*E(ATO)
Where E(CQ+ATO),
E(CQ) and E(ATO) are the
inhibition rates of the combination treatment, CQ monotherapy and
ATO monotherapy, respectively. Q<0.85 indicates antagonism,
0.85≤Q≤1.15 indicates additive effects and Q>1.15 indicates
synergy. Briefly, 1×104 NB4 cells were seeded into each
well of a 96-well plate and were treated with various
concentrations of CQ and ATO. The combined effect was evaluated by
calculating the Q values of each treatment at 48 and 72 h.
Apoptosis assay
The rate of apoptosis in NB4 cells was determined
using the Annexin V-fluorescein isothiocyanate (FITC)/propidium
iodide (PI) apoptosis kit (BD Biosciences, Franklin Lakes, NJ, USA)
according to the manufacturer's instructions. Following a 48 h
incubation with CQ, the cells were harvested and washed twice with
PBS, prior to being incubated with 5 µl Annexin V-FITC and 5 µl PI
for 10 min in the dark at room temperature. Analysis of the cell
apoptotic rate was performed using a flow cytometer (FC 500;
Beckman Coulter, Inc., Brea, CA, USA) with CellQuest Pro software
program (version 5.1; BD Biosciences). The early apoptotic (Annexin
V-FITC-positive, PI-negative) and late apoptotic (Annexin
V-FITC-positive, PI-positive) cells were quantified.
Cell cycle analysis
Following a 48 h incubation with CQ, cells were
harvested, washed twice with cold PBS and fixed in cold 70% ethanol
at 4°C for 24 h. The cells were then centrifuged at a speed of
1,000 × g at 4°C for 5 min. The fixed cells were resuspended in PBS
containing 50 mg/ml PI, 100 mg/ml RNase and 0.2% Triton X-100 (both
from Sigma-Aldrich; Merck KGaA) at 37°C for 15 min. The cell cycle
profiles of the treated cells were subsequently analyzed using a
flow cytometer (Beckman Coulter, Inc.). The percentage of cells in
each phase of the cell cycle was determined using Modfit LT
software (version 3.2; Verity Software House, Inc., Topsham, ME,
USA).
Western blot analysis
After 48 h of incubation with CQ, ATO or CQ+ATO, the
cells were collected and the total cell lysates were prepared with
cell lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China). Western blot analysis was performed as previously described
(21), using rabbit monoclonal
antibodies against B-cell lymphoma 2 (Bcl-2; cat. no. 2872S),
Bcl-2-like protein X (Bax; cat. no. 5023S), Bcl-2-like protein 11
(Bim; cat. no. 2933S), myeloid cell leukemia 1 (Mcl-1; cat. no.
39224S), cleaved caspase-9 (cat. no. 20750S), cleaved caspase-3
(cat. no. 9661S), cell division cycle 25A (CDC25A) (cat. no.
3652S), cyclin-dependent kinase 2 (CDK2) (cat. no. 2546S), cyclin
A, light chain 3B (LC3B; cat. no. 2775S), p62 (cat. no. 39749S) and
GAPDH (cat. no. 2118S) (all diluted to 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), followed by incubation with a
IRDye® 680RD conjugated goat anti-rabbit IgG secondary
antibody (1:20,000, cat. no. P/N 926–68071; LI-COR Biosciences,
Lincoln, NE, USA) at room temperature for 2 h in the dark. The
bands were then detected using the Odyssey® CLx Infrared
Imaging System (LI-COR Biosciences).
