Introduction
Von Hippel-Lindau (VHL) syndrome is an autosomal
dominant genetic disorder with an incidence of 1/36,000 to 1/45,500
(1). VHL syndrome is characterized by
hemangioblastoma of the central nervous system (CNS) and retina,
and multiple cysts and tumors in visceral organs. VHL syndrome may
occur at any age, and the penetrance is >90% prior to the age of
65 (1). Without treatment, most
patients succumb to the disease by the age of 50 years, and the
most common causes of VHL syndrome-associated mortality are
comorbidities of cerebellar hemangioblastoma or metastasis of renal
cell carcinoma (2). In 1993, Latif
et al (3) reported that the
VHL gene was located on chromosome 3p25-26 by linkage analysis, and
for the first time the VHL gene was successfully cloned. The VHL
gene is a tumor suppressor. The encoded protein is able to regulate
the activity of elongin, a transcription elongation factor, and
suppress tumor growth by inhibiting mRNA synthesis (4).
Mutation, deletion or methylation of the VHL gene
may lead to abnormal protein synthesis. Therefore, VHL syndrome is
associated with disruption of VHL gene structure or function
(4). According to whether the patient
develops pheochromocytoma or renal carcinoma, VHL syndrome may be
divided into types I and II. Pheochromocytoma is only present in
type II VHL syndrome (5). These
subtypes may be further divided. Type IA is characterized by
retinal and CNS hemangioblastoma, pancreatic cysts, neuroendocrine
tumors and renal carcinoma. Type IB is characterized by retinal and
CNS hemangioblastoma, pancreatic cysts and neuroendocrine tumors
without the presence of renal carcinoma. Type IIA is characterized
by pheochromocytoma and retinal and CNS hemangioblastoma without
the presence of renal carcinoma. Pheochromocytoma, retinal and CNS
hemangioblastoma, as well as renal carcinoma, are present in type
IIB VHL syndrome. Patients presenting only with pheochromocytoma
are classified as type 2C (5). Based
on the classification criteria, the 3 cases of VHL syndrome treated
at the Affiliated Hospital of Zunyi Medical College (Zunyi, China)
between September 2014 and October 2015 were characterized as type
