Introduction
Breast cancer is the most commonly diagnosed cancer
in women and approximately accounts for 29% of all new cancers in
women, and it is the leading cause of cancer-related death in women
worldwide (1). Breast cancer is
considered as a heterogeneous disease but not a single disease at
molecular and clinical levels (2,3). The
well-known characteristics of breast cancer-associated factors
include pathologic and clinical characteristics of the primary
tumor, tumor histology, axillary lymph node (ALN) status, estrogen
receptor (ER) content, progesterone receptor (PR) content, content,
tumor HER2 status, detectable metastatic disease, patient age,
patient comorbid conditions, and menopausal status (3,4). Based on
the determination of ER, PR, HER2, and Ki-67, breast cancer is
often to be accepted as four subtypes, namely Luminal A, Luminal B,
Erb-B2 overexpression and Basal-like (also known as Triple negative
breast cancer) according to St Gallen (3). Luminal A (ER positive, PR positive,
HER2/neu negative) is the most common subtype, accounting for more
than 50% of all breast cancer patients (5,6).
Multigene predictors have been introduced by various
technologies, including immunohistochemistry (IHC), reverse
transcription-quantitative polymerase chain reaction (RT-qPCR),
fluorescence in situ hybridization (FISH) and genomic
microarrays (7). Nowadays, some
microarray-based multigene predictors have been developed as
predictors of response to hormonal therapy (8,9),
predictors of response to multiagent cytotoxic chemotherapy
(10–13) and independent prognostic biomarkers
(14–16).
Studies have shown that the recurrence score based
on a 21-gene assay is a recurrence predictor for breast cancer
patients receiving adjuvant endocrine therapy (17–19).
Recurrence score is an independent predict factor for the response
to adjuvant chemotherapy (20,21).
Patients with high scores could benefit from adjuvant treatments,
whereas those with low scores could not regardless of the
pathologic and clinical characteristics. In ATAC trial (22), the risk of recurrence score obtained
using a 50-gene assay was seen to have an obvious relationship with
the 10 years distant recurrence risk in postmenopausal breast
cancer women treated with tamoxifen or aromatase inhibitors. In
addition, a first commercialized microarray-based multigene assay
containing 70 genes, primarily associated with proliferation,
metastasis, invasion, stromal integrity, and angiogenesis, is
approved by the FDA's new in diagnostic multivariate index assay
classification (7).
Though multigene predictors have been widely
investigated and used for the breast cancer, there are still rare
studies focusing on the prognosis of subtypes. According to
retrospective analyses and authoritative guidelines, these subtypes
are associated with different relapse-free survival and overall
survival and the patients with different subtypes should be
administrated with different systemic treatment strategies
(3). In this study, we aimed to
utilize microarray profiling to investigate potential biomarkers
that are differentially expressed in women with Luminal A-like
breast cancer based on significant pathways analysis through gene
expression profiles analysis. To validate the ability of the
candidate multigene assay for the prediction of clinical outcomes,
the gene expression data and survival data of Luminal A breast
cancer patients were downloaded from Gene Expression Omnibus for
analysis.
Materials and methods
Gene expression data
The gene expression profiles of breast cancer
patients were downloaded from The Cancer Genome Atlas (TCGA,
https://cancergenome.nih.gov/) database
with the deadline of December 27, 2016, including 20,501 genes
obtained from 1,160 samples (1,041 tumor tissue samples, 112 normal
tissue samples and 7 peripheral blood samples). According to the
clinical information of ER, PR and HER2 information (23), 370 Luminal A breast cancer samples
were screened.
Data processing and differentially
expressed genes (DEGs) identifying
The expression profile data of Luminal A patients
were normalized. Z-score correction method was utilized to rule out
the difference at gene expression level (24). A total of 249 Luminal A samples from
alive patients were assigned as good outcome group; whereas a total
of 47 Luminal A samples from dead patients were assigned as poor
outcome group. P<0.01 and |log2 Fold-Change (FC)|
>1 were regarded as the cut-off criteria to screen out DEGs
between the good and poor outcome groups using LIMMA package
(25).
Hierarchical clustering analysis
Hierarchical clustering analysis was conducted for
the DEGs using heatmap2 package in R language (26) and the result was visualized using the
form of heatmap.
Identifying statistically significant
pathways
The pathway information were download from Kyoto
Encyclopedia of Genes and Genomes (KEGG) (http://www.kegg.jp/kegg/pathway.html) database on
March 1, 2017. The KEGG pathway enrichment analyses were performed
based on pathway feature vector calculation (27) and nearest shrunken centroids (28). Briefly, the KEGG pathway was scored
using the expression values of the DEGs in all samples. The sample
was projected by taking the upregulated score and downregulated
score as coordinates. The accuracy of the good group and the poor
group was evaluated by calculating the geometric center of the same
sample and specifying the radius (27). The pathways with accuracy more than
80% in these two groups were screened. The statistically
significant pathways were recognized by calculating the Ratio of
rank and converting to P-value according to the random 10,000 times
perturbation of the background library (train set samples)
(28).
Identifying prognostic biomarkers and
training
Survival analysis for Luminal A breast cancer
samples in TCGA were performed using DEGs involved in the obtained
significant pathways (29). The
support vector machine (SVM) classification model was constructed
using these DEGs. Meanwhile, the model was trained using the
Luminal A samples and the receiver operating characteristic (ROC)
curve was drawn.
Verification of multigene prognostic
assay
The reliability and repeatability of the multigene
assay were verified using the gene expression profiles of GSE2034
(https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE2034)
and the survival data of Luminal A breast cancer patients was
downloaded from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) database.
Besides, the SVM model was also utilized to test the multigene
assay, and the accuracy of the model was analyzed using the ROC
curve.