Statistical analysis
The data are presented as the mean ± standard
deviation. Comparison between two groups was performed using a
two-tailed Student's t-test, and pairwise multiple comparisons
among the groups were performed using a one-way analysis of
variance test together with the Student-Newman-Keuls method with
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
CQ inhibits the proliferation of NB4
cells
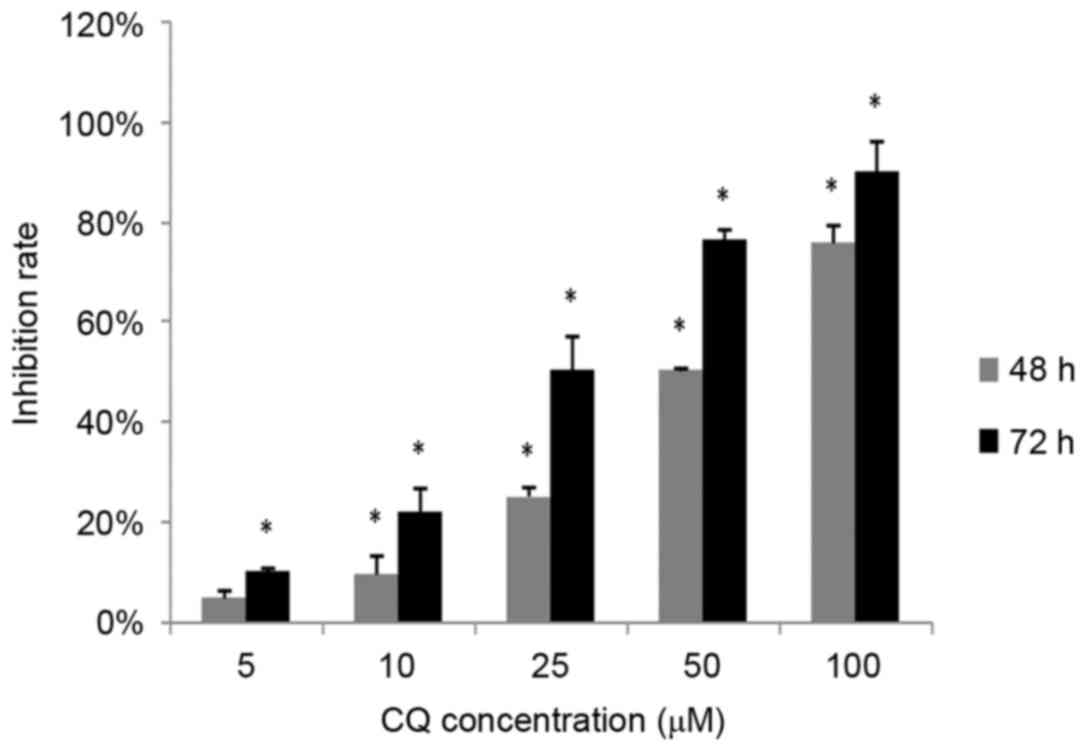
The inhibitory effect of CQ on NB4 cell
proliferation was measured using an MTS assay. NB4 cells were
exposed to 0, 5, 10, 25, 50 and 100 µM CQ for 48 and 72 h,
respectively, followed by an MTS assay. As demonstrated in Fig. 1, the proliferation of NB4 cells
decreased significantly with CQ treatment in a time- and
dose-dependent manner (P<0.05). The highest inhibition rates
observed were 75.88±3.5% at 48 h and 90.32±5.89% at 72 h at a
concentration of 100 µM CQ.
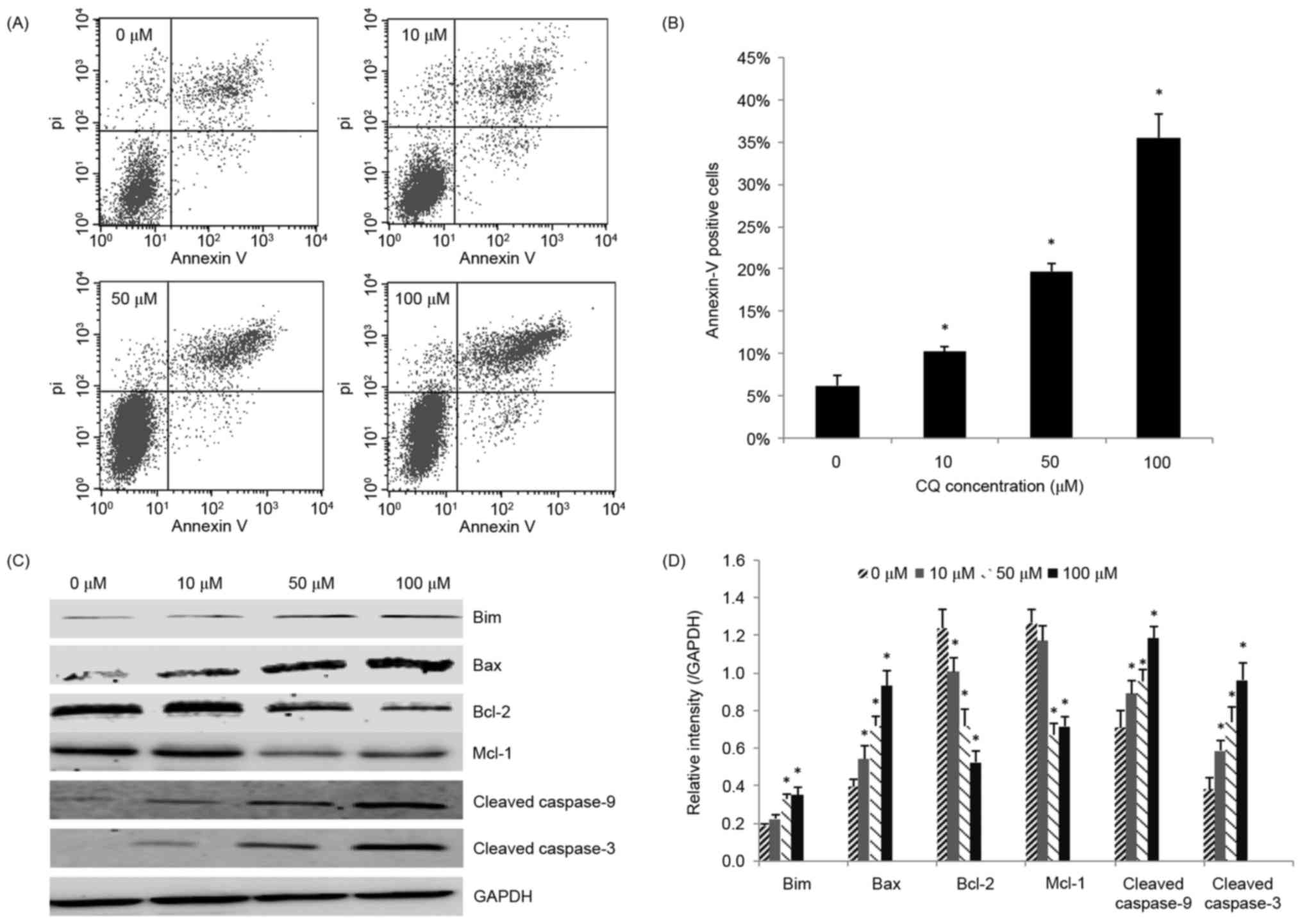
CQ induces apoptosis in NB4 cells
Next, the role of apoptosis in the antitumor
activity of CQ was assessed. NB4 cells that had been exposed to CQ
