Introduction
Prostate cancer is the sixth leading cause of
cancer-associated mortality in males worldwide, and is the second
leading cause among males in the United States (1). The morbidity rates of prostate cancer
vary widely across the world and are less frequent in South and
East Asia, compared with that in Europe and United States (2). In spite of the high incidence, the
etiology of prostate cancer remains unknown (3). Furthermore, early prostate cancer is
typically asymptomatic, with ~66% of patients diagnosed with
prostate cancer exhibiting no symptoms, preventing the early
control of prostate cancer (4). The
identification of novel therapeutic targets is required to diagnose
early prostate cancer.
Established risk factors of prostate cancer include
age, race and exhibiting a family history of prostate cancer
(5). Genetic factors, which may
increase the risk of developing prostate cancer, are associated
with ethnicity and family history of the disease (6). A number of different genes have been
implicated in the development of prostate cancer (7,8). A
previous study validated that prostate cancer is prone to recurrent
gene fusions of androgen-regulated genes, including transmembrane
protease serine 2 (TMPRSS2), solute carrier family 45 member
3 and N-myc downstream regulated 1, and E26 transformation-specific
(ETS) transcription factors, including ERG ETS transcription factor
(ERG) and ETS variant 1 (9).
In addition, loss of tumor suppressor genes [including Kruppel-like
factor 6 (10), BTG
anti-proliferation factor 3 (11) and
phosphatase and tensin homolog (12)]
and activation of oncogenes [including nuclear receptor coactivator
2 (NCOA2) and MYC proto-oncogene bHLH transcription factor]
in the carcinogenesis of prostate cancer have been identified
(13). Although previous studies have
revealed a number of candidate genes, the causes of prostate cancer
remain unclear.
Next-generation RNA sequencing (RNA-seq) enables the
identification of genes that may be susceptible to prostate cancer
(14). On the basis of RNA-seq data,
the transcriptome profiles of primary prostate cancer are
identified, including gene fusions, long non-coding RNAs,
alternative splicing and somatic mutations (9). Ren et al (15) used RNA-seq to profile genetic
aberrations in Chinese patients with prostate cancer and identify
three recurrent gene fusions, including TMPRSS2-ERG,
ubiquitin specific peptidase 9 Y-linked-testis-specific
transcripts, Y-linked 15 on chromosome Y and the interchromosomal
translocation of CTAGE family member 5 ER export factor-KH RNA
binding domain containing signal transduction associated 3. Kannan
et al (16) investigated
chimeric RNAs expressed in human prostate cancer and obtained 1.3
billion sequence reads, which enabled the identification of 2,369
chimeric RNA candidates for distinguishing of prostate cancer. On
the basis of this, Xu et al (17) identified 116 disruptive mutations in
92 genes with high confidence, including a frameshift
insertion/deletion in the coding region of the TNF superfamily
member 10 gene associated with apoptosis. However,
differentially-expressed genes (DEGs) between prostate cancer
samples and normal control samples were not investigated, and a
number of somatic mutations remain unknown.
In the present study, raw RNA-seq data from the
study by Kannan et al (16)
was downloaded from the National Center for Biotechnology
Information database and analyzed by a number of bioinformatics
methods. First, DEGs between prostate cancer samples and normal
control samples were identified. Subsequently, functional
enrichment analysis of DEGs was performed, to understand the
underlying molecular mechanisms of prostate cancer. Furthermore,
the upstream regulatory elements of DEGs were identified and
analyzed. In addition, single nucleotide variations (SNVs) were
determined and the somatic mutation sites were annotated. The risk
level of SNVs was assessed for selection of SNVs with high risk and
high frequency. These candidate genes may contribute to further the
understanding of prostate cancer.
Materials and methods
Raw RNA-seq data
The RNA-seq data GSE22260 (16) were downloaded from the Gene Expression
Omnibus (www.ncbi.nlm.nih.gov) database,
including 4 prostate cancer samples (GSM554078, GSM554082,
GSM554086 and GSM554088) and 4 matched normal tissues samples
(GSM554118, GSM554120, GSM554122 and GSM554124), respectively. The
Gleason score of the four cancer samples were 7, 7, 7 and 6
(18). The sequencing platform was
Illumina Genome Analyzer IIx (Illumina, Inc., San Diego, CA, USA).
Reads were generated via a paired-end approach.
Read alignment
All RNA-Seq reads were mapped to the reference human
genome (hg19) of the University of California Santa Cruz (Santa
Cruz, CA, USA) by using Tophat software (19). Only the reads that mapped to specific
genome locations were retained. A maximum of two mismatches in each
read were permitted. Other parameters were set up according to the
default settings of Tophat.
DEG analysis
On the basis of Refseq gene annotation (20), transcripts were assembled using
Cufflinks (version 0.9.3) (21).