Results
DEGs for Luminal A breast cancer with
good and poor outcome groups
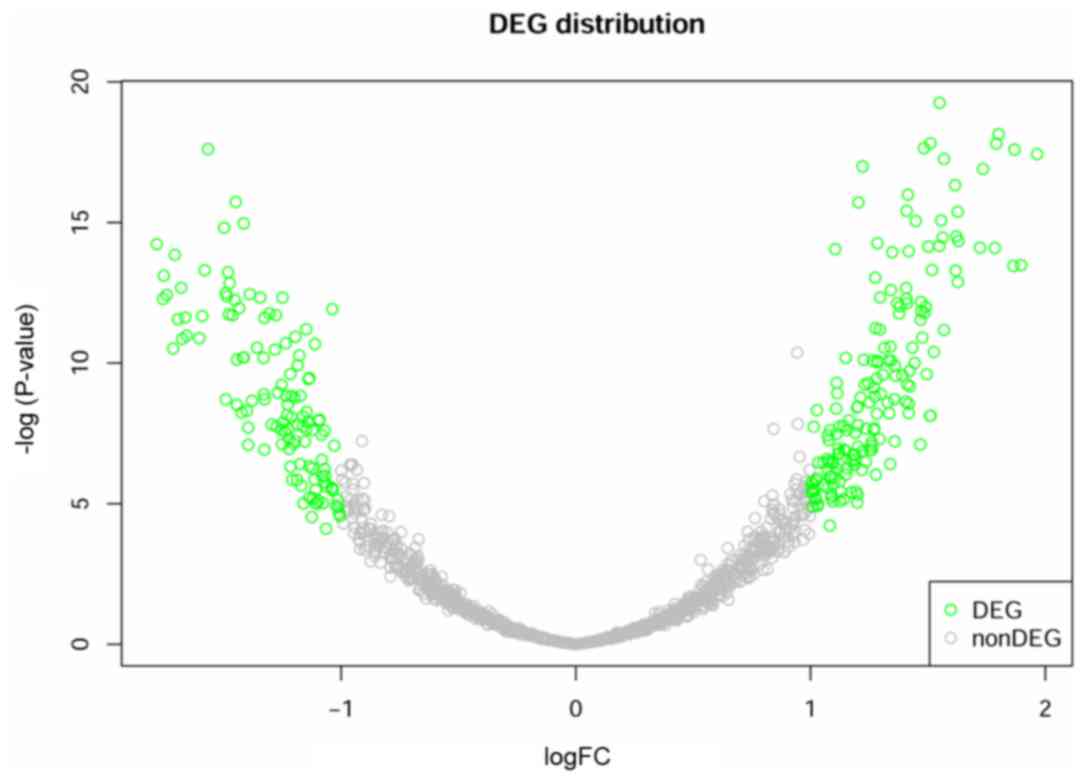
Using LIMMA package, a total of 300 DEGs were
identified between 249 samples from good outcome group and 47
samples from poor outcome group, including 176 upregulated genes
and 124 downregulated genes (Fig. 1).
It can be observed from the figure that the data are homogenized to
eliminate the deviation and the deviation scores of most genes were
concentrated in −1 to 1. The genes distributed in the two branches
were the most significant DEGs.
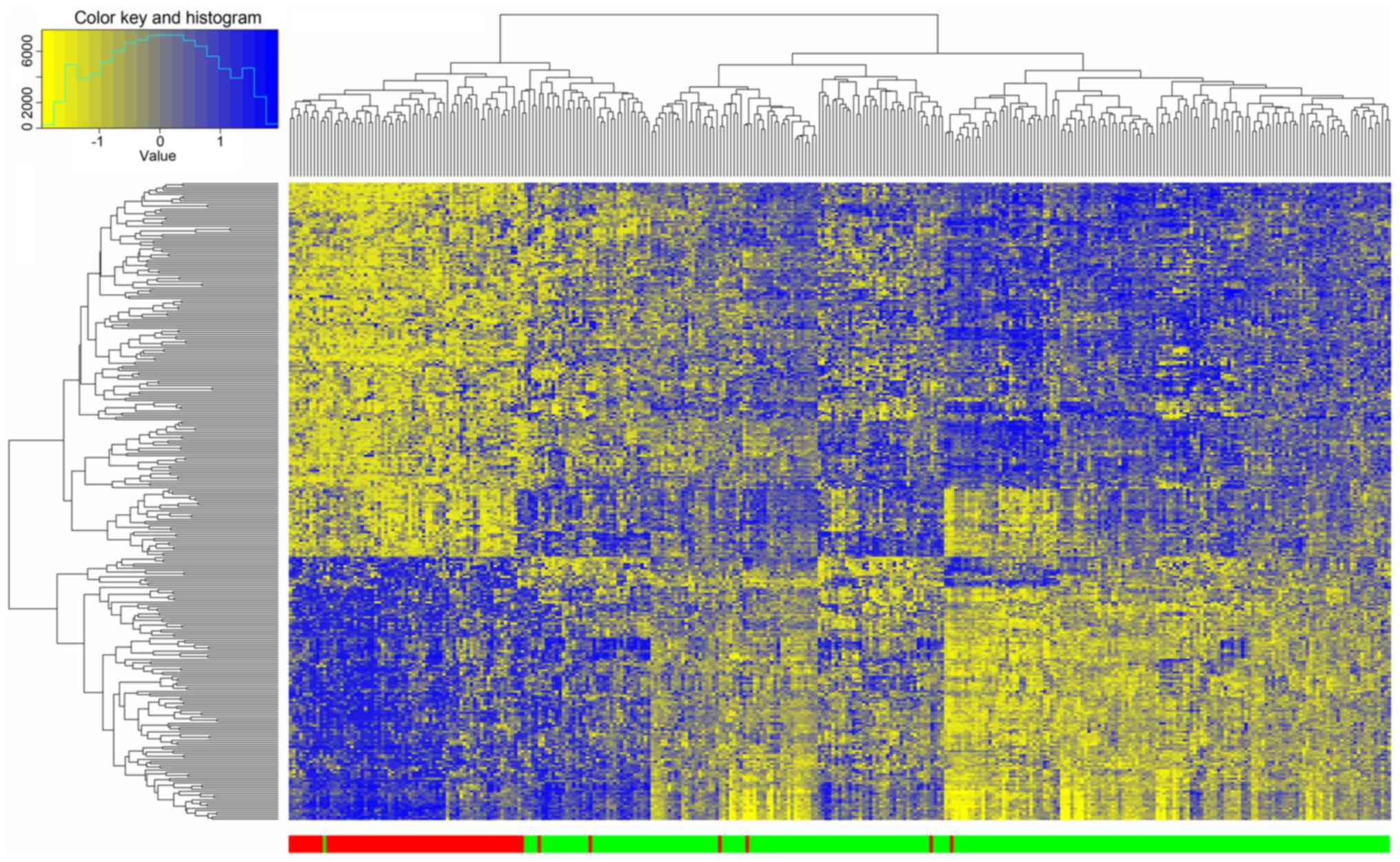
Clustering of DEGs
The 300 DEGs identified between good and poor
outcome groups were selected for hierarchical clustering analysis.