for 48 h were collected for the apoptosis assay. As demonstrated in
Fig. 2A and B, CQ treatment caused a
significant increase in the rate of apoptosis (P<0.05),
particularly in the rate of late apoptosis, in a dose-dependent
manner. The highest proportion of apoptotic (Annexin V-positive)
cells was 35.51±2.79% at 100 µM.
To further explore the underlying mechanisms of
CQ-induced apoptosis, the effect of CQ on the expression of
apoptosis-related proteins was investigated. Increasing
concentrations of CQ upregulated the level of cleaved caspase-3,
thereby confirming CQ-induced apoptosis. The pro-apoptotic proteins
Bax, Bim and cleaved caspase-9 were significantly upregulated,
whereas the anti-apoptotic proteins Bcl-2 and Mcl-1 were
significantly downregulated in the CQ-treated groups in a
dose-dependent manner (P<0.05; Fig. 2C
and D).
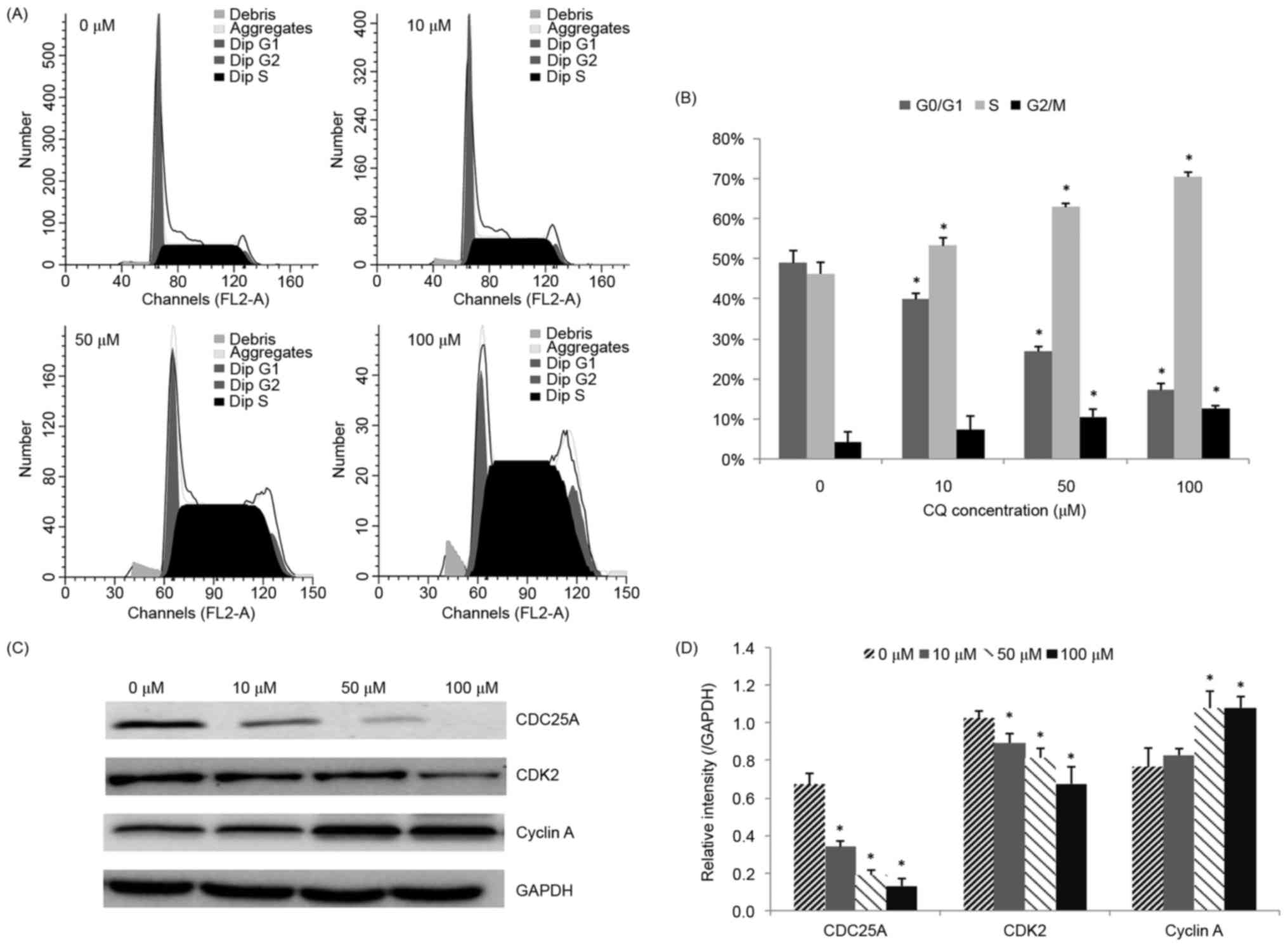
CQ induces NB4 cell S phase
arrest
The influence of CQ on cell cycle progression was
examined in order to illustrate another potential mechanism of its
anti-proliferative activity. The effects of various concentrations
of CQ on the cell cycle distribution in NB4 cells were determined.