Subsequently, the gene expression levels of transcripts were
calculated using Cuffdiff (part of the Cufflinks package), on the
basis of the fragments/kilobase/million reads method (22). The differentially expressed
transcripts were identified by calculating the fold change in the
transcript and Student's t-tests in Cufflinks were performed to
assess the difference. Transcripts with a log2 fold
change >2 and P<0.05 were considered DEGs.
Function enrichment analysis of
DEGs
Gene Ontology (GO) (23) enrichment analysis was performed for
the aforementioned DEGs, on the basis of the Database for
Annotation, Visualization and Integrated Discovery database
(www.david.niaid.nih.gov) (24). GO categories were classified into
biological process (BP), molecular function and cellular component
(CC) GO-terms. P<0.05 was set as the threshold criterion.
DEGs which demonstrated regulatory function were
selected, labeled and inputted into the tumor suppressor gene
(25) database (bioinfo.mc.vanderbilt.edu/TSGene) and
the tumor-associated gene (26)
database (www.binfo.ncku.edu.tw/TAG/GeneDoc.php) to select
cancer-related genes (tumor suppressor genes or oncogenes) for
additional analysis.
Upstream regulatory elements of
DEGs
In the present study, the upstream region (1.5 kb)
of the transcription start site was defined as the promoter region.
For the promoter region of up- and downregulated DGEs, motif
identification was performed using Seqpos (27) to identify transcription factors.
P<0.00001 and the frequency of motif targeted sequences >50%
of up- and downregulated DEGs were set as the threshold
criteria.
Identification of SNVs
The Genome Analysis Toolkit (28) software (available at www.broadinstitute.org/gatk) was used to identify
the SNVs. The coverage of each credible SNV was >5×. The minimum
quality score of reliability SNV was 30 and SNVs with a quality
score >50 were defined as high reliability SNVs. Based on the
single nucleotide polymorphism (SNP) sites documented in dbSNP137
and 1,000 genome databases, the already known SNV callings in tumor
tissues were removed. To remove the interference of RNA editing in
the transcriptome, SNV calling was optimized by combining with
RNA-seq data of normal controls.
Somatic mutation sites annotation and
high-risk mutation sites assessment
The VarioWatch (29)
software (genepipe.ncgm.sinica.edu.tw/variowatch/main.do)
was used to annotate the SNVs in coding sequence and assess the
functional impact of gene products, on the basis of the risk
assessment of the software. The SNVs with high-risk level of
abnormal protein function were selected.
Results
Differential expression analysis and
functional enrichment analysis
According to the differential expression analysis of
paired RNA-seq data between prostate cancer samples and normal
control samples, a total of 150 upregulated and 211 downregulated
DEGs were selected (Table I). On the
basis of the annotation information of transcription factors, 8
differentially-expressed transcription factors were identified, 4
of which were upregulated [interferon-γ inducible protein 16
(IFI16), neurogenin 3 (NEUROG3), retinoic acid
receptor-γ (RARG) and single-minded family bHLH
transcription factor 1 (SIM1)] and 4 were downregulated
[diencephalon/mesencephalon homeobox 1 (DMBX1), nuclear
receptor coactivator 2 (NCOA2), one cut homeobox 2
(ONECUT2) and zing finger protein 83 (ZNF83)]
(Table I).
 | Table I.Comparison between identified DEGs
from prostate cancer samples and normal control samples. |
Table I.
Comparison between identified DEGs
from prostate cancer samples and normal control samples.
| Expression | DEGs | TF counts | TF genes |
|---|
| Downregulated | 211 | 4 | IFI16, NEUROG3,
RARG, SIM1 |
| Upregulated | 150 | 4 | DMBX1, NCOA2,
ONECUT2, ZNF83 |
The most significantly enriched BP GO-terms of
upregulated DEGs were chromatin assembly or disassembly (P=0.0016)
and nucleosome assembly (P=0.0024) (Table II). In addition, the most significant
CC GO-terms identified were nucleosome (P=0.0006) and protein-DNA
complex (P=0.0020). The results of the present study suggested that
genes associated with chromatin structure stability may be highly
expressed in prostate cancer.
| Table II.Ten most significant GO terms of up-
and downregulated DEGs in prostate cancer. |
Table II.