IA, 2A and IA, repectively. The present study was approved by the
Medical Ethics Committee at the Affiliated Hospital of Zunyi
Medical College and written informed consent was obtained from all
patients.
Case report
Case 1
On September 10, 2014, a 38-year-old male patient
visited the Department of Urology at the Affiliated Hospital of
Zunyi Medical College due to the detection of lesions in his right
kidney during a physical examination. In November 2011, the patient
visited Chongqing Southwest Hospital (Chongqing, China) due to
dizziness and headache. Lesions were detected in the left side of
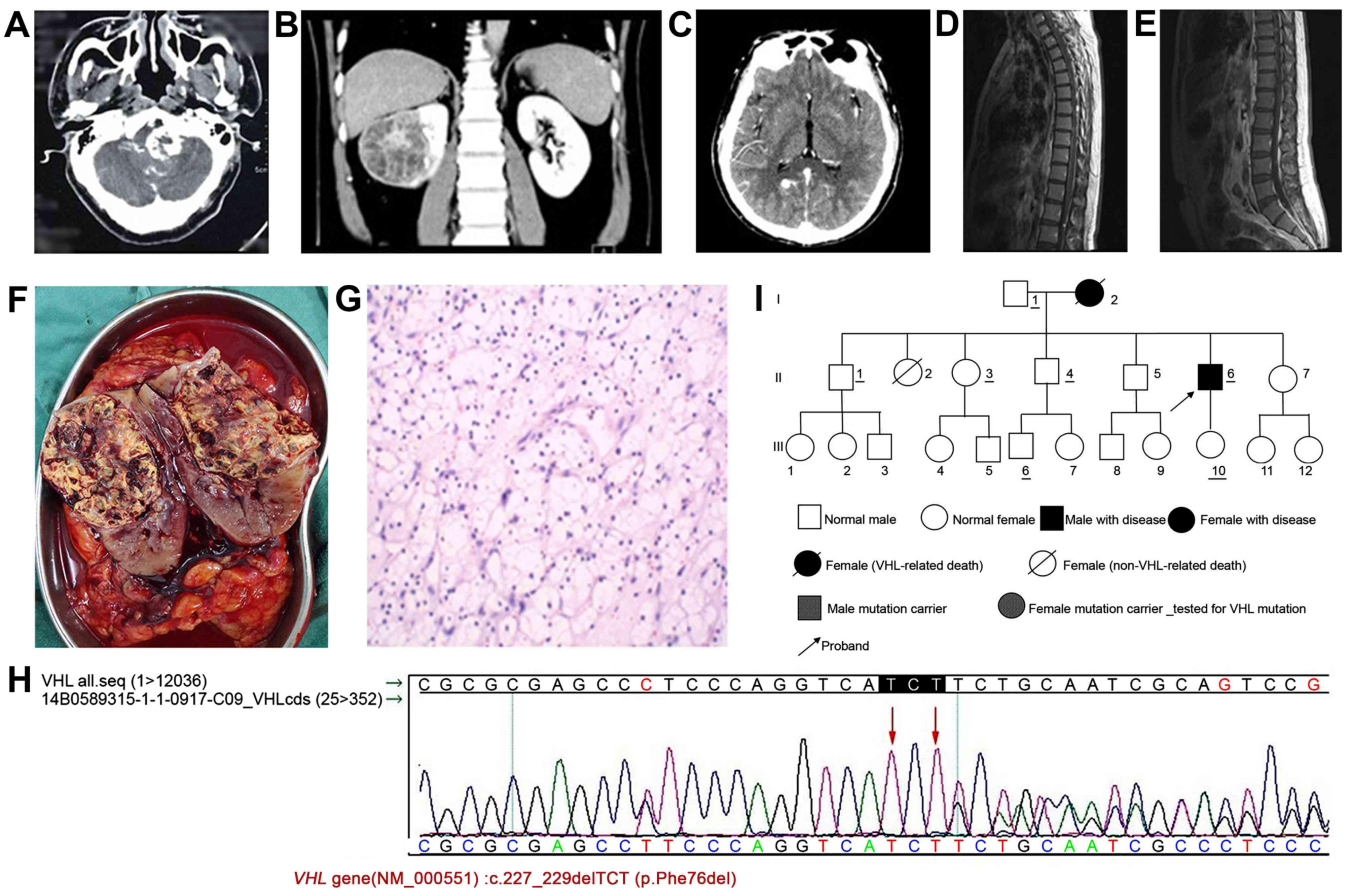
the brain stem (Fig. 1A). The tumor
was removed by surgery. Postoperative pathological examination
confirmed that the lesions were hemangioblastomas. VHL syndrome was
not considered as a diagnosis at that time. The patient had no
previous history of hypertension, infectious diseases or allergy.
The patient's mother was deceased, and the cause may have been
associated with VHL. The patient's father suffered from
hypertension and diabetes. One of the patient's sisters (II-3)
suffered from diabetes, and the remaining siblings had no history
of hypertension, diabetes or tumor.
Physical examination after admission demonstrated
that the body temperature of the patient was 36.5°C, and the heart
rate was 81 beats/min. The respiration rate of the patient was 18
times/min, and blood pressure was 132/94 mmHg. The patient
exhibited normal development, slow gait and poor balance. Old
surgical scars were observed on the left side of the occipital
region. Muscle atrophy was observed to be marginally more on the
left side of the limbs compared with the contralateral limbs. The
patient responded well in terms of sensation, pain and
physiological reflex. No abnormalities were observed in the heart,
lung or abdomen. Bilateral renal computed tomography (CT) indicated
the presence of a 64×59 mm mass in the right renal parenchyma, with
a clear edge. The mass protruded into the renal capsule and
suppressed the renal pelvis. Low-density areas with multiple cysts
and shadows of line-like partitions were observed inside the mass.