As presented in Fig. 2, 68 of 74 dead
samples were clustered together and only 6 samples were clustered
to the alive group with a precision of 92%. Meanwhile, 248 of 249
alive samples were clustered together and only 1 sample was
clustered to the dead group with a precision of 99.6%. This result
indicated that the DEGs could be used to effectively distinguish
Luminal A samples with different prognoses.
Statistically significant
pathways
Total 9 pathways with accuracy more than 80% in the
two groups were screened. The significance analysis for these 9
pathways was conducted using Nearest Shrunken Centroids and the
results are shown in Table I. It was
observed that all of the 9 pathways significantly distinguished
cancer samples with good outcome from cancer samples with poor
outcome (P<0.05).
 | Table I.Nine significant pathways according
to Kyoto Encyclopedia of Genes and Genomes analysis nearest
shrunken centroids. |
Table I.
Nine significant pathways according
to Kyoto Encyclopedia of Genes and Genomes analysis nearest
shrunken centroids.
| Pathway | Initial
precision | Average value | P-value |
|---|
| Colorectal
cancer | 0.7814 | 0.5775 | 1.27E-12 |
| Oocyte meiosis | 0.8050 | 0.6291 | 9.24E-09 |
| Wnt signaling
pathway | 0.7033 | 0.5088 | 3.38E-06 |
| Terpenoid backbone
biosynthesis | 0.6268 | 0.4298 | 5.80E-04 |
| Endometrial
cancer | 0.5582 | 0.4769 | 7.90E-04 |
| Cell cycle | 0.5843 | 0.4648 | 2.81E-03 |
| Proteasome | 0.5422 | 0.4178 | 1.13E-02 |
| Basal cell
carcinoma | 0.5715 | 0.4104 | 1.50E-02 |
| Small cell lung
cancer | 0.5567 | 0.3995 | 1.72E-02 |
Identifying prognostic biomarkers
based on the significant pathways
The DEGs involved in these 9 significant pathways
were collected and a total of 18 DEGs were identified as prognostic
biomarkers (Table II). Three genes
[transcription factor 7-like 2 (TCF7L2), anterior parietal
cortex (APC), and lymphocyte enhancer factor-1
(LEF1)], were involved in four pathways, one gene [cyclin E1
(CCNE1)] in three pathways, four [S-phase kinase-associated
protein 2 (SKP2), human frizzled-7 (FZD7), polo-like
kinase 1 (PLK1), and B cell lymphoma 2 (BCL2)] in two
pathways and ten [proteasome activator subunit 4 (PSME4),
prenyldiphosphate synthase, subunit 1 (PDSS1), promoters for
human DNA-PK cs (PRKDC), TTK protein kinase (TTK),
minichromosome maintenance deficient 4 (MCM4), progesterone
receptor (PGR), proteasome subunit alpha 7 (PSMA7),
MDM2 proto-oncogene (MDM2), laminin subunit beta 2
(LAMB2), and proteasome 26S subunit, non-ATPase 7
(PSMD7)] in one pathway. Meanwhile, the annotation results
for the 18 genes based on the TCGA database are shown in Fig. 3A. Simultaneously, the heat map was
shown for the changes of the 18 DEGs in TCGA breast cancer samples
(Fig. 3B). It can be seen that the
expression of these 18 DEGs in TCGA breast cancer samples are
almost upregulated.