Ten most significant GO terms of up-
and downregulated DEGs in prostate cancer.
| A, Upregulated
DEGs |
|---|
|
|---|
| Category | Term | Count, n | P-value |
|---|
| CC | GO:0000786;
nucleosome | 5 | 0.0006 |
| BP | GO:0006333;
chromatin assembly or disassembly | 6 | 0.0016 |
| CC | GO:0032993;
protein-DNA complex | 5 | 0.0020 |
| BP | GO:0006334;
nucleosome assembly | 5 | 0.0024 |
| BP | GO:0031497;
chromatin assembly | 5 | 0.0027 |
| BP | GO:0065004;
protein-DNA complex assembly | 5 | 0.0032 |
| BP | GO:0034728;
nucleosome organization | 5 | 0.0034 |
| BP | GO:0051223;
regulation of protein transport | 5 | 0.0070 |
| BP | GO:0006323; DNA
packaging | 5 | 0.0077 |
| CC | GO:0000785;
chromatin | 6 | 0.0083 |
|
| B, Downregulated
DEGs |
|
| Category | Term | Count, n | P-value |
|
| CC | GO:0005902;
microvillus | 4 | 0.0033 |
| CC | GO:0031226;
intrinsic to plasma membrane | 20 | 0.0055 |
| BP | GO:0009100;
glycoprotein metabolic process | 7 | 0.0082 |
| BP | GO:0009101;
glycoprotein biosynthetic process | 6 | 0.0120 |
| CC | GO:0044459; plasma
membrane part | 29 | 0.0129 |
| CC | GO:0046658;
anchored to plasma membrane | 3 | 0.0144 |
| BP | GO:0006955; immune
response | 13 | 0.0162 |
| CC | GO:0005887;
integral to plasma membrane | 18 | 0.0196 |
| CC | GO:0005886; plasma
membrane | 43 | 0.0196 |
| BP | GO:0043413;
biopolymer glycosylation | 5 | 0.0250 |
Of the downregulated DEGs, BP GO terms were
significantly enriched in glycoprotein metabolic process
(P=0.0082), glycoprotein biosynthetic process (P=0.0120) and immune
response (P=0.0162); whereas, CC GO terms were significantly
enriched in the microvillus (P=0.0033), intrinsic to plasma
membrane (P=0.0055) and plasma membrane part (P=0.0129) (Table II).
Identification of the upstream
regulatory elements of DEGs
Motif scanning of the upstream regulatory elements
of up-regulated DEGs revealed 25 transcription factors that may
regulate these DEGs (Table III).