Contrast-enhanced scanning revealed uneven enhancement, and the
partitions demonstrated significant enhancement. In delayed-phase
imaging, the contrast was strengthened and patch-like intensified
shadows were observed in the middle of the mass (Fig. 1B).
Brain CT revealed that the left occipital bone was
altered following surgery. Malacia was observed in the left side of
the cerebellum. Multiple enhanced nodules were observed in the
brain. Brain magnetic resonance imaging (MRI) indicated that bone
defect and the shadow of the internal fixation were observed in the
left occipital bone. Weak long T2 signals were observed in the left
cerebellar hemisphere. Enhanced nodules were observed in the right
occipital lobe, right cerebellar hemisphere and foramen magnum.
Weak long T2 signals could be observed in the surrounding areas.
The largest nodule was located in the right cerebellar hemisphere
with a size of ~8×10 mm (Fig. 1C).
MRI of the thoracolumbar spinal cord revealed that multiple
nodule-like shadows were present on the surface of the
thoracolumbar spinal cord and nerve plexus in the cauda equina. The
larger nodules had a diameter of ~6 mm. The contrast was markedly
enhanced in the MRI images. Abnormal enlarged blood vessels were
observed in the surrounding areas, which appeared to indicate
hemangioma. Multiple nodules with signals of a similar intensity
were observed next to the lumbar spinal canal and cauda equina. The
larger nodules had a diameter of ~5.5 mm. The contrast was markedly
enhanced in the images, which indicated the presence of multiple
neurofibromas (Fig. 1D and E).
Electrocardiogram, chest radiograph, liver and kidney function, and
the coagulation profile all appeared normal. The patient was
revealed to have diabetes during the intial examination following
hospitalization. Following preoperative preparation, the patient
underwent radical tumor resection of the right kidney under general
anesthesia. The sample was dissected following surgery, and the
tumor was observed in the middle and upper portions of the right
kidney with a size of ~8×7×6 cm. The tumor displayed
fish-flesh-like lesions accompanied by necrosis and had
metastasized into the surrounding renal pelvis (Fig. 1F).
Postoperative pathological examination confirmed the
diagnosis of renal clear cell carcinoma in the right kidney. The
tumor did not invade the surrounding adipose tissue or the residual
ureter and blood vessels (Fig.
1G).
Hematoxylin and eosin staining was performed. Tissue
paraffin sections (slice thickness, 5µm) were prepared and dewaxed
with xylene for 15 min. Sections were then placed in 100% ethanol
for 5 sec, 95% ethanol for 5 sec, 80% ethanol for 5 sec and 70%
ethanol for 5 sec, separately, to hydrate. Subsequently, tissue
sections were rinsed with running tap water for 5 sec. The tissue
sections were then stained with hematoxylin (BA-4097; Baso
Biotechnology Co. Ltd., Zhuhai, China) for 8 min, following by
rinsing with running tap water for 15 sec. The tissue sections were
placed in hydrochloric acid alcohol for 5 sec, and then rinsed with
running tap water for 8 min. Eosin (1%; BA-4098; Baso Biotechnology
Co. Ltd.) was used for staining for 15 sec. The tissue sections
were placed under running tap water to remove excess dye for 20
sec, and then placed in 80% ethanol for 5 sec, 95% ethanol for 5
sec, 100% ethanol for 10 sec and xylene for 2 min, separately.
Finally, the tissue sections were sealed. The aforementioned steps
were performed at room temperature. Genetic analysis revealed a
mutation in the VHL gene of the patient, c.227_229delTCT
(p.Phe76del), which resulted in the deletion of phenylalanine at
position 76 of the encoded protein. This genetic analysis result
was confirmed by Sanger sequencing (Fig.