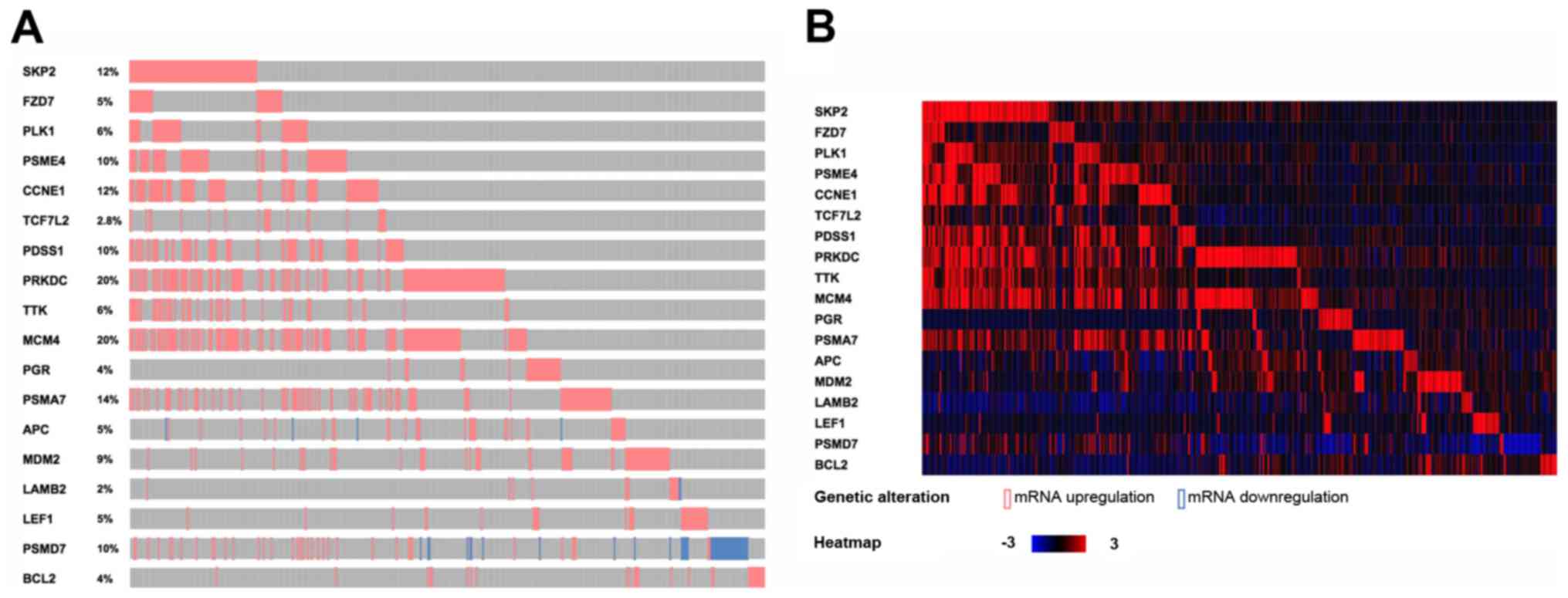 | Figure 3.The annotation results and the heat
map of 18 biomarkers in Luminal A breast cancer samples from the
TCGA database. (A) The annotation results of 18 biomarkers in the
TCGA database. Red represents upregulation and blue represents
downregulation. The proportion of each gene mutation is also
marked. (B) The heat map for the changes of the 18 genes in TCGA
breast cancer samples. Red represents upregulation and blue
represents downregulation. TCGA, The Cancer Genome Atlas;
TCF7L2, transcription factor 7-like 2; APC, anterior
parietal cortex; LEF1, lymphocyte enhancer factor-1;
CCNE1, cyclin E1; SKP2, S-phase kinase-associated
protein 2; FZD7, human frizzled-7; PLK1, polo-like
kinase 1; BCL2, B cell lymphoma 2; PSME4, proteasome
activator subunit 4; PDSS1, prenyldiphosphate synthase,
subunit 1; PRKDC, promoters for human DNA-PK cs; TTK,
TTK protein kinase; MCM4, minichromosome maintenance
deficient 4; PGR, progesterone receptor; PSMA7,
proteasome subunit α7; MDM2, MDM2 proto-oncogene;
LAMB2, laminin subunit β2; PSMD7, proteasome 26S
subunit, non-ATPase 7. |
| Table II.DEGs identified as prognostic
biomarkers based on the significant pathways. |
Table II.
DEGs identified as prognostic
biomarkers based on the significant pathways.
| Biomarkers | Pathway | Counts |
|---|
| TCF7L2 | Wnt signaling
pathway, colorectal cancer, endometrial cancer, basal cell
carcinoma | 4 |
| APC | Wnt signaling
pathway, colorectal cancer, endometrial cancer, basal cell
carcinoma | 4 |
| LEF1 | Wnt signaling
pathway, colorectal cancer, endometrial cancer, B cell
carcinoma | 4 |
| CCNE1 | Cell cycle, oocyte
meiosis, small cell lung cancer | 3 |
| SKP2 | Cell cycle, small
cell lung cancer | 2 |