Additionally, 17 transcription factors were identified to regulate
downregulated DEGs (Table III).
| Table III.Potential upstream regulatory
elements of DEGs in prostate cancer. |
Table III.
Potential upstream regulatory
elements of DEGs in prostate cancer.
| Expression | Count, n | Candidate TFs |
|---|
| Upregulated | 25 | BARHL1, CD200,
CEBPA, CUX1, EN1, ESX1, HNF4A, HOXA5, IRF8, LHX6, MEOX1, NOX1,
NR1H4, PAX6, PBX1, PHOX2A, PITX2, PRKRA, RAX, RORA, SIX4, TCF7L2,
VAX2, VSX2, ZEB1 |
| Downregulated | 17 | CDX1, EPAS1,
FOXC1, HMX1, HMX3, HOXA10, HOXA3, HOXC12, HSF1, HSF2, IKZF2, IRF1,
IRF2, MSX1, NR3C1, POU2F1, ZEB1 |
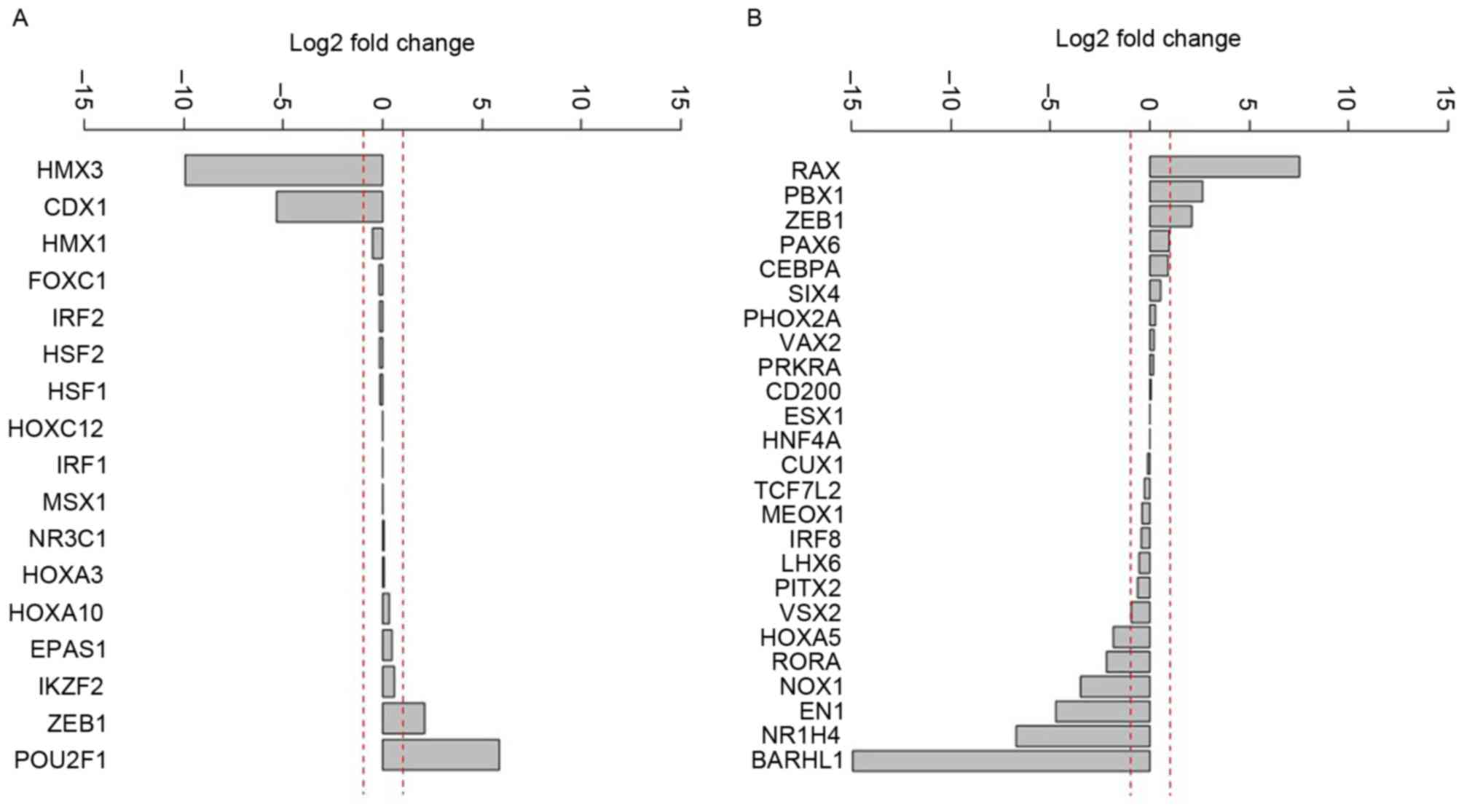
Differential expression analysis of the potential
upstream regulatory elements demonstrated that the expression of H6
family homeobox 3 and caudal type homeobox 1 were markedly
decreased (Fig. 1A). In addition, the
following genes were activated in different degrees: Retina and
anterior neural fold homeobox, PBX homeobox 1, zinc finger E-box
binding homeobox 1, paired box 6 and CCAAT/enhancer binding
protein-α (Fig. 1B).
Detection of SNVs in prostate
cancer
Following the removal of polymerase chain reaction
duplicates and data processing of RNA-seq in 4 prostate cancer
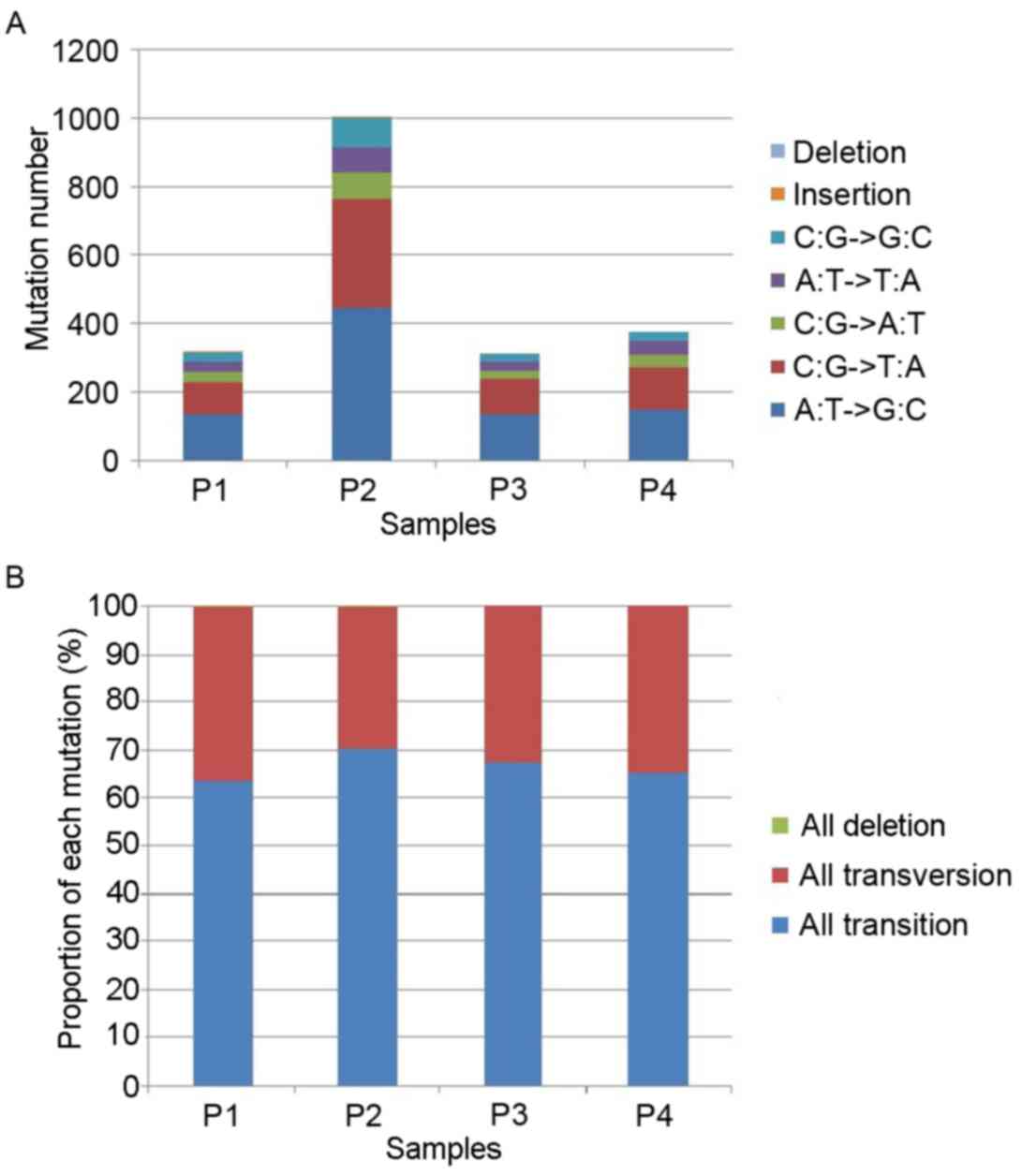
samples, 317, 1,004, 310 and 373 somatic mutation sites were
identified in 4 prostate cancer samples particularly (Fig. 2). In each sample, base transversion
(C:G → A:T; A:T → T:A; C:G → G:C) and base transition (A:T → G:C;
C:G → T:A) were the most common types of base mutation (Fig. 2A). Only one insertion site was
identified in the 4 prostate cancer samples. The proportions of
each mutation type in the 4 prostate cancer samples were similar,
suggesting that 65% of mutations were base transition and 35% were
base transversion (Fig. 2B).
Mutation sites that occurred in >2 prostate
cancer samples were defined as a high-frequency site (frequency
≥2). A total of 17 mutation sites were identified as high-frequency