1H). Pedigree analysis was performed following surgery
(Fig. 1I). Further genetic tests and
physical examinations were performed on relatives of the patients
with informed consent. The tests included abdominal Doppler
ultrasonography and brain MRI. Although all family members had no
abnormal findings in genetic tests and physical examinations,
follow-up visits were scheduled for all family members. The
follow-up for the patient continued until September 2015, at which
point the patient was in good condition without recurrence or novel
tumor development elsewhere.
Case 2
A 41-year-old male patient was admitted to the
Department of Ophthalmology and Endocrinology at the Affiliated
Hospital of Zunyi Medical College between 24 September and 5
October 2014 due to blurred vision and elevated blood glucose
level. During hospital admission, adrenal masses were identified on
the left and right sides, and the patient was transferred to the
Department of Urology on 23 October 2014. Vision was lost in the
right eye due to a previous injury. The patient had hypertension
and diabetes for >2 months. In September 2010, the patient
underwent resection of the lesions in the right cerebellar
hemisphere. Postoperative pathological analysis confirmed that the
lesion was a hemangioblastoma. The mother of the patient was
deceased, and the cause was unknown. The mother exhibited a brain
tumor and coronary heart disease whilst alive. Multiple nodules
were detected in the thyroid gland of the patient's eldest sister
(II-1), but were not treated. The rest of the family members were
healthy.
Physical examination after admission demonstrated
that the body temperature of the patient was 36.8°C, and the heart
rate was 84 beats/min. His respiration rate was 19 times/min, and
blood pressure was 154/112 mmHg. An old surgical scar was observed
in the right occipital area. Opacity of the lens was observed in