As demonstrated in Fig. 3A and B, the
population of cells in the S phase was significantly increased
(P<0.05), whereas the number of cells in the
G0/G1 phase was reduced, following CQ
treatment, in a concentration-dependent manner. Treatment of NB4
cells with 100 µM CQ resulted in a significant increase in the
percentage of cells in the S phase, from 46.20±2.86% (at 0 µM) to
70.43±1.17% (P<0.05).
Since CQ was observed to arrest NB4 cells in the S
phase, western blot analysis was subsequently performed in order to
evaluate its effect on S phase cell cycle regulators, including
CDC25A, CDK2 and cyclin A. The results presented in Fig. 3C and D demonstrate that CDC25A and
CDK2 were downregulated, whereas cyclin A was upregulated,
following CQ treatment, in a dose-dependent manner.
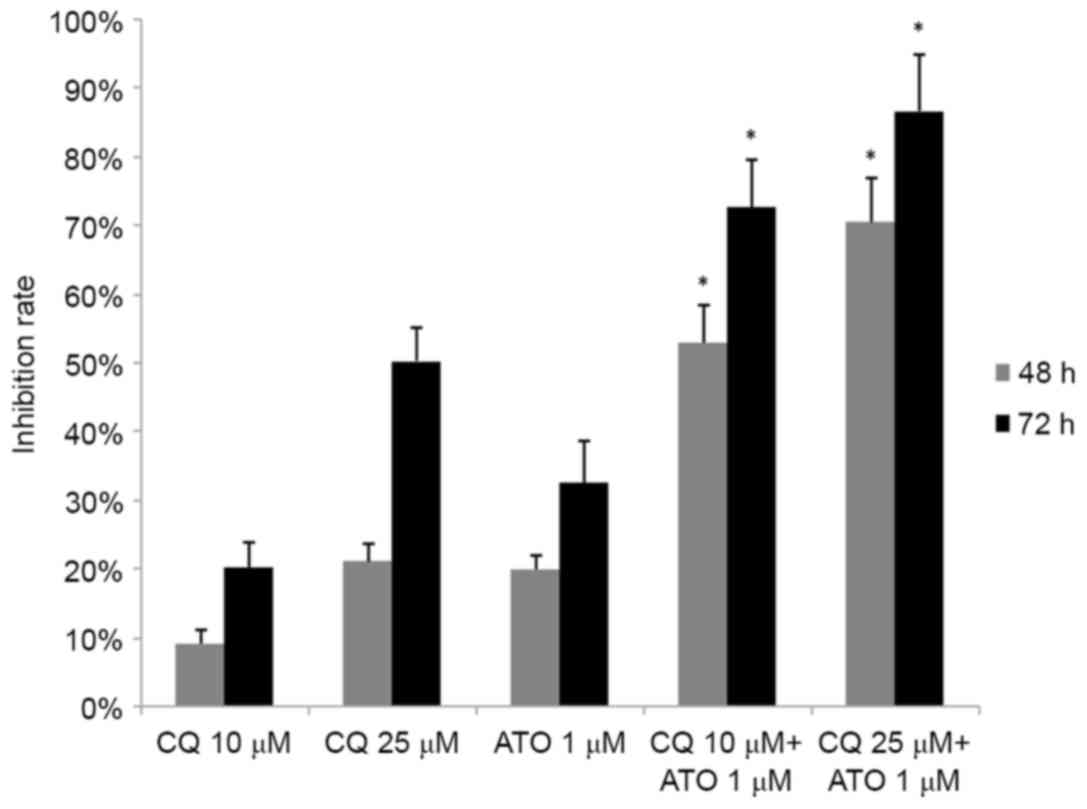
CQ synergizes with ATO in inhibiting
the growth of NB4 cells
As ATO is a typical drug for the treatment of APL,
the combined effect of CQ and ATO was subsequently examined. NB4
cells were exposed to 1 µM ATO and 10 or 25 µM CQ simultaneously
for 48 or 72 h. Jin's modified Burgi's formula was used to evaluate
the combined effect of ATO and CQ. There was a significant increase
in the inhibition rate in the combined treatment groups (P<0.05;
Fig. 4). As demonstrated in Fig. 4, the Q values were all >1.15 at 48
and 72 h, indicating a synergistic effect between the two drugs in
inhibiting cell proliferation.