sites (Table IV). The T → A base
transversion of chromosome 1189431695 was identified in 3 cancer
samples and its loci was located in the folate hydrolase 1B
(FOLH1B) gene. An additional 16 mutation sites were detected
in 2 cancer samples and loci were all located in the coding
sequence (CDS) region of genes.
| Table IV.High-frequency mutation sites in
prostate cancer. |
Table IV.
High-frequency mutation sites in
prostate cancer.
|
|
| Base |
|
|---|
|
|
|
|
|
|---|
| Chr | Position | Reference | Mutated | Frequency, n |
|---|
| Chr11 | 89431695 | T | A | 3 |
| ChrY | 22941450 | A | C | 2 |
| ChrY | 22921933 | A | G | 2 |
| ChrX | 51807602 | G | A | 2 |
| ChrX | 43634502 | T | C | 2 |
| Chr9 | 96214432 | G | C | 2 |
| Chr7 | 73245681 | A | G | 2 |
| Chr6 | 29692069 | C | A | 2 |
| Chr6 | 10749928 | A | G | 2 |
| Chr19 | 51325077 | G | T | 2 |
| Chr18 | 3253963 | C | G | 2 |
| Chr18 | 21057145 | C | G | 2 |
| Chr17 | 60631088 | A | G | 2 |
| Chr16 | 2149965 | G | A | 2 |
| Chr15 | 28421681 | A | G | 2 |
| Chr1 | 89474720 | T | C | 2 |
| Chr1 | 147580839 | A | G | 2 |
The frequency of high-risk mutant genes was
calculated using VarioWatch (Table
V). A total of 20 high-risk mutant genes were identified, of
which ribosomal protein S4 Y-linked 2 (RPS4Y2), polycystin 1
transient receptor potential channel interacting (PKD1) and
FOLH1B were identified in 3 cancer samples. Furthermore,
kallikrein 1 (KLK1) and PKD1 were known tumor
suppressor genes (30,31). Although the expression level of the
KLK1 gene was increased in prostate cancer, the high
frequency of mutation located in the CDS region may lead to
dysfunction of the tumor suppressor. However, the expression levels
of other 19 high-risk genes were not markedly different in prostate
cancer, indicating that the abnormal function of these genes,
induced by base mutation, may be associated with the development of
prostate cancer.
| Table V.Frequency of high-risk genes in
prostate cancer. |
Table V.