the right eye, which led to weak vision, while the left eye
retained blurry vision. No abnormality was observed in the heart,
lung or abdomen. Abdominal CT indicated the presence of irregular
bumps in the left (size, ~72×54 mm) and right (size, ~92×73 mm)
adrenal glands. The edge was clear, and the density was uneven.
Low-density areas and shadows of calcification were observed.
Contrast-enhanced scanning revealed that the solid portion of the
mass was markedly enhanced, but the cystic region was not enhanced
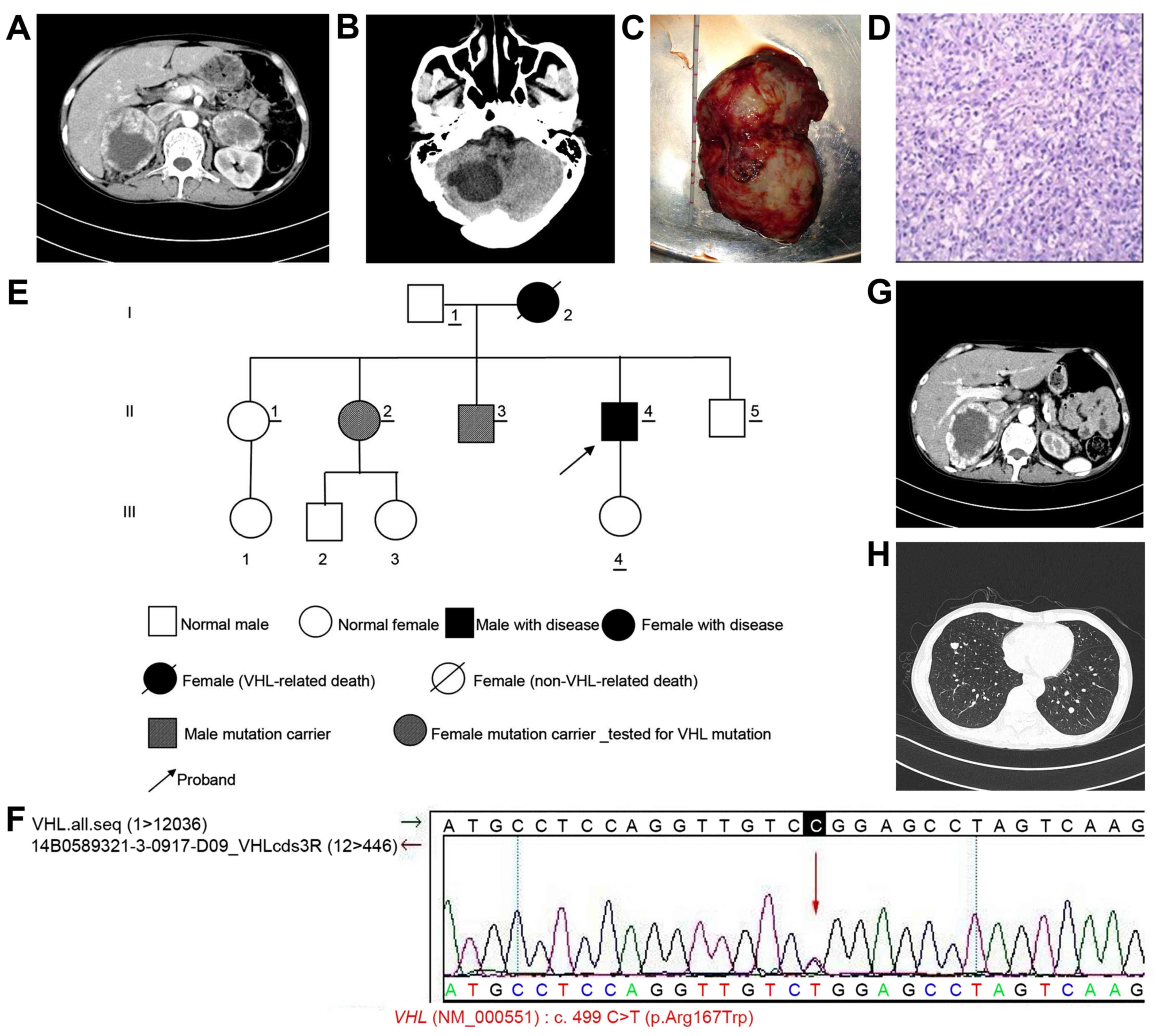
(Fig. 2A). No swollen lymph nodes
were observed in the abdomen, pelvis or retroperitoneal area;
therefore, bilateral adrenal pheochromocytoma was considered. Brain
CT indicated that the right occipital area was altered following
surgery. A cystic low-density area (34×44 mm) with clear edges was
observed in the right cerebellar hemisphere, which was indicative
of a cystic lesion or malacia in the right cerebellar hemisphere
(Fig. 2B). Cortisol and aldosterone
levels were in the normal range. Electrocardiogram, chest
radiograph, liver and kidney function, and coagulation profile all
appeared normal. Following preoperative preparation, the patient
underwent resection of the left adrenal tumor under general
anesthesia. The gross sample is presented in Fig. 2C.
Postoperative pathological examination confirmed
that the tumor was an adrenal pheochromocytoma (Fig. 2D). Pedigree analysis was performed
following surgery (Fig. 2E). Genetic
tests and physical examinations were performed for a number of
relatives of the patient (who had not succumbed to disease) with
informed consent. The same missense mutation, c.499C>T
(p.Arg167Trp), in the VHL gene was found in two family members
(II-2 and II-3) and the patient, which led to the substitution of
arginine with tryptophan at position 167 of the encoded protein.