CQ regulates ATO-induced
autophagy
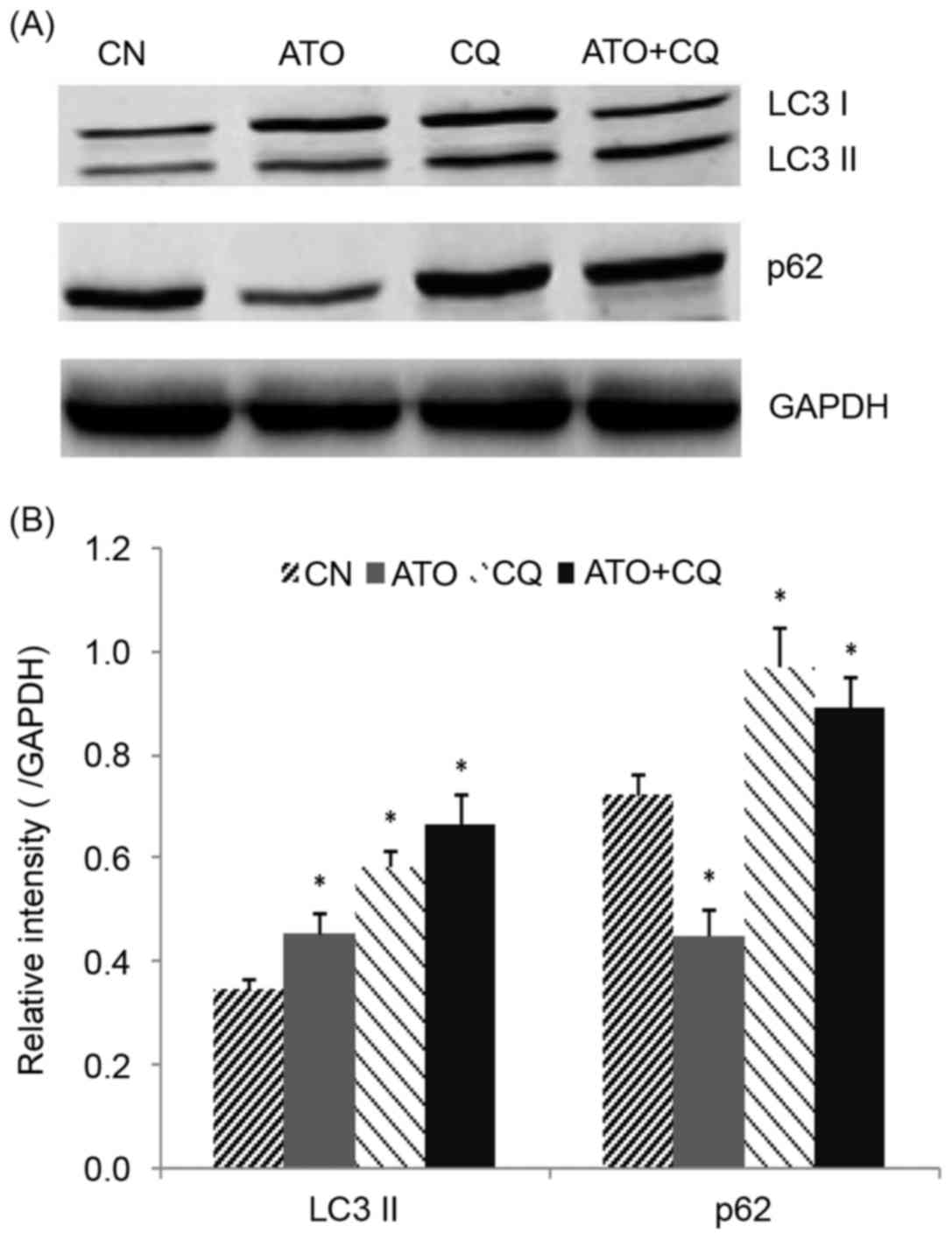
LC3 and p62 were detected as markers for autophagy
using western blot analysis. As demonstrated in Fig. 5A and B, the level of LC3-II was
increased, and p62 was decreased, following treatment with ATO
alone, indicating that ATO induced autophagy in NB4 cells. CQ
upregulated LC3-II and p62 levels, confirming that CQ inhibited
autophagy in its later phase. A significant increase in LC3-II and
a moderate increase in p62 were observed in the combined treatment
group (P<0.05), indicating that CQ inhibited the ATO-induced
autophagy in NB4 cells.
Discussion
CQ exerts antitumor effects on a variety of cancer
cells; however, whether it has the same effect on the NB4 APL cell
line remains unknown. Therefore, the present study was designed to
explore the effect of CQ on NB4 cells. The present study indicated
that CQ has a potent antitumor effect and functions synergistically
with ATO in NB4 cells.
Previous studies have demonstrated that the
potential mechanisms for the antitumor effects of CQ may include
the inhibition of autophagy (22),
the induction of apoptosis (23,24), the
elimination of cancer stem cells (25), the normalization of vasculature
(26), the enhancement of the immune
response (27) and the arrest of cell
cycle progression (22). The focus of
the present study was primarily on the apoptosis, cell cycle
distribution and autophagy induced by CQ treatment. CQ treatment
was revealed to induce apoptosis, to upregulate the pro-apoptosis
proteins Bax and Bim, and to downregulate the anti-apoptosis
proteins Bcl-2 and Mcl-1 in a dose-dependent manner. Furthermore,
the expression of cleaved caspase-9, an important component of the
intrinsic mitochondrial pathway, was also increased. These data are
consistent with the results of previous studies (24,28). Based
on these data, we hypothesized that the mitochondrial apoptotic
pathway and members of the Bcl-2 family are involved in the
CQ-induced apoptosis of NB4 cells.
CQ has been reported to induce cell cycle
alterations in cancer cells, but the specific results have varied
for different types of cancer; CQ has been demonstrated to cause
G2/M cell cycle arrest in breast cancer cells (29), G0/G1 cell cycle
arrest in liver cancer cells (28), S
phase arrest in choriocarcinoma cells (30) and no obvious change in colon cancer
cells (22). The inconsistency in
cell cycle alterations may be due to the intrinsic differences
between the tumor cell lines. In the present study, CQ treatment
induced a significant increase in the number of cells in the S
phase and a decrease in the number of cells in the
G0/G1 phase in a concentration-dependent
manner. S phase arrest accompanied with a decrease in the number of
cells in G0/G1 phase following treatment with
antitumor drugs has also been reported in a number of other studies
(31–33), and does not appear to affect the
antitumor effect of these drugs. One potential explanation for this
is that the inhibition of cell proliferation is associated with
cell cycle arrest, but the phase at which the cells are arrested
depends on the anticancer drugs and the antitumor cells involved.
Tumor cell proliferation relies on the progression from one cell
cycle phase to the next; on the introduction of antitumor drugs,
the cell cycle progression is disrupted. In the present study, a
relatively high number of cells had progressed from the
G0/G1 phase to the S phase, whereas
relatively fewer cells had progressed from the S phase to the
G2/M phase, thereby indicating that the cell cycle was
blocked at the S phase. The normal growth process was disrupted and
cell proliferation was partly suppressed.
Cell cycle regulatory proteins, including cyclins
and CDKs, are considered to serve an important role in cell cycle
progression (34). Cyclin A, CDK2 and
CDC25A are critical factors associated with the S phase of the cell
cycle (35). CDC25A may activate
CDK2, which in turn leads to the activation of the cyclin-CDK
complex and causes cell cycle progression (36,37). The
present study demonstrated that CQ reduced the expression of CDC25A
and CDK2, and increased the expression of cyclin A. It was deduced
that CQ downregulated CDC25A, suppressing the activation of CDK2,
which therefore decreased the formation of the cyclin A-CDK2
complex. The reduced formation of the cyclin-CDK complex arrested
the NB4 cells in the S phase. Furthermore, the increased expression
of cyclin A may result from the reduced formation of the cyclin-CDK
complex.
Despite being a frequently used anticancer agent in
APL, ATO may also induce unwanted or fatal side effects, including
differentiation syndrome, QT interval prolongation, hepatotoxicity,
the incidence of secondary malignancies and damage to the nervous
system (38,39). In the present study, CQ functioned
synergistically with ATO, indicating that a lower dose of ATO is
required to achieve the same curative effect when combined with CQ,
therefore reducing the risks associated with ATO. As mentioned
previously, autophagy may promote survival in NB4 cells (17). CQ, a widely used autophagy inhibitor,
was also confirmed to effectively inhibit the late phase of
autophagy in the present study, which may be a possible explanation
for the antitumor effect of CQ on NB4 cells. A previous study
demonstrated that ATO induces autophagy in NB4 cells and that the
suppression of autophagy may enhance its effect (40). In the present study, the level of
autophagy induced by ATO was significantly reduced when used in
combination with CQ in NB4 cells, which may explain why CQ
functioned synergistically with ATO. Another potential reason for
this synergy is that the two drugs exhibit different effects on
cell cycle distribution, with CQ inducing S phase arrest, and ATO
inducing G2/M phase arrest (41).
Taken together, the results of the present study
demonstrated that CQ effectively suppressed the growth of NB4 cells
by inducing apoptosis, inducing S phase arrest and inhibiting
autophagy. Furthermore, CQ appears to function synergistically with
ATO, indicating that the combined use of CQ and ATO maybe a
promising approach for future APL therapy. However, the true
potential of this treatment option requires further
investigation.
Acknowledgements
The present study was funded by the Medical Science
and Technology Research Foundation of Guangdong Province (grant no.
A2016181) and the Natural Science Foundation of Guangdong Province
(grant no. 2017A030310337).
References
|
1
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe cancer
therapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Augustijns P, Geusens P and Verbeke N:
Chloroquine levels in blood during chronic treatment of patients
with rheumatoid arthritis. Eur J Clin Pharmacol. 42:429–433.
1992.PubMed/NCBI
|
|
3
|
Bezerra EL, Vilar MJ, da Trindade Neto PB
and Sato EI: Double-blind, randomized, controlled clinical trial of
clofazimine compared with chloroquine in patients with systemic
lupus erythematosus. Arthritis Rheum. 52:3073–3078. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang X, Tang J, Liang Y, Jin R and Cai X:
Suppression of autophagy by chloroquine sensitizes
5-fluorouracil-mediated cell death in gallbladder carcinoma cells.
Cell Biosci. 4:102014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li
C, Zhang P, Wei D, Li P, Jeong LS, et al: Synergistic inhibition of
autophagy and neddylation pathways as a novel therapeutic approach
for targeting liver cancer. Oncotarget. 6:9002–9017. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mei L, Chen Y, Wang Z, Wang J, Wan J, Yu
C, Liu X and Li W: Synergistic anti-tumour effects of tetrandrine
and chloroquine combination therapy in human cancer: A potential
antagonistic role for p21. Br J Pharmacol. 172:2232–2245. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rahim R and Strobl JS: Hydroxychloroquine,
chloroquine, and all-trans retinoic acid regulate growth, survival,
and histone acetylation in breast cancer cells. Anticancer Drugs.
20:736–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin NY, Beyer C, Giessl A, Kireva T,
Scholtysek C, Uderhardt S, Munoz LE, Dees C, Distler A, Wirtz S, et
al: Autophagy regulates TNFα-mediated joint destruction in
experimental arthritis. Ann Rheum Dis. 72:761–768. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan HX, Russell RC and Guan KL:
Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient
stress-induced autophagy. Autophagy. 9:1983–1995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu LL, Long ZJ, Wang LX, Zheng FM, Fang
ZG, Yan M, Xu DF, Chen JJ, Wang SW, Lin DJ and Liu Q: Inhibition of
mTOR pathway sensitizes acute myeloid leukemia cells to aurora
inhibitors by suppression of glycolytic metabolism. Mol Cancer Res.
11:1326–1336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiavarina B, Whitaker-Menezes D, Migneco
G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB,
Casimiro MC, Wang C, Pestell RG, et al: HIF1-alpha functions as a
tumor promoter in cancer associated fibroblasts, and as a tumor
suppressor in breast cancer cells: Autophagy drives
compartment-specific oncogenesis. Cell Cycle. 9:3534–3551. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morselli E, Galluzzi L, Kepp O, Mariño G,
Michaud M, Vitale I, Maiuri MC and Kroemer G: Oncosuppressive
functions of autophagy. Antioxid Redox Signal. 14:2251–2269. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gañán-Gómez I, Estañ-Omaña MC, Sancho P,
Aller P and Boyano-Adánez MC: Mechanisms of resistance to apoptosis
in the human acute promyelocytic leukemia cell line NB4. Ann
Hematol. 94:379–392. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tasdemir E, Galluzzi L, Maiuri MC, Criollo
A, Vitale I, Hangen E, Modjtahedi N and Kroemer G: Methods for
assessing autophagy and autophagic cell death. Methods Mol Biol.
445:29–76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fan Y, Chen M, Meng J, Yu L, Tu Y, Wan L,
Fang K and Zhu W: Arsenic trioxide and resveratrol show synergistic
anti-leukemia activity and neutralized cardiotoxicity. PLoS One.
9:e1058902014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu SS, Wang XP, Li XB, Liang JY, Liu LL,
Lu Y, Zhong XY and Chen YX: Zoledronic acid exerts antitumor
effects in NB4 acute promyelocytic leukemia cells by inducing
apoptosis and S phase arrest. Biomed Pharmacother. 68:1031–1036.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi JH, Yoon JS, Won YW, Park BB and Lee
YY: Chloroquine enhances the chemotherapeutic activity of
5-fluorouracil in a colon cancer cell line via cell cycle
alteration. APMIS. 120:597–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng Y, Zhao YL, Deng X, Yang S, Mao Y,
Li Z, Jiang P, Zhao X and Wei Y: Chloroquine inhibits colon cancer
cell growth in vitro and tumor growth in vivo via induction of
apoptosis. Cancer Invest. 27:286–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang PD, Zhao YL, Deng XQ, Mao YQ, Shi W,
Tang QQ, Li ZG, Zheng YZ, Yang SY and Wei YQ: Antitumor and
antimetastatic activities of chloroquine diphosphate in a murine
model of breast cancer. Biomed Pharmacother. 64:609–614. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Choi DS, Blanco E, Kim YS, Rodriguez AA,
Zhao H, Huang TH, Chen CL, Jin G, Landis MD, Burey LA, et al:
Chloroquine eliminates cancer stem cells through deregulation of
Jak2 and DNMT1. Stem Cells. 32:2309–2323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maes H, Kuchnio A, Peric A, Moens S, Nys
K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et
al: Tumor vessel normalization by chloroquine independent of
autophagy. Cancer Cell. 26:190–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ratikan JA, Sayre JW and Schaue D:
Chloroquine engages the immune system to eradicate irradiated
breast tumors in mice. Int J Radiat Oncol Biol Phys. 87:761–768.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu T, Li P, Luo Z, Chen X, Zhang J, Wang
C, Chen P and Dong Z: Chloroquine inhibits hepatocellular carcinoma
cell growth in vitro and in vivo. Oncol Rep. 35:43–49. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang PD, Zhao YL, Shi W, Deng XQ, Xie G,
Mao YQ, Li ZG, Zheng YZ, Yang SY and Wei YQ: Cell growth
inhibition, G2/M cell cycle arrest, and apoptosis induced by
chloroquine in human breast cancer cell line Bcap-37. Cell Physiol
Biochem. 22:431–440. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nilkaeo A, Bhuvanath S, Praputbut S and
Wisessombat S: Induction of cell cycle arrest and apoptosis in JAR
trophoblast by antimalarial drugs. Biomed Res. 27:131–137. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Romani AA, Desenzani S, Morganti MM, La
Monica S, Borghetti AF and Soliani P: Zoledronic acid determines
S-phase arrest but fails to induce apoptosis in cholangiocarcinoma
cells. Biochem Pharmacol. 78:133–141. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ohnuki H, Izumi K, Terada M, Saito T, Kato
H, Suzuki A, Kawano Y, Nozawa-Inoue K, Takagi R and Maeda T:
Zoledronic acid induces S-phase arrest via a DNA damage response in
normal human oral keratinocytes. Arch Oral Biol. 57:906–917. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tan H, Gao S, Zhuang Y, Dong Y, Guan WH,
Zhang K, Xu J and Cui J: R-Phycoerythrin induces SGC-7901 apoptosis
by arresting cell cycle at S phase. Mar Drugs. 14:E1662016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hsu YL, Uen YH, Chen Y, Liang HL and Kuo
PL: Tricetin, a dietary flavonoid, inhibits proliferation of human
breast adenocarcinoma mcf-7 cells by blocking cell cycle
progression and inducing apoptosis. J Agric Food Chem.
57:8688–8695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
George Rosenker KM, Paquette WD, Johnston
PA, Sharlow ER, Vogt A, Bakan A, Lazo JS and Wipf P: Synthesis and
biological evaluation of 3-aminoisoquinolin-1(2H)-one based
inhibitors of the dual-specificity phosphatase Cdc25B. Bioorg Med
Chem. 23:2810–2818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tilaoui M, Mouse HA, Jaafari A and Zyad A:
Differential effect of artemisinin against cancer cell lines. Nat
Prod Bioprospect. 4:189–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Au WY, Kumana CR, Lam CW, Cheng VC, Shek
TW, Chan EY, Liu R and Kwong YL: Solid tumors subsequent to arsenic
trioxide treatment for acute promyelocytic leukemia. Leuk Res.
31:105–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ghavamzadeh A, Alimoghaddam K, Rostami S,
Ghaffari SH, Jahani M, Iravani M, Mousavi SA, Bahar B and Jalili M:
Phase II study of single-agent arsenic trioxide for the front-line
therapy of acute promyelocytic leukemia. J Clin Oncol.
29:2753–2757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ren Y, Xie Y, Chai L, Wang S and Cheng M:
Autophagy modification augmented the treatment effects initiated by
arsenic trioxide in NB4 cells. Med Oncol. 28:231–236. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Qu X, Qu J, Zhang Y, Liu J, Teng Y,
Hu X, Hou K and Liu Y: Arsenic trioxide induces apoptosis and G2/M
phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and
thereby regulate p53 activation. Cancer Lett. 284:208–215. 2009.
View Article : Google Scholar : PubMed/NCBI
|