Frequency of high-risk genes in
prostate cancer.
| High-risk mutated
gene | Gene name | Frequency | Expression
level | Tumor-associated
gene |
|---|
| PPP4R2 | Protein phosphatase
4, regulatory subunit 2 | 2 | – | – |
| CLDN4 | Claudin 4 | 2 |
Upregulateda |
Tumor-associateda |
| FOLH1B | Folate hydrolase
1B | 3 |
Downregulateda |
Tumor-associateda |
| KIF27 | Kinesin family
member 27 | 2 | – | – |
| MAOB | Monoamine oxidase
B | 2 |
Upregulateda |
Tumor-associateda |
| DUXA | Double homeobox
A | 2 | – | – |
| KLK1 | Kallikrein 1 | 2 | Upregulated | Tumor suppressor
gene |
| MYL12A | Myosin, light chain
12A, regulatory, non-sarcomeric | 2 | – | – |
| HERC2 | Hect domain and RLD
2 | 2 | – | – |
| HLA-E | Major
histocompatibility complex, class I, E | 2 |
Downregulateda | Tumor suppressor
genea |
| HLA-G | Major
histocompatibility complex, class I, G | 2 |
Downregulateda |
Tumor-associateda |
| RIOK3 | RIO kinase 3 | 2 | – | – |
| RPS4Y2 | Ribosomal protein
S4, Y-linked 2 | 3 | – | – |
| PKD1 | Polycystic kidney
disease 1 (autosomal dominant) | 3 |
Upregulateda | Tumor suppressor
gene |
| USP10 | Ubiquitin specific
peptidase 10 | 2 | – | – |
| TLK2 | Tousled-like kinase
2 | 2 | – | – |
| TMEM14B | Transmembrane
protein 14D; transmembrane protein 14B | 2 | – | – |
| TUBA1A | Tubulin, α 1a | 2 | – | – |
| GBP3 | Guanylate binding
protein 3 | 2 | – | – |
| MAGED4B | Melanoma antigen
family D, 4B; melanoma antigen family D, 4 | 2 | – | Tumor-associated
gene |
Discussion
The present study provided a survey of DEGs and
mutant genes in human prostate cancer. RNA-seq data between 4
prostate cancer samples and 4 matched normal samples were analyzed.
A total 150 upregulated and 211 downregulated DEGs were identified,
4 of which were upregulated transcription factors (DMBX1, NCOA2,
ONECUT2 and ZNF83), and 4 were downregulated
transcription factors (IFI16, NEUROG3, RARG and
SIM1).
NCOA2 has been suggested as an oncogene in
primary tumors by increasing androgen receptor signaling, which is
known to have a function in early and late-stage prostate cancer
(13). Furthermore, the upregulated
expression of transcription factor ONECUT2 was revealed in
breast and prostate cancer cell lines (32), and increased IFI16 protein in normal
human prostate epithelial cells was associated with cellular
senescence-associated cell growth arrest (33). NEUROG3 was identified to be
expressed in metastatic neuroendocrine prostate cancer cells
(34). However, mechanism of abnormal
regulation of DMBX1, ZNF83, RARG and SIM1 in prostate
cancer remains unknown. For example, the abnormal regulation of
ZNF83 has been identified in hepatocellular carcinoma (35) and colorectal cancer (36), but not in prostate cancer. DMBX1 is a
paired-class homeodomain transcription factor and may be determined
in the brain, stomach and testis in adult normal tissues (37). A previous study focused on the
expression pattern of DMBX1 in the development of the neural
network (38). Therefore, these
aforementioned transcription factors may have important functions
in the progress of prostate cancer.
Determining the SNVs in prostate cancer identified
17 mutation sites with a frequency ≥2. The mutation site with the
highest frequency was located in FOLH1B. Furthermore, 20
high-risk mutant genes with highest frequency were identified using
VarioWatch and included RPS4Y2, PKD1 and FOLH1B.
FOLH1B originates from the duplication of folate hydrolase 1
(FOLH1) (39), which is an
established biomarker for prostate cancer (40). FOLH1, also known as prostate-specific
membrane antigen 1, is used as a diagnostic and prognostic
indicator for prostate cancer, and is associated with
aggressiveness and metastasis of prostate cancer (41). Of the identified high-risk mutant
genes exhibiting the highest frequency, KLK1 and PKD1
are known tumor suppressor genes and MAGE family member D4B is a
tumor-associated gene (30,31). PKD1 has been identified to be
downregulated in advanced-stage prostate cancer and was present as
a protein complex, combined with the androgen receptor, in prostate
cancer cells (42). The
kallikrein-related peptidases have been identified in a number of
types of cancer, including prostate and ovarian, and a combination
of the dysregulation of KLK1, 5 and 13 was associated
with poorer disease-free survival for prostate cancer (43). The high frequency of mutation located
in KLK1, PKD1 and D4B may lead to dysfunction of the
tumor suppressor functions and consequently contribute to the
progress of prostate cancer.
There were a number of limitations in the present
study; for example, the sample size used for the analysis was
small. Furthermore, the results of the present study require
validation using other RNA-seq data or microarray data of prostate
cancer.
Combined with bioinformatics methods, the RNA-seq
data were analyzed to determine candidate genes for diagnosing
and/or treating of prostate cancer. The identified DEGs (DMBX1,
ZNF83, RARG and SIM1) and mutant genes (RPS4Y2,
KLK1 and FOLH1B) may have important functions in the
progression of prostate cancer. The results of the present study
may enable an improved understanding of the molecular mechanisms
that underlie prostate cancer pathogenesis.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsing AW and Chokkalingam AP: Prostate
cancer epidemiology. Front Biosci. 11:1388–1413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller DC, Hafez KS, Stewart A, Montie JE
and Wei JT: Prostate carcinoma presentation, diagnosis and staging:
An update form the National Cancer Data Base. Cancer. 98:1169–1178.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leitzmann MF and Rohrmann S: Risk factors
for the onset of prostatic cancer: Age, location, and behavioral
correlates. Clin Epidemiol. 4:1–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schaid DJ: The complex genetic
epidemiology of prostate cancer. Hum Mol Genet 13 Spec No.
1:R103–R121. 2004. View Article : Google Scholar
|
|
7
|
Mahmoud AM, Yang W and Bosland MC: Soy
isoflavones and prostate cancer: A review of molecular mechanism. J
Steroid Biochem Mol Biol. 140:116–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watson PA, Arora VK and Sawyers CL:
Emerging mechanisms of resistance to androgen receptor inhibitors
in prostate cancer. Nat Rev Cancer. 15:701–711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pflueger D, Terry S, Sboner A, Habegger L,
Esgueva R, Lin PC, Svensson MA, Kitabayashi N, Moss BJ, MacDonald
TY, et al: Discovery of non-ETS gene fusions in human prostate
cancer using next-generation RNA sequencing. Genome Res. 21:56–67.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Narla G, Heath KE, Reeves HL, Li D, Giono
LE, Kimmelman AC, Glucksman MJ, Narla J, Eng FJ, Chan AM, et al:
KLF6, a candidate tumor suppressor gene mutated in prostate cancer.
Science. 294:2563–2566. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Majid S, Dar AA, Shahryari V, Hirata H,
Ahmad A, Saini S, Tanaka Y, Dahiya AV and Dahiya R: Genistein
reverses hypermethylation and induces active histone modifications
in tumor suppressor gene B-Cell translocation gene 3 in prostate
cancer. Cancer. 116:66–76. 2010.PubMed/NCBI
|
|
12
|
Carver BS, Tran J, Gopalan A, Chen Z,
Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino
PT, et al: Aberrant ERG expression cooperates with loss of PTEN to
promote cancer progression in the prostate. Nat Genet. 41:619–624.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor BS, Schultz N, Hieronymus H,
Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva
B, et al: Integrative genomic profiling of human prostate cancer.
Cancer cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X,
Cui Z, Zhang J, Yi K, Xu W, et al: RNA-seq analysis of prostate
cancer in the Chinese population identifies recurrent gene fusions,
cancer-associated long noncoding RNAs and aberrant alternative
splicings. Cell Res. 22:806–821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kannan K, Wang L, Wang J, Ittmann MM, Li W
and Yen L: Recurrent chimeric RNAs enriched in human prostate
cancer identified by deep sequencing. Proc Natl Acad Sci USA.
108:9172–9177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu X, Zhu K, Liu F, Wang Y, Shen J, Jin J,
Wang Z, Chen L, Li J and Xu M: Identification of somatic mutations
in human prostate cancer by RNA-Seq. Gene. 519:343–347. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Makarov DV, Trock BJ, Humphreys EB,
Mangold LA, Walsh PC, Epstein JI and Partin AW: Updated nomogram to
predict pathological stage of prostate cance given
prostate-specific antigen level, clinical stage, and biopsy Gleason
score (Partin tables) based on cases from 2000 to 2005. Urology.
69:1095–1101. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trapnell C, Pachter L and Salzberg SL:
TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics.
25:1105–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
reference sequences (RefSeq): A curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res 35
(Database Issue). D61–D65. 2007. View Article : Google Scholar
|
|
21
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beane J, Vick J, Schembri F, Anderlind C,
Gower A, Campbell J, Luo L, Zhang XH, Xiao J, Alekseyev YO, et al:
Characterizing the impact of smoking and lung cancer on the airway
transcriptome using RNA-Seq. Cancer Prev Res (Phila). 4:803–817.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hulsegge I, Kommadath A and Smits MA:
Globaltest and GOEAST: Two different approaches for Gene Ontology
analysis. BMC Proc. 3 Suppl 4:S102009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT and Lempicki R:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
25
|
Zhao M, Sun J and Zhao Z: TSGene: A web
resource for tumor suppressor genes. Nucleic Acids Res.
41:(Database Issue). D970–D976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen JS, Hung WS, Chan HH, Tsai SJ and Sun
HS: In silico identification of oncogenic potential of fyn-related
kinase in hepatocellular carcinoma. Bioinformatics. 29:420–427.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He HH, Meyer CA, Shin H, Bailey ST, Wei G,
Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al: Nucleosome dynamics
define transcriptional enhancers. Nat Genet. 42:343–347. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng YC, Hsiao FC, Yeh EC, Lin WJ, Tang
CY, Tseng HC, Wu HT, Liu CK, Chen CC, Chen YT and Yao A:
VarioWatch: Providing large-scale and comprehensive annotations on
human genomic variants in the next generation sequencing era.
Nucleic Acids Res. 40:(Web Server Issue). W76–W81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Raea F, Bulmera B, Nicolb D and Clementsa
J: The human tissue kallikreins (KLKs 1–3) and a novel KLK1 mRNA
transcript are expressed in a renal cell carcinoma cDNA library.
Immnuopharmacology. 45:83–88. 1999. View Article : Google Scholar
|
|
31
|
Shabelnik MY, Kovalevska LM, Yurchenko MY,
Shlapatska LM, Rzepetsky Y and Sidorenko S: Differential expression
of PKD1 and PKD2 in gastric cancer and analysis of PKD1 and PKD2
function in the model system. Exp Oncol. 33:206–211.
2011.PubMed/NCBI
|
|
32
|
Yamamoto F and Yamamoto M: Scanning copy
number and gene expression on the 18q21-qter chromosomal region by
the systematic multiplex PCR and reverse transcription-PCR methods.
Electrophoresis. 28:1882–1895. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alimirah F, Chen J, Davis FJ and Choubey
D: IFI16 in human prostate cancer. Mol Cancer Res. 5:251–259. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Telesca D, Inoue LY, Neira M, Etzioni R,
Gleave M and Nelson C: Differential expression and network
inferences through functional data modeling. Biometrics.
65:793–804. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong H, Ge X, Shen Y, Chen L, Kong Y,
Zhang H, Man X, Tang L, Yuan H, Wang H, et al: Gene expression
profile analysis of human hepatocellular carcinoma using SAGE and
LongSAGE. BMC Med Genomics. 2:52009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jovov B, Araujo-Perez F, Sigel CS,
Stratford JK, McCoy AN, Yeh JJ and Keku T: Differential gene
expression between African American and European American
colorectal cancer patients. PLoS One. 7:e301682012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ohtoshi A, Nishijima I, Justice MJ and
Behringer RR: Dmbx1, A novel evolutionarily conserved paired-like
homeobox gene expressed in the brain of mouse embryos. Mech Dev.
110:241–244. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fujimoto W, Shiuchi T, Miki T, Minokoshi
Y, Takahashi Y, Takeuchi A, Kimura K, Saito M, Iwanaga T and Seino
S: Dmbx1 is essential in agouti-related protein action. Proc Natl
Acad Sci USA. 104:15514–15519. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
De Lorenzi L, Genualdo V, Gimelli S, Rossi
E, Perucatti A, Iannuzzi A, Zannotti M, Malagutti L, Molteni L,
Iannuzzi L and Parma P: Genomic analysis of cattle rob (1;29).
Chromosome Res. 20:815–823. 2012. View Article : Google Scholar
|
|
40
|
Zhang T, Song B, Zhu W, Xu X, Gong QQ,
Morando C, Dassopoulos T, Newberry RD, Hunt SR and Li E: An Ileal
Crohn's disease gene signature based on whole human genome
expression profiles of disease unaffected ileal mucosal biopsies.
PLoS One. 7:e371392012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ergün A, Lawrence CA, Kohanski MA, Brennan
TA and Collins JJ: A network biology approach to prostate cancer.
Mol Syst Biol. 3:822007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mak P, Jaggi M, Syed V, Chauhan SC, Hassan
S, Biswas H and Balaji KC: Protein kinase D1 (PKD1) influences
androgen receptor (AR) function in prostate cancer cells. Biochem
Biophys Res Commun. 373:618–623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Girgis AH, Bui A, White NM and Yousef GM:
Integrated genomic characterization of the kallikrein gene locus in
cancer. Anticancer Res. 32:957–963. 2012.PubMed/NCBI
|