This genetic analysis result was confirmed by Sanger sequencing
(Fig. 2F). Physical examinations were
also performed, including abdominal Doppler ultrasonography and
brain MRI, on several family members. MRI of the brain of the
patient's brother (II-3) indicated a nodule in the left cerebellar
tonsil with a size of ~9.0×10.5 mm, which indicated the presence of
a hemangioblastoma, astrocytoma or ependymoma. However, the
diagnosis could not be confirmed. Therefore, patient II-3 was
regarded as a carrier of the mutant gene. Doppler ultrasound
examination of the patient's eldest sister (II-1) suggested the
presence of multiple nodules with calcification and liquefaction in
the left and right thyroid glands. The size of the larger nodules
was ~34×17 mm on the left side and ~29×16 mm on the right side of
the thyroid gland. A lymph node (size, 14×4 mm) was detected on the
right side of the neck, and another lymph node (size, 10×3 mm) was
detected on the left side. Spot-like signals corresponding to blood
flow were observed inside the node. Another sister of the patient
(II-2) carried the mutated VHL gene. However, no VHL-associated
lesions were detected, therefore II-2 was considered to be a
mutation carrier. Routine follow-ups were suggested.
The daughter and father of the patient declined
testing due to personal reasons. The remaining family members had
no abnormalities according to the abdominal Doppler ultrasound and
brain MRI scan. The family members with abnormal results were
advised to arrange for routine follow-ups. Due to physical and
economic reasons, the patient returned to the Affiliated Hospital
of Zunyi Medical College. On 2 June 2015, for resection of the
right adrenal gland tumor. Postoperative abdominal CT is presented
in Fig. 2G. However, preoperative
chest CT indicated that multiple pulmonary metastases were present
on the left and right sides (Fig.
2H), which was indicative of malignant pheochromocytoma. Since
surgery could not be performed, the patient was advised for a
transfer to the Department of Oncology. The patient succumbed
following 6 months.
Case 3
A 38-year-old male patient visited the Department of
Urology at the Affiliated Hospital of Zunyi Medical College on 27
April 2015, due to the detection of a mass in the right kidney