| FZD7 | Wnt signaling
pathway, basal cell carcinoma | 2 |
| PLK1 | Cell cycle, oocyte
meiosis | 2 |
| BCL2 | Colorectal cancer,
small cell lung cancer | 2 |
| PSME4 | Proteasome | 1 |
| PDSS1 | Terpenoid backbone
biosynthesis | 1 |
| PRKDC | Cell cycle | 1 |
| TTK | Cell cycle | 1 |
| MCM4 | Cell cycle | 1 |
| PGR | Oocyte meiosis | 1 |
| PSMA7 | Proteasome | 1 |
| MDM2 | Cell cycle | 1 |
| LAMB2 | Small cell lung
cancer | 1 |
| PSMD7 | Proteasome | 1 |
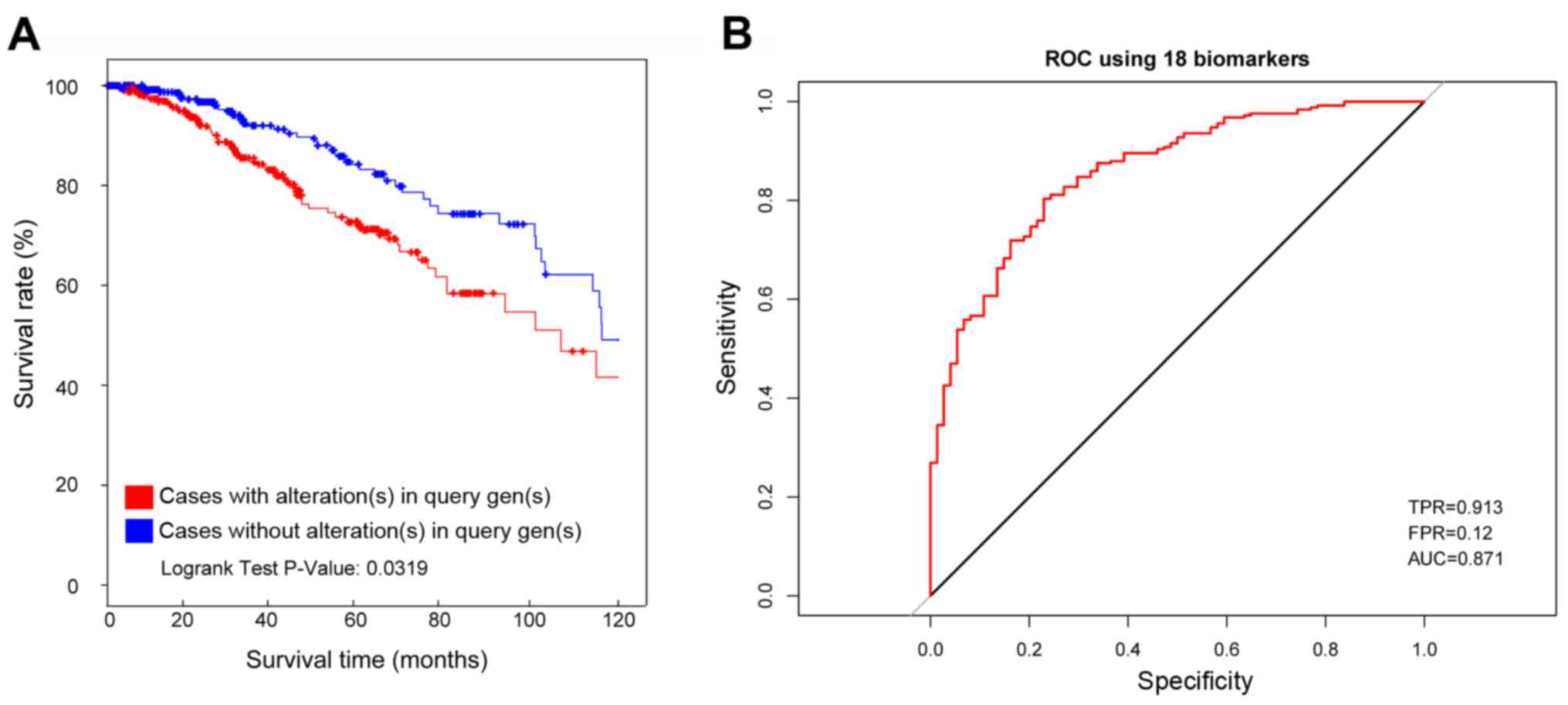
Survival analysis for the 18 DEGs in
train set
The survival analysis was conducted for the Luminal
A breast patient samples with abnormal expression of these 18 genes
and samples with normal expression of these 18 genes in TCGA
database. As a result, the samples with abnormal expression of
these 18 genes showed a significantly lower survival rate than
samples with normal expression of these 18 genes (Fig. 4A, P=0.0319).
Additionally, the average AUC (area under curve) was
0.871 for the ROC of these 18 genes calculated from the random
train set in TCGA database. The sensitivity was 0.913 and the
specificity was 0.88 (Fig. 4B).
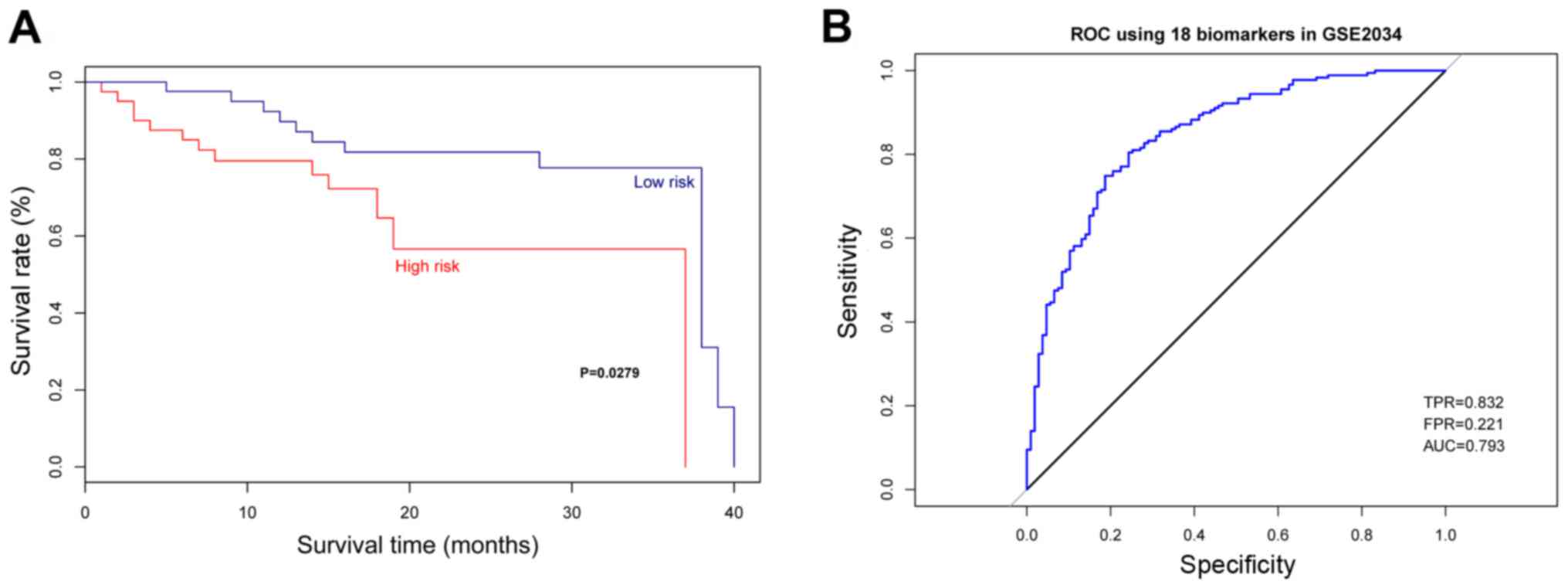
The verification of multigene
prognostic assay in test set
The multigene assay was verified using the Luminal A
breast patient samples with abnormal expression of these 18 genes
and samples with normal expression of these 18 genes obtained from
GSE2034 database. As a result, the samples with abnormal expression
of these 18 genes showed a significantly lower survival rate than
samples with normal expression of these 18 genes (Fig. 5A, P=0.0279). According to the ROC for
the test set, the average AUC was 0.793 with sensitivity of 0.832
and specificity of 0.779 (Fig.
5B).
Discussion
With the promotion and put forward of precision
medical, studies focusing on special subtypes of breast cancer are
particularly meaningful. Regardless of the development of the
multigene prognostic assay for breast cancer, our study still has a
critical necessary for the prognostic study of breast cancer by
focusing on the Luminal A subtype. According to our results, a
total of 300 DEGs were identified between good prognosis group and
poor prognosis group, including 176 upregulated genes and 124
downregulated genes. Based on the hierarchical clustering analysis,
these DEGs could clearly distinguish the samples of the two groups.
Meanwhile, the 18 genes predictors were involved in 9 significant
pathways, including cancer-related pathways (colorectal cancer,
endometrial cancer, basal cell carcinoma and small cell lung
cancer), oocyte meiosis, Wnt signaling pathway, Terpenoid backbone
biosynthesis, cell cycle and proteasome were selected. According to
the survival analysis and ROC curve, the obtained 18-gene
prognostic assay exhibited a good prognostic function with high
sensitivity and specificity for the train set samples and
verification set samples.
TCF7L2 was identified as a key gene in the
multigene prognostic assay and it was involved in four significant
pathways, namely Wnt signaling pathway, colorectal cancer,
endometrial cancer and basal cell carcinoma, based on the pathway
analysis. TCF7L2, located on chromosome 10q25.2, plays a
critical role in cancer cell growth, apoptosis, invasion and
metastasis by regulating Wnt signalling (30,31). The
regulation roles of TCF7L2 gene and related Wnt signaling
pathway in breast cancer and its special subtypes have been widely
confirmed (32–34). Several studies have exploited the
association between the gene polymorphisms of TCF7L2 and the
risk of breast cancer. Naidu et al (34) and Burwinkel et al (35) reported that TCF7L2 variants
induced an increased breast cancer risk, and might be a potential
candidate for the susceptibility of breast cancer. Additionally,
the TCF7L2 protein is involved in the homeostasis of blood glucose
and the gene polymorphisms of TCF7L2 are identified to
increase the risk of type 2 diabetes (36). Diabetes have been reported to be
associated with the increased risk of breast cancer and the similar
results were seen in Luminal A and B subtypes (37). Consistent with these previous studies,
TCF7L2 gene was screened as a key DEG between patients with
good outcomes and poor outcomes in Luminal A breast cancer.
In addition to TCF7L2, APC and LEF1
are also involved in the above mentioned four significant pathways.
The association between APC and the prognosis of breast
cancer has also been reported. In a study conducted by Müller et
al, the methylated APC DNA indicated the worst prognosis
in breast cancer samples from the train set and the independent
test set (P<0.001) and it was considered as an potentially
independent prognostic factor for breast cancer with poor outcomes
(38). The prognostic importance of
APC was also been confirmed by a study which discovered that
the deletion of APC was associated with a poor overall
survival of breast cancer patients (39). According to the hierarchical analysis,
the alterations of APC were significantly higher in
ER-/PR-breast cancer compared with ER+/PR+ breast cancer samples
(39). In general, patients with
ER-/PR-have a worse outcome than patients with ER+/PR+. Thus, it is
reasonable to speculate that the abnormal expression of APC
might be associated poor outcome in Luminal A (ER+/PR+) breast
cancer. LEF1 has been widely reported to promote cancer cell
metastasis, mediate chemotactic invasion, and is associated with
cancer progression (40). The LEF1
overexpression has been identified as a prognostic factor for poor
outcome and increased risk of liver metastasis in primary
colorectal cancer (41). Delaunay
et al reported that the depletion of LEF1 strongly
decreased the chemotactic potential of breast cancer cells and the
expression level of LEF1 was associated with the risk of
developing metastasis in breast cancer patients (42). As expected, the expression of
LEF1 was significantly different between good and poor
outcome groups and it was screened as a key DEG according to our
analysis.
The 18-gene prognostic assay including the three key
genes, TCF7L2, APC and LEF1, was also demonstrated
with an accurate ability to distinguish good outcomes and poor
outcomes in Luminal A breast cancer. To meet the background of the
precision treatment of our study would enrich the research field of
specific multi-gene prognosis for breast cancer subtypes. Further
study with large samples should be conducted to verify the
prognostic value of this 18-gene prognostic assay and prospective
study is also needed.
By conducting survival analysis, the 18-multigene
assay showed effective distinguish effect on patients with
different prognosis status in the low risk and high risk groups.
However, the prognosis in these two groups was extraordinarily
poor. The 18-gene prognostic assay should be verified in more
Luminal A breast cancer samples with more typical prognosis status
in further experiment.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aure MR, Vitelli V, Jernström S, Kumar S,
Krohn M, Due EU, Haukaas TH, Leivonen SK, Vollan HK, Lüders T, et
al: Integrative clustering reveals a novel split in the Luminal A
subtype of breast cancer with impact on outcome. Breast Cancer Res.
19:442017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goldhirsch A, Winer EP, Coates AS, Gelber
RD, Piccart-Gebhart M, Thürlimann B and Senn HJ Panel members:
Personalizing the treatment of women with early breast cancer:
Highlights of the St Gallen international expert consensus on the
primary therapy of early breast cancer 2013. Ann Oncol.
24:2206–2223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jagsi R, Raad RA, Goldberg S, Sullivan T,
Michaelson J, Powell SN and Taghian AG: Locoregional recurrence
rates and prognostic factors for failure in node-negative patients
treated with mastectomy: Implications for postmastectomy radiation.
Int J Radiat Oncol Biol Phys. 62:1035–1039. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carey LA, Perou CM, Livasy CA, Dressler
LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S,
et al: Race, breast cancer subtypes, and survival in the Carolina
breast cancer study. JAMA. 295:2492–2502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McDermott AM, Miller N, Wall D, Martyn LM,
Ball G, Sweeney KJ and Kerin MJ: Identification and validation of
oncologic miRNA biomarkers for Luminal A-like breast cancer. PLoS
One. 9:e870322014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ross JS, Hatzis C, Symmans WF, Pusztai L
and Hortobágyi GN: Commercialized multigene predictors of clinical
outcome for breast cancer. Oncologist. 13:477–493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van 't Veer LJ, Dai H, van de Vijver MJ,
He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ,
Witteveen AT, et al: Gene expression profiling predicts clinical
outcome of breast cancer. Nature. 415:530–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma XJ, Wang Z, Ryan PD, Isakoff SJ,
Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT,
et al: A two-gene expression ratio predicts clinical outcome in
breast cancer patients treated with tamoxifen. Cancer Cell.
5:607–616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang JC, Wooten EC, Tsimelzon A,
Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK,
Chamness GC, Allred DC and O'Connell P: Gene expression profiling
for the prediction of therapeutic response to docetaxel in patients
with breast cancer. Lancet. 362:362–369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang JC, Wooten EC, Tsimelzon A,
Hilsenbeck SG, Gutierrez MC, Tham YL, Kalidas M, Elledge R, Mohsin
S, Osborne CK, et al: Patterns of resistance and incomplete
response to docetaxel by gene expression profiling in breast cancer
patients. J Clin Oncol. 23:1169–1177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cleator S, Tsimelzon A, Ashworth A,
Dowsett M, Dexter T, Powles T, Hilsenbeck S, Wong H, Osborne CK,
O'Connell P and Chang JC: Gene expression patterns for doxorubicin
(Adriamycin) and cyclophosphamide (cytoxan) (AC) response and
resistance. Breast Cancer Res Treat. 95:229–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peintinger F, Anderson K, Mazouni C,
Kuerer HM, Hatzis C, Lin F, Hortobagyi GN, Symmans WF and Pusztai
L: Thirty-gene pharmacogenomic test correlates with residual cancer
burden after preoperative chemotherapy for breast cancer. Clin
Cancer Res. 13:4078–4082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu R, Wang X, Chen GY, Dalerba P, Gurney
A, Hoey T, Sherlock G, Lewicki J, Shedden K and Clarke MF: The
prognostic role of a gene signature from tumorigenic breast-cancer
cells. N Engl J Med. 356:217–226. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Desmedt C, Piette F, Loi S, Wang Y,
Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y,
d'Assignies MS, et al: Strong time dependence of the 76-gene
prognostic signature for node-negative breast cancer patients in
the TRANSBIG multicenter independent validation series. Clin Cancer
Res. 13:3207–3214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM,
Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu
J, et al: Gene-expression profiles to predict distant metastasis of
lymph-node-negative primary breast cancer. Lancet. 365:671–679.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paik S, Shak S, Tang G, Kim C, Baker J,
Cronin M, Baehner FL, Walker MG, Watson D, Park T, et al: A
multigene assay to predict recurrence of tamoxifen-treated,
node-negative breast cancer. N Engl J Med. 351:2817–2826. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dowsett M, Cuzick J, Wale C, Forbes J,
Mallon EA, Salter J, Quinn E, Dunbier A, Baum M, Buzdar A, et al:
Prediction of risk of distant recurrence using the 21-gene
recurrence score in node-negative and node-positive postmenopausal
patients with breast cancer treated with anastrozole or tamoxifen:
A TransATAC study. J Clin Oncol. 28:1829–1834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mamounas EP, Tang G, Fisher B, Paik S,
Shak S, Costantino JP, Watson D, Geyer CE Jr, Wickerham DL and
Wolmark N: Association between the 21-gene recurrence score assay
and risk of locoregional recurrence in node-negative, estrogen
receptor-positive breast cancer: Results from NSABP B-14 and NSABP
B-20. J Clin Oncol. 28:1677–1683. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang G, Shak S, Paik S, Anderson SJ,
Costantino JP, Geyer CE Jr, Mamounas EP, Wickerham DL and Wolmark
N: Comparison of the prognostic and predictive utilities of the
21-gene recurrence score assay and Adjuvant! for women with
node-negative, ER-positive breast cancer: Results from NSABP B-14
and NSABP B-20. Breast Cancer Res Treat. 127:133–142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Albain KS, Barlow WE, Shak S, Hortobagyi
GN, Livingston RB, Yeh IT, Ravdin P, Bugarini R, Baehner FL,
Davidson NE, et al: Prognostic and predictive value of the 21-gene
recurrence score assay in postmenopausal women with node-positive,
oestrogen-receptor-positive breast cancer on chemotherapy: A
retrospective analysis of a randomised trial. Lancet Oncol.
11:55–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dowsett M, Sestak I, Lopez-Knowles E,
Sidhu K, Dunbier AK, Cowens JW, Ferree S, Storhoff J, Schaper C and
Cuzick J: Comparison of PAM50 risk of recurrence score with
oncotype DX and IHC4 for predicting risk of distant recurrence
after endocrine therapy. J Clin Oncol. 31:2783–2790. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu X, Ying J, Wang F, Wang J and Yang H:
Estrogen receptor, progesterone receptor, and human epidermal
growth factor receptor 2 status in invasive breast cancer: A 3,198
cases study at National cancer center, China. Breast Cancer Res
Treat. 147:551–555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomas JG, Olson JM, Tapscott SJ and Zhao
LP: An efficient and robust statistical modeling approach to
discover differentially expressed genes using genomic expression
profiles. Genome Res. 11:1227–1236. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Xie J, Yang J, Fennell A, Zhang C
and Ma Q: QUBIC: A bioconductor package for qualitative
biclustering analysis of gene co-expression data. Bioinformatics.
33:450–452. 2017.PubMed/NCBI
|
|
27
|
Chen X, Liu L, Wang Y, Liu B, Zeng D, Jin
Q, Li M, Zhang D, Liu Q and Xie H: Identification of breast cancer
recurrence risk factors based on functional pathways in tumor and
normal tissues. Oncotarget. 8:20679–20694. 2017.PubMed/NCBI
|
|
28
|
Choi BY, Bair E and Lee JW: Nearest
shrunken centroids via alternative genewise shrinkages. PLoS One.
12:e01710682017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ravindranath A, O'Connell A, Johnston PG
and El-Tanani MK: The role of LEF/TCF factors in neoplastic
transformation. Curr Mol Med. 8:38–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dey N, Barwick BG, Moreno CS,
Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C,
Kerstann KF, Sledge GW Jr, et al: Wnt signaling in triple negative
breast cancer is associated with metastasis. BMC Cancer.
13:5372013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vijaya Kumar A, Salem Gassar E, Spillmann
D, Stock C, Sen YP, Zhang T, Van Kuppevelt TH, Hülsewig C,
Koszlowski EO, Pavao MS, et al: HS3ST2 modulates breast cancer cell
invasiveness via MAP kinase- and Tcf4 (Tcf7l2)-dependent regulation
of protease and cadherin expression. Int J Cancer. 135:2579–2592.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Naidu R, Yip CH and Taib NA: Genetic
variations in transcription factor 7-like 2 (TCF7L2) gene:
Association of TCF7L2 rs12255372(G/T) or rs7903146(C/T) with breast
cancer risk and clinico-pathological parameters. Med Oncol.
29:411–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Burwinkel B, Shanmugam KS, Hemminki K,
Meindl A, Schmutzler RK, Sutter C, Wappenschmidt B, Kiechle M,
Bartram CR and Frank B: Transcription factor 7-like 2 (TCF7L2)
variant is associated with familial breast cancer risk: A
case-control study. BMC Cancer. 6:2682006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bodhini D, Radha V, Dhar M, Narayani N and
Mohan V: The rs12255372(G/T) and rs7903146(C/T) polymorphisms of
the TCF7L2 gene are associated with type 2 diabetes mellitus in
Asian Indians. Metabolism. 56:1174–1178. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Crispo A, Augustin LS, Grimaldi M,
Nocerino F, Giudice A, Cavalcanti E, Di Bonito M, Botti G, De
Laurentiis M, Rinaldo M, et al: Risk differences between
prediabetes and diabetes according to breast cancer molecular
subtypes. J Cell Physiol. 232:1144–1150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Müller HM, Widschwendter A, Fiegl H,
Ivarsson L, Goebel G, Perkmann E, Marth C and Widschwendter M: DNA
methylation in serum of breast cancer patients: An independent
prognostic marker. Cancer Res. 63:7641–7645. 2003.PubMed/NCBI
|
|
39
|
Mukherjee N, Bhattacharya N, Alam N, Roy
A, Roychoudhury S and Panda CK: Subtype-specific alterations of the
Wnt signaling pathway in breast cancer: Clinical and prognostic
significance. Cancer Sci. 103:210–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang WJ, Yao Y, Jiang LL, Hu TH, Ma JQ,
Liao ZJ, Yao JT, Li DF, Wang SH and Nan KJ: Knockdown of lymphoid
enhancer factor 1 inhibits colon cancer progression in vitro and in
vivo. PLoS One. 8:e765962013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin AY, Chua MS, Choi YL, Yeh W, Kim YH,
Azzi R, Adams GA, Sainani K, van de Rijn M, So SK and Pollack JR:
Comparative profiling of primary colorectal carcinomas and liver
metastases identifies LEF1 as a prognostic biomarker. PLoS One.
6:e166362011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Delaunay S, Rapino F, Tharun L, Zhou Z,
Heukamp L, Termathe M, Shostak K, Klevernic I, Florin A, Desmecht
H, et al: Elp3 links tRNA modification to IRES-dependent
translation of LEF1 to sustain metastasis in breast cancer. J Exp
Med. 213:2503–2523. 2016. View Article : Google Scholar : PubMed/NCBI
|