Introduction
In 2012, lung cancer was the leading cause of
cancer-associated mortality in males according to global cancer
statistics (1). The 5-year survival
rate for patients with non-small cell lung cancer (NSCLC), which
accounts for 85–90% of all the lung cancer diagnoses, was <20%
(2,3).
The first-line standard treatment for NSCLC, including surgery,
chemotherapy and radiotherapy, is selected depending on the disease
and patient characteristics (4).
Currently, platinum-based regimens are first-line chemotherapies;
single-agent docetaxel, pemetrexed or erlotinib are prominent
second-line therapies (5). However,
no response or resistance to conventional chemotherapies is a
problem when studying the pathological processes and new
therapeutic strategies for lung tumors (6). Therefore, current therapies have been
demonstrated to be inadequate, and novel strategies are required
(7).
Sorafenib is a multi-tyrosine kinase inhibitor that
blocks Raf kinases, platelet-derived growth factor receptors and
vascular endothelial growth factor receptors (VEGFR) (8). Sorafenib is frequently used in the
clinical treatment of unresectable hepatocellular carcinoma and
advanced renal cell carcinoma (9,10). Ongoing
clinical trials are studying its activity in other types of
malignancy, including NSCLC (11,12). The
effects of Sorafenib have been confirmed effect in preclinical
models of NSCLC (13,14). However, Sorafenib does not improve the
overall survival time of patients with advanced NSCLC (15). Furthermore, Sorafenib has a number of
side effects, including hand-foot syndrome, rash, diarrhea,
hypertension and fatigue, which limit its application in the
treatment of NSCLC. Generally, the majority of these aforementioned
side effects are dose-dependent (16). Therefore, increasing the sensitivity
of tumor tissues to Sorafenib and thus reducing the required dose,
warrants further investigation.
c-Met is another important member of the receptor
tyrosine kinase superfamily (17). It
has been demonstrated that c-Met is upregulated or partially
activated by mutation in NSCLC (18).
Phosphatidylinositol-3-kinase (PI3K)-AKT and mitogen-activated
protein kinase (MAPK) are common downstream pathways of the
hepatocyte growth factor (HGF)/c-Met and VEGF signaling pathways
(19). The c-Met/HGF pathway serves
an important role in proliferation, apoptosis, invasion, metastasis
of tumor cells and angiogenesis (20). Furthermore, treatments targeting the
HGF/c-Met signaling pathway have been proposed for multiple types
of cancer (21). Specific c-Met small
molecule inhibitors, including SU-11274 and PHA-665752, have
emerged as treatments for malignant tumors (22). PF-2341066 is an effective and orally
available ATP-competitive small molecule compound that targets
c-Met, blocking its phosphorylation (23).
Previous studies have demonstrated that selective
c-Met inhibitors can attenuate or promote the antitumor activities
of different molecularly-targeted therapies, including the
VEGFR-inhibitor, pazopanib, and the epidermal growth factor
receptor (EGFR)-inhibitor erlotinib (24,25).
Furthermore, c-Met amplification is one of the most important
mechanisms that facilitate resistance to gefitinib by re-activating
the AKT and extracellular signal-regulated kinase (ERK) pathways in
NSCLC (26); however, whether
PF-2341066 can affect the antitumor activities of Sorafenib remains
unknown. In the present study, whether or not PF-2341066 can
effectively inhibit the phosphorylation of c-Met in NSCLC cells was
investigated. Following this, the antitumor potential of the
combined PF-2341066 and Sorafenib treatment in human NSCLC cells
was studied as a potential comprehensive treatment for this
disease.
Materials and methods
Materials
Sorafenib,
(4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-methylpyridine-2-carboxamide)
was purchased from Bayer AG (Leverkusen, Germany); PF-2341066 was
obtained from Pfizer, Inc. (New York, NY USA). Dimethyl sulfoxide
(DMSO) was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany).
Cell culture
The human NSCLC cell line NCI-H1993 was provided by
the Cell Resource Center of the Shanghai Institutes for Biological
Sciences of the Chinese Academy of Sciences (Shanghai, China).
NCI-H1993 cells were maintained in RPMI 1640 medium (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% HyClone fetal bovine serum (FBS; GE Healthcare Life Sciences,
Logan, UT, USA), penicillin (100 U/ml; Sigma-Aldrich; Merck KGaA)
and streptomycin (100 mg/ml; Sigma-Aldrich; Merck KGaA). The cells
were cultured in a humidified incubator at 37°C with 5%
CO2.
Cell proliferation and colony
formation assays
NCI-H1993 cells (8×103/well) were seeded
into 96-well plates and cultured in RPMI-1640 medium with the
designated concentrations of Sorafenib and/or PF-2341066 dissolved
in DMSO for 24, 48, 72 and 96 h at 37°C and 5% CO2. Cell
proliferation assays were performed using a Cell Counting Kit-8
assay kit (Beyotime Institute of Biotechnology, Haimen, China),
according to the manufacturer's instructions. The absorbance value
in each well was measured at a wavelength of 490 nm. DMSO was used
as negative control. The experiments were repeated three times.
For the soft agar assay, each well of a 6-well plate
was setup containing a bottom layer of 1.2% agarose, a middle layer
of 0.7% agarose with 2,000 cells and a top layer of RPMI-1640
medium with designated concentrations of Sorafenib and/or
PF-2341066 dissolved in DMSO (0.5 µM PF, 2.5 µM Sora, 0.5 µM PF +
2.5 µM Sora). The medium in the top layer was changed every 6 days.
After 28 days, the colonies were counted.
Apoptosis assay
Apoptosis was assessed with an Annexin V-FITC/PI
Apoptosis Detection kit (Nanjing Keygen Biotech. Co., Ltd.,
Nanjing, China), according to the manufacturer's protocol. Briefly,
1×105 cells were seeded into 6-well plates and incubated
with designated concentrations of Sorafenib and/or PF-2341066
dissolved in DMSO (0.5 µM PF, 2.5 µM Sora, 0.5 µM PF + 2.5 µM Sora)
for 24 h at 37°C and 5% CO2. Then, the cells were
harvested. The cells were re-suspended in 100 µl 1X binding buffer
from the Apoptosis Detection kit and incubated for 15 min at room
temperature. A total of 4 µl Annexin V-fluorescein isothiocyanate
(FITC) and 1 µl propidium iodide were added and cultured for 10 min
at room temperature. Finally, 400 µl 1X binding buffer was added to
each sample. A CytoFLEX flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA) was used to analyze Annexin V-FITC-positive cells
and FlowJo (v 9.3.2; Tree Star, Inc., Ashland, OR, USA) was used
for the data analysis. DMSO was used as negative control. The
experiments were repeated three times.
Migration assay
Migration assays were performed using polycarbonate
membrane chambers (pore size, 8.0 µm; EMD Millipore, Billerica, MA,
USA) as previously demonstrated (27). Briefly, NCI-H1993 cells
(1×105) were suspended in 100 µl RPMI-1640 medium
containing 1.5% FBS were added to the upper chambers. The lower
compartments of the chamber were filled with RPMI-1640 medium
containing 15% FBS and the designated concentrations of Sorafenib
and/or PF-2341066 dissolved in DMSO (0.5 µΜ PF, 2.5 µΜ Sora, 0.5 µΜ
PF + 2.5 µΜ Sora). Following incubation for 24 h at 37°C and 5%
CO2, the cells that had migrated through the filters
were fixed using 4% paraformaldehyde and stained with 0.05% crystal
violet 5 min at room temperature. The migrated cells were counted
under a light microscope at magnification, ×200. A total of 6
fields of cells were counted using ImageJ Software (National
institutes of Health, Bethesda, MD, USA).
Western blotting
Cells were lysed in Triton lysing buffer
(Sigma-Aldrich; Merck KGaA) and centrifuged at 12,000 × g at 4°C
for 15 min. Protein concentrations were measured using a BCA kit.
Total protein lysates (10 µg) were separated by 10% SDS-PAGE and
transferred onto Immobilon-P polyvinylidene difluoride membranes
(EMD Millipore), subsequently blocked with 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) at 37°C for 1 h. The blots were probed
with anti-c-Met (cat. no. 3127; dilution, 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-phospho (p)-Met
(Tyr1234/1235, Tyr1349; cat. no. 3077; dilution, 1:1,000; Cell
Signaling Technology, Inc.), anti-p-AKT (473; cat. no. 4060;
dilution, 1:1,000; Cell Signaling Technology, Inc.), anti-AKT (cat.
no. 4691; dilution, 1:1,000; Cell Signaling Technology, Inc.),
anti-p-ERK (Thr202/Tyr204) (cat. no. 4370; dilution, 1:1,000; Cell
Signaling Technology, Inc.), anti-ERK (cat. no. 4695; dilution,
1:1,000; Cell Signaling Technology, Inc.), anti-p-c-Jun N-terminal
kinase (JNK; Thr183/Tyr185) (cat. no. 9255; dilution, 1:1,000; Cell
Signaling Technology, Inc.), anti-JNK (cat. no. 9252; dilution,
1:1,000; Cell Signaling Technology, Inc.), anti-p-p38 (cat. no.
4511; Thr180/Tyr182; dilution, 1:1,000; Cell Signaling Technology,
Inc.), anti-p38 (cat. no. 8690; dilution, 1:1,000; Cell Signaling
Technology, Inc.) and anti-poly-ADP-ribose polymerase (cat. no.
9532; PARP 1;1000; Cell Signaling Technology, Inc., Danvers, MA,
USA) incubating overnight at 4°C followed by either anti-rabbit or
anti-mouse horseradish peroxidase-conjugated IgG secondary
antibodies (cat. no. 4410; dilution, 1:2,000; Cell Signaling
Technology, Inc.) incubating for 1 h at room temperature.
Immunoreactive bands were detected using an ECL system (Pierce;
Thermo Fisher Scientific, Inc.). β-actin (cat. no. SAB3500350;
dilution, 1:5,000; Sigma-Aldrich; Merck KGaA) was used as an
internal control.
Statistical analysis
Data are expressed as the mean ± standard error and
were analyzed using the GraphPad Prism software (version 5.0;
GraphPad Software, Inc., La Jolla, CA, USA). The datasets were
compared using the two-tailed unpaired Student's t-test. Multiple
comparisons were tested with two-way analysis of variance, followed
by Bonferroni's post hoc test. A value of P<0.05 was considered
to indicate statistically significance.
Results
PF-2341066 inhibits the
phosphorylation of c-Met in NCI-H1993 cells
A previous study demonstrated that c-Met and its
receptor are overexpressed by ~70 and 40% of human lung cancer
tissues, respectively (28). However,
c-Met gene amplification and activation was exhibited in NCI-H1993
cells but not NCI-H1975 or A549 cells (29,30). In
the present study, the expression levels of c-Met in NCI-H1993
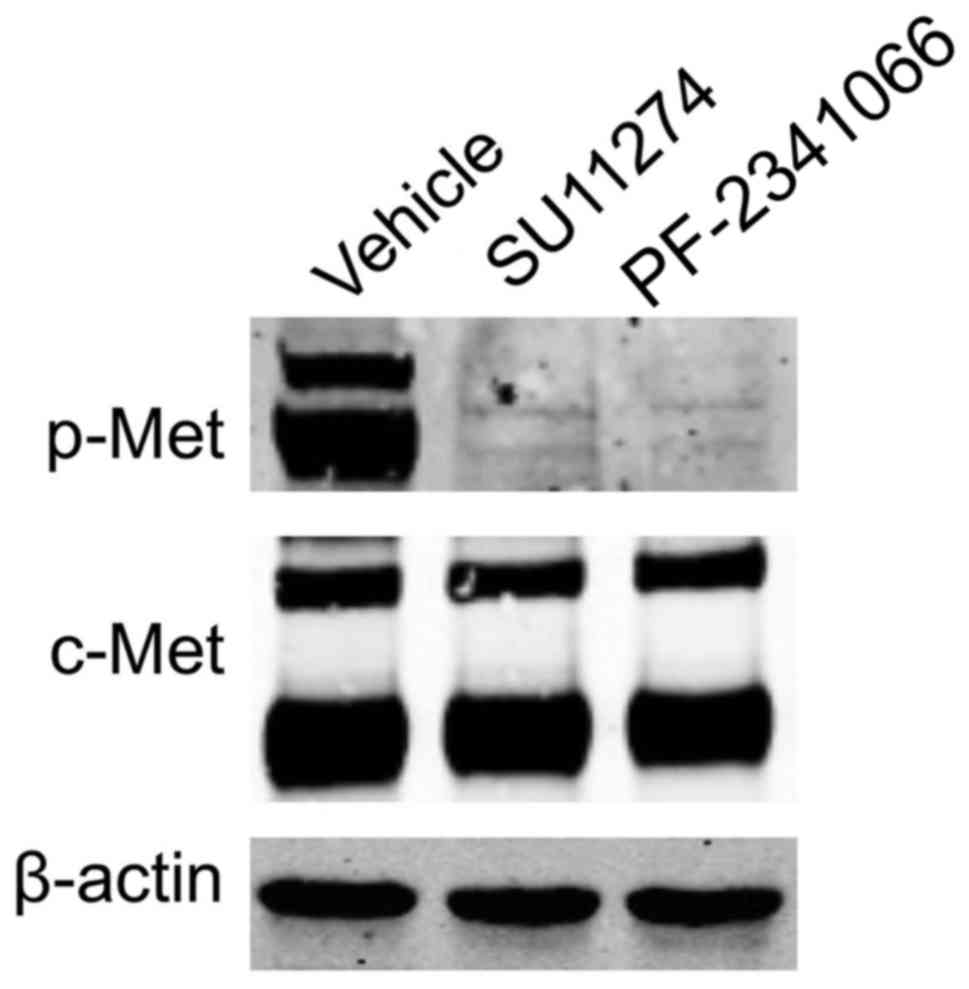
cells were examined by western blot analysis (Fig. 1). The specific c-Met inhibitors,
PF-2341066 and SU-11274, were able to inhibit the phosphorylation
of c-Met in NCI-H1993 cells (Fig.
1).
c-Met inhibitors increase the effects
of Sorafenib on proliferation of NSCLC cells
To determine whether increased c-Met phosphorylation
following treatment with Sorafenib mediates the growth of NSCLC
cells, the effects of Sorafenib and/or PF-2341066 treatment on the
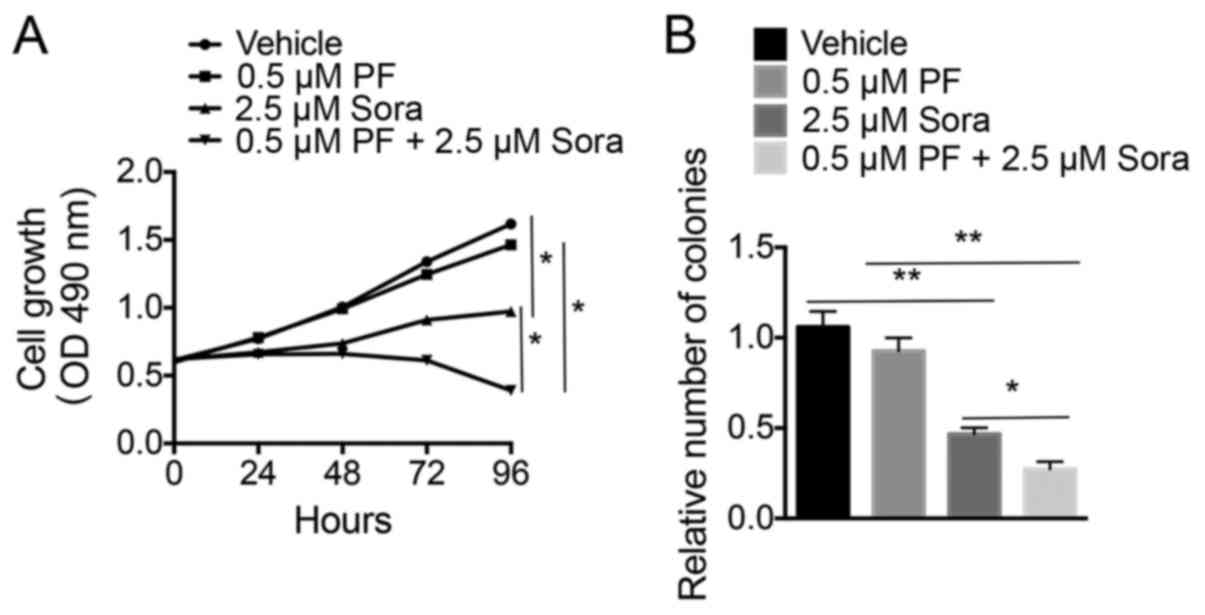
proliferation of NCI-H1993 cells were analyzed. As presented in
Fig. 2A, blocking c-Met activation
with a low-dose of PF-2341066 did not significantly increase the
Sorafenib-induced inhibition of proliferation of NCI-H1993 cells
compared with the vehicle-treated cells. However, the colony
formation ability of NCI-H1993 cells was notably blocked following
the combined treatment of Sorafenib and a low-dose of PF-2341066
compared with the vehicle-treated cells (Fig. 2B). This indicated that treatment with
a combination of Sorafenib and a low-dose of PF-2341066 was able to
inhibit the growth of NSCLC cells.
A low-dose of c-Met inhibitor
facilitates Sorafenib-induced apoptosis of NSCLC cells
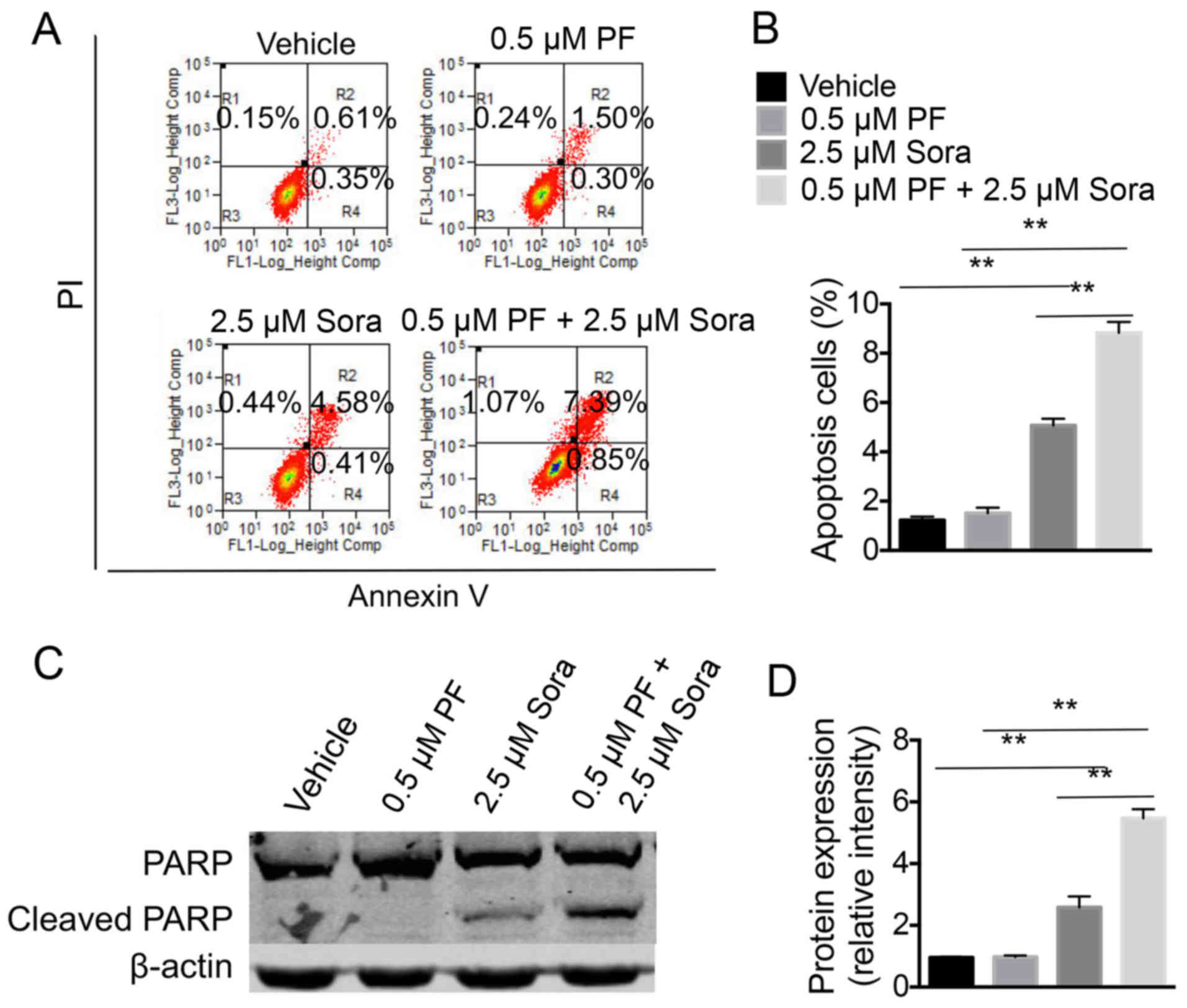
Next, the efficiency of a combination of PF-2341066
and Sorafenib in inducing the apoptosis of NCI-H1993 cells was
examined. The percentage of apoptotic cells was higher in the cells
that were treated with a combination of Sorafenib and a low-dose of
PF-2341066 compared with cells treated with Sorafenib alone or a
low-dose of PF-2341066 alone (Fig. 3A and
B). In addition, the cell extracts were analyzed for the
expression of PARP (an endogenous substrate of caspase-3 and −7)
and of cleaved caspase-3, which is associated with programmed cell
death. Notably, the addition of the c-Met inhibitor markedly
increased the level of cleaved PARP in NCI-H1993 cells that were
treated with Sorafenib (Fig. 3C and
D). The data indicated that PF2341066 and Sorafenib act
synergistically, at least in part through inducing cell
apoptosis.
Blocking c-Met signaling enhances the
inhibitory effects of Sorafenib on the migration of NSCLC
cells
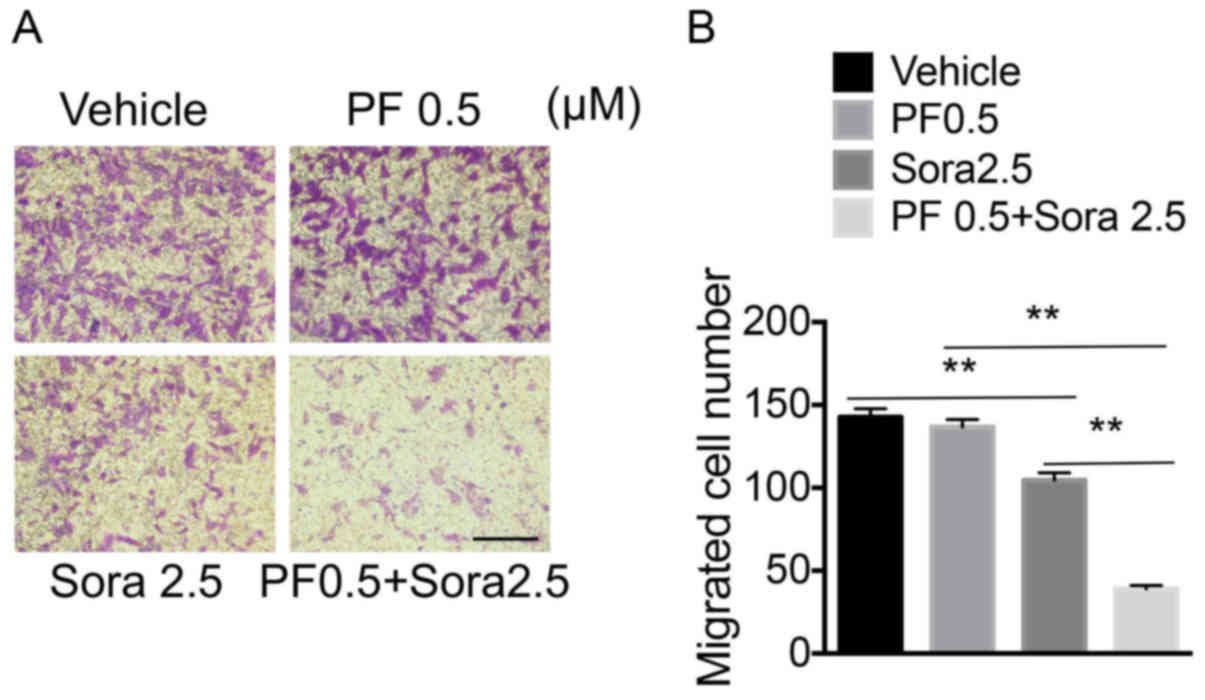
The combined effect of low-dose PF-2341066 and
Sorafenib on the migration of NSCLC cells was further investigated.
The treatment of cells with a low-dose of PF-2341066 and Sorafenib
significantly inhibited the migration of NCI-H1993 cells, compared
with the groups treated with a low-dose of PF-2341066 or Sorafenib
alone (Fig. 4A and B). Therefore,
blocking c-Met signaling increased the sensitivity of NSCLC cells
to Sorafenib by decreasing cell migration.
c-Met mediates Sorafenib sensitivity
through the PI3K and MAPK signaling pathways in NSCLC
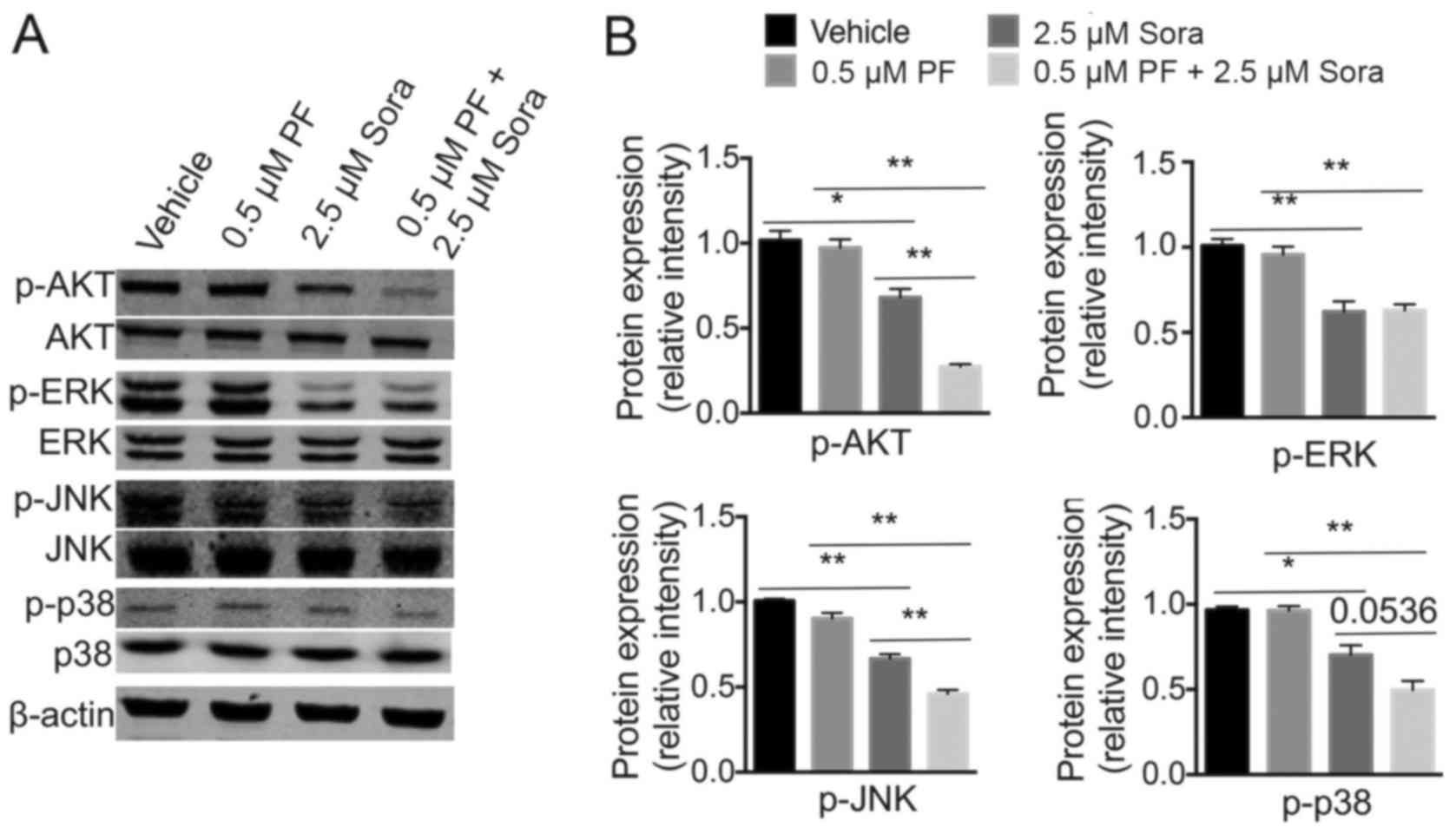
PI3K-AKT and MAPK are key downstream targets of the
VEGFR and c-Met signaling pathways, both of which regulate the
proliferation, apoptosis and migration of tumor cells (31). To investigate whether intracellular
AKT, JNK, p38 MAPK or ERK signaling were involved in the antitumor
ability of Sorafenib following the addition of low-dose PF-2341066,
the expression levels of these proteins were analyzed (Fig. 5A). Notably, when compared with single
Sorafenib treatment, the phosphorylation of AKT, JNK and p38 MAPK
was markedly inhibited following the addition of low-dose
PF-2341066.; However, ERK phosphorylation was not significantly
blocked by the combined treatment (Fig.
5B). These findings indicated that the PI3K-AKT and MAPK
signaling pathways are required for c-Met-mediated Sorafenib
sensitivity in NSCLC.
Discussion
NSCLC is one of the prevalent causes of
cancer-associated mortality globally. Additionally, drug resistance
in NSCLC further reduces the survival rate of patients (28,32).
Sorafenib has been demonstrated to have great effects on advanced
stages of liver and renal cancer (33). However, the efficacy of Sorafenib in
NSCLC is frequently limited by its unfavorable pharmacokinetics,
low tumor accumulation and other adverse effects (34).
In the present study, it was demonstrated that
Sorafenib in combination with a low-dose of PF-2341066 was able to
significantly inhibit the proliferation and migration, and promote
the apoptosis, of NCI-H1993 cells, compared with treatment with
Sorafenib or a low dose of PF-2341066 alone, indicating that a low
dose of c-Met inhibitor is able to effectively increase the
sensitivity of NSCLC cells to Sorafenib and subsequently decrease
the dose requirement. Further experiments indicated that the
PI3K-AKT and MAPK signaling pathways are biologically important for
c-Met-mediated sensitivity to Sorafenib. Therefore, data
demonstrated the success of combination therapy with a c-Met
inhibitor and Sorafenib in the treatment of NSCLC in an initial
in vitro study.
Raf is an important mediator of the small G-protein
signaling step within the MAPK signaling pathway that is frequently
activated in malignant tumors (35).
Raf amplification or mutation leads to activation of the ERK
signaling pathway, and therefore affects multiple tumor
characteristics, including uncontrolled proliferation, evasion from
apoptosis, invasion, distant metastasis, angiogenesis and immune
evasion (36,37). Sorafenib is able to effectively block
the Ras/Raf/mitogen-activated protein kinase kinase (MEK)/ERK
signaling pathway. Furthermore, Sorafenib inhibits the VEGF
signaling pathway, as it is a multikinase inhibitor (38). It has been confirmed that
molecular-targeting drugs act in a dose-dependent manner in NSCLC.
This is because development of drug resistance patients with NSCLC
is easy as a result of activation of alternative pathways (39–41).
Previous studies have demonstrated that combined EGFR and a
low-dose of c-Met inhibitors may be an effective treatment for
NSCLC (42–44). The data from the present study are
consistent with these previous reports, and further validate this
conclusion at the cellular and molecular levels. Therefore,
treatment with a low dose of c-Met inhibitor may increase the
sensitivity to Sorafenib in the patients with NSCLC.
In the present study, the selective c-Met inhibitor
PF-2341066 was used at a low concentration (0.5 µM) in combination
with Sorafenib (2.5 µM), compared with as a single agent. This may
markedly reduce side effects of Sorafenib in patients. Although a
high dose (>10 µM) of Sorafenib may achieve the same results as
combined a low dose of Sorafenib and PF-2341066, this may result in
more side effects for the patients. A combination of Sorafenib and
a low-dose of PF-2341066 was able to significantly reduce the
phosphorylation of AKT, JNK and p38 MAPK. However, the
phosphorylation of ERK was not significantly blocked by the
combined treatment, compared with treatment with a low dose of
Sorafenib. Sorafenib may exerts its effects on ERK via the
Ras/Raf/MEK/ERK signaling pathway (45), which may account for the similar
levels of phosphorylated ERK observed between the groups treated
with Sorafenib.
There were a number of limitations to the present
study. Firstly, the study was conducted using only one cell line,
and animal models were not used. Furthermore, a more detailed
mechanism must be investigated in order to explain the effects of
the combination of Sorafenib and low-dose c-Met inhibitor. It has
also been demonstrated that tumor stem cells serve important roles
in drug resistance in NSCLC (46).
Future studies will be required to focus on the effect of this
combined treatment on tumor stem cells.
In conclusion, the present study demonstrated that
treatment with a combination of Sorafenib and a low dose of c-Met
inhibitor was able to significantly inhibit the proliferation and
migration as well as promote the apoptosis of NCI-H1993 cells,
compared with treatment with Sorafenib alone. Furthermore, the
present study indicated there are more therapeutic choices for
patients with NSCLC who exhibit high c-Met and Raf/VEGFR expression
levels, indicating the importance of individualized therapy for
NSCLC.
Acknowledgements
The authors would like to thank Dr Yin Chen and Dr
Fei Chen at Shanghai Xinhua Hospital for assistance with
experiments. The authors also thank Dr James P. Mahaffey for
editing the English text of a draft of this manuscript.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ott DE and Marcu KB: Molecular
requirements for immunoglobulin heavy chain constant region gene
switch-recombination revealed with switch-substrate retroviruses.
Int Immunol. 1:582–591. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ettinger DS, Wood DE, Akerley W, Bazhenova
LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D'Amico TA,
Demmy TL, et al: Non-small cell lung cancer, version 1.2015. J Natl
Compr Canc Netw. 12:1738–1761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu CY, Wang CL, Li SH, Hsu PC, Chen CH,
Lin TY, Kuo CH, Fang YF, Ko HW, Yu CT, et al: The efficacy of 40 mg
versus dose de-escalation to less than 40 mg of afatinib (Giotrif)
as the first-line therapy for patients with primary lung
adenocarcinoma harboring favorable epidermal growth factor
mutations. Oncotarget. 8:97602–97612. 2017.PubMed/NCBI
|
|
6
|
Lategahn J, Keul M and Rauh D: Lessons to
be learned: The molecular basis of kinase-targeted therapies and
drug resistance in non-small cell lung cancer. Angew Chem Int Ed
Engl. Nov 27–2017.(Epub ahead of print).
|
|
7
|
Thabitha A, Dravid AA, Tripathi R and Lulu
SS: Database of transcription factors in lung cancer (DBTFLC): A
novel resource for exploring transcription factors associated with
lung cancer. J Cell Biochem. Dec 13–2017.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kharaziha P, Chioureas D, Baltatzis G,
Fonseca P, Rodriguez P, Gogvadze V, Lennartsson L, Björklund AC,
Zhivotovsky B, Grandér D, et al: Sorafenib-induced defective
autophagy promotes cell death by necroptosis. Oncotarget.
6:37066–37082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bruix J, Cheng AL, Meinhardt G, Nakajima
K, De Sanctis Y and Llovet J: Prognostic factors and predictors of
sorafenib benefit in patients with hepatocellular carcinoma:
Analysis of two phase III studies. J Hepatol. 67:999–1008. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tafreshi A, Thientosapol E, Liew MS, Guo
Y, Quaggiotto M, Boyer M and Davis ID: Efficacy of sorafenib in
advanced renal cell carcinoma independent of prior treatment,
histology or prognostic group. Asia Pac J Clin Oncol. 10:60–65.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kudo M: Immune checkpoint inhibition in
hepatocellular carcinoma: Basics and ongoing clinical trials.
Oncology. 92 Suppl 1:S50–S62. 2017. View Article : Google Scholar
|
|
12
|
Rautenberg C, Nachtkamp K, Dienst A,
Schmidt PV, Heyn C, Kondakci M, Germing U, Haas R, Kobbe G and
Schroeder T: Sorafenib and azacitidine as salvage therapy for
relapse of FLT3-ITD mutated AML after allo-SCT. Eur J Haematol.
98:348–354. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gridelli C, Maione P, Del Gaizo F,
Colantuoni G, Guerriero C, Ferrara C, Nicolella D, Comunale D, De
Vita A and Rossi A: Sorafenib and sunitinib in the treatment of
advanced non-small cell lung cancer. Oncologist. 12:191–200. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou Q, Guo X and Choksi R: Activation of
focal adhesion kinase and Src mediates acquired sorafenib
resistance in A549 human lung adenocarcinoma xenografts. J
Pharmacol Exp Ther. 363:428–443. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Degen A, Weichenthal M, Ugurel S, Trefzer
U, Kilian K, Garbe C, Egberts F, Poppe LM, Hauschild A and Gutzmer
R: Cutaneous side effects of combined therapy with sorafenib and
pegylated interferon alpha-2b in metastatic melanoma (phase II
DeCOG trial). J Dtsch Dermatol Ges. 11:846–853. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang H and Wang M: Mechanism of c-Met in
non-small cell lung cancer and its treatment and testing. Zhongguo
Fei Ai Za Zhi. 18:745–751. 2015.(In Chinese). PubMed/NCBI
|
|
18
|
Ma PC, Jagadeeswaran R, Jagadeesh S,
Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K,
Lader A, et al: Functional expression and mutations of c-Met and
its therapeutic inhibition with SU11274 and small interfering RNA
in non-small cell lung cancer. Cancer Res. 65:1479–1488. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deying W, Feng G, Shumei L, Hui Z, Ming L
and Hongqing W: CAF-derived HGF promotes cell proliferation and
drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78
signalling in ovarian cancer cells. Biosci Rep. 37(pii):
BSR201604702017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gohda E, Tsubouchi H, Nakayama H, Hirono
S, Sakiyama O, Takahashi K, Miyazaki H, Hashimoto S and Daikuhara
Y: Purification and partial characterization of hepatocyte growth
factor from plasma of a patient with fulminant hepatic failure. J
Clin Invest. 81:414–419. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Konstorum A and Lowengrub JS: Activation
of the HGF/c-Met axis in the tumor microenvironment: A multispecies
model. J Theor Biol. 439:86–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kucerova L, Demkova L, Skolekova S,
Bohovic R and Matuskova M: Tyrosine kinase inhibitor SU11274
increased tumorigenicity and enriched for melanoma-initiating cells
by bioenergetic modulation. BMC Cancer. 16:3082016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zou HY, Li Q, Lee JH, Arango ME, McDonnell
SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, et al: An
orally available small-molecule inhibitor of c-Met, PF-2341066,
exhibits cytoreductive antitumor efficacy through antiproliferative
and antiangiogenic mechanisms. Cancer Res. 67:4408–4417. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cascone T, Xu L, Lin HY, Liu W, Tran HT,
Liu Y, Howells K, Haddad V, Hanrahan E, Nilsson MB, et al: The
HGF/c-Met pathway is a driver and biomarker of VEGFR-inhibitor
resistance and vascular remodeling in non-small cell lung cancer.
Clin Cancer Res. 23:5489–5501. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tarhini AA, Rafique I, Floros T, Tran P,
Gooding WE, Villaruz LC, Burns TF, Friedland DM, Petro DP, Farooqui
M, et al: PPhase 1/2 study of rilotumumab (AMG 102), a hepatocyte
growth factor inhibitor, and erlotinib in patients with advanced
non-small cell lung cancer. Cancer. 123:2936–2944. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bignold LP, Ferrante A and Haynes DR:
Studies of chemotactic, chemotactic movement-inhibiting and random
movement-inhibiting effects of interleukin-1 alpha and beta, tumour
necrosis factor alpha and beta and interferon gamma on human
neutrophils in assays using ‘sparse-pore’ polycarbonate (Nuclepore)
membranes in the Boyden chamber. Int Arch Allergy Appl Immunol.
91:1–7. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamaoka T, Ohmori T, Ohba M, Arata S,
Murata Y, Kusumoto S, Ando K, Ishida H, Ohnishi T and Sasaki Y:
Distinct afatinib resistance mechanisms identified in lung
adenocarcinoma harboring an EGFR mutation. Mol Cancer Res.
15:915–928. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao J, Fang L, Zhang X, Liang Y and Gou
S: Synthesis and biological evaluation of new
[1,2,4]triazolo[4,3-a]pyridine derivatives as potential c-Met
inhibitors. Bioorg Med Chem. 24:3483–3493. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li D, Yang H, Li R, Wang Y, Wang W, Li D,
Ma S and Zhang X: Antitumor activity of gambogic acid on NCI-H1993
xenografts via MET signaling pathway downregulation. Oncol Lett.
10:2802–2806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao L, Zhu Z, Yao C, Huang Y, Zhi E, Chen
H, Tian R, Li P, Yuan Q, Xue Y, et al: VEGFC/VEGFR3 signaling
regulates mouse spermatogonial cell proliferation via the
activation of AKT/MAPK and cyclin D1 pathway and mediates the
apoptosis by affecting caspase 3/9 and Bcl-2. Cell Cycle. 1–50.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Katayama R: Therapeutic strategies and
mechanisms of drug resistance in anaplastic lymphoma kinase
(ALK)-rearranged lung cancer. Pharmacol Ther. 177:1–8. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu YJ, Zheng B, Wang HY and Chen L: New
knowledge of the mechanisms of sorafenib resistance in liver
cancer. Acta Pharmacol Sin. 38:614–622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murray M, Gillani TB, Ghassabian S,
Edwards RJ and Rawling T: Differential effects of hepatic cirrhosis
on the intrinsic clearances of sorafenib and imatinib by CYPs in
human liver. Eur J Pharm Sci. 114:55–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bahrami A, Hassanian SM, ShahidSales S,
Farjami Z, Hasanzadeh M, Anvari K, Aledavood A, Maftouh M, Ferns
GA, Khazaei M and Avan A: Targeting RAS signaling pathway as a
potential therapeutic target in the treatment of colorectal cancer.
J Cell Physiol. 233:2058–2066. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu H, Zhang Q, Li K, Gong Z, Liu Z, Xu Y,
Swaney MH, Xiao K and Chen Y: Prognostic significance of USP33 in
advanced colorectal cancer patients: New insights into
β-arrestin-dependent ERK signaling. Oncotarget. 7:81223–4020. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li XL, Chen XQ, Zhang MN, Chen N, Nie L,
Xu M, Gong J, Shen PF, Su ZZ, Weng X, et al: SOX9 was involved in
TKIs resistance in renal cell carcinoma via Raf/MEK/ERK signaling
pathway. Int J Clin Exp Pathol. 8:3871–3881. 2015.PubMed/NCBI
|
|
38
|
Ranieri G, Gadaleta-Caldarola G, Goffredo
V, Patruno R, Mangia A, Rizzo A, Sciorsci RL and Gadaleta CD:
Sorafenib (BAY 43–9006) in hepatocellular carcinoma patients: From
discovery to clinical development. Curr Med Chem. 19:938–944. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bonanno L, Jirillo A and Favaretto A:
Mechanisms of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors and new therapeutic
perspectives in non small cell lung cancer. Curr Drug Targets.
12:922–933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Polverino A, Coxon A, Starnes C, Diaz Z,
DeMelfi T, Wang L, Bready J, Estrada J, Cattley R, Kaufman S, et
al: AMG 706, an oral, multikinase inhibitor that selectively
targets vascular endothelial growth factor, platelet-derived growth
factor, and kit receptors, potently inhibits angiogenesis and
induces regression in tumor xenografts. Cancer Res. 66:8715–8721.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stabile LP, Rothstein ME, Keohavong P,
Lenzner D, Land SR, Gaither-Davis AL, Kim KJ, Kaminski N and
Siegfried JM: Targeting of both the c-Met and EGFR pathways results
in additive inhibition of lung tumorigenesis in transgenic mice.
Cancers (Basel). 2:2153–2170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu YL, Soo RA, Locatelli G, Stammberger U,
Scagliotti G and Park K: Does c-Met remain a rational target for
therapy in patients with EGFR TKI-resistant non-small cell lung
cancer? Cancer Treat Rev. 61:70–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Martinez-Marti A, Felip E, Matito J, Mereu
E, Navarro A, Cedrés S, Pardo N, Martinez de Castro A, Remon J,
Miquel JM, et al: Dual MET and ERBB inhibition overcomes intratumor
plasticity in osimertinib-resistant-advanced non-small-cell lung
cancer (NSCLC). Ann Oncol. 28:2451–2457. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen JC, Chuang HY, Hsu FT, Chen YC, Chien
YC and Hwang JJ: SSorafenib pretreatment enhances radiotherapy
through targeting MEK/ERK/NF-κB pathway in human hepatocellular
carcinoma-bearing mouse model. Oncotarget. 7:85450–85463. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pisanu ME, Noto A, De Vitis C, Morrone S,
Scognamiglio G, Botti G, Venuta F, Diso D, Jakopin Z, Padula F, et
al: Blockade of Stearoyl-CoA-desaturase 1 activity reverts
resistance to cisplatin in lung cancer stem cells. Cancer Lett.
406:93–104. 2017. View Article : Google Scholar : PubMed/NCBI
|