during physical examination. The patient had previously undergone
extracorporeal shock wave lithotripsy in February 2010 at
Changzheng Hospital (Zunyi, China) for the treatment of kidney
stones (details are unknown). There was no history of hypertension,
infectious diseases or allergy.
The mother of the patient had pancreatic cancer. She
did not undergo surgery and subsequently succumbed to disease. The
father of the patient is alive. The eldest sister of the patient
(II-1) underwent surgery for a brain tumor in June 2007, and had
pancreatic and renal cysts (details are unknown). Another sister
(II-2) underwent surgery for breast cancer on the left side in
April 2014 and was treated with regular chemotherapy following
surgery (details are unknown). The rest of the siblings reported no
history of hypertension, diabetes or cancer.
Physical examination after admission demonstrated
that the body temperature of the patient was 36.9°C, and the heart
rate was 62 beats/min. The respiration rate was 18 times/min, and
blood pressure was 121/85 mmHg. No abnormalities were observed in
the heart, lung or abdomen. Upper abdominal CT revealed the
presence of multiple nodules and lumpy low-density areas in the
left and right kidneys with uneven density. The diameter of the
larger nodule in the right kidney was ~34 mm. Contrast-enhanced
imaging indicated that the lesions were enhanced unevenly in a
progressive manner with clear edges. Multiple round low-density
lesions were observed in the left and right kidneys, and in the
pancreas. Contrast-enhanced scanning revealed no significant
enhancement. Based on the presence of multiple lesions in the left
and right kidneys and the pancreas, as well as the patient's
medical history, VHL syndrome was considered as a possible
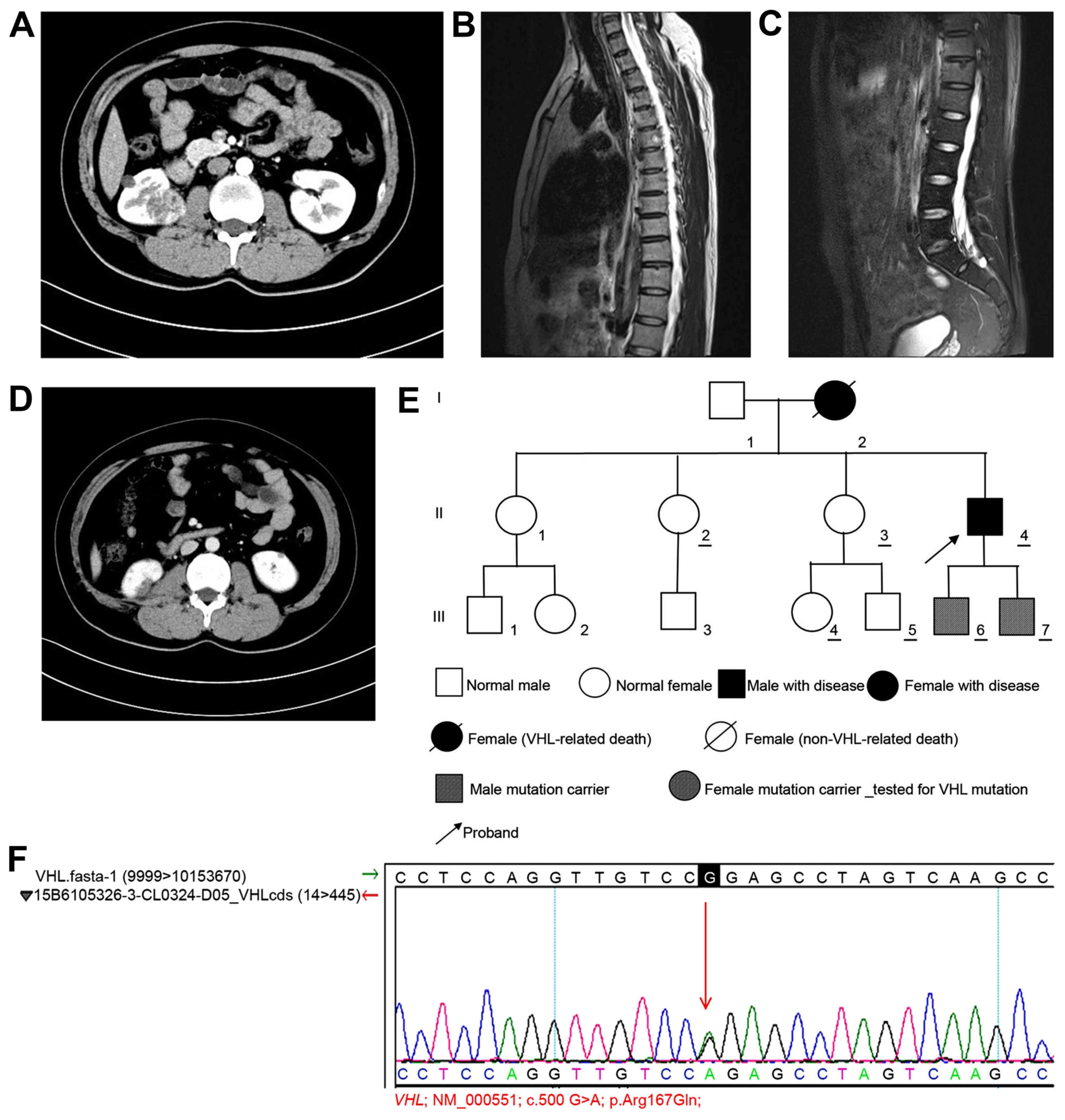
diagnosis (Fig. 3A). Brain CT
revealed no abnormalities, but sphenoiditis was observed on the
right side. Thoracic MRI revealed a round-shaped mixed signal
(diameter, 12.0 mm) in the sixth thoracic vertebra.
Contrast-enhanced scanning revealed minor enhancement, which
appeared to indicate the presence of a hemangioma (Fig. 3B). Lumbar MRI revealed reduced T2
signals in the L5/S1 intervertebral disc, which was marginally
protruding. Contrast-enhanced scanning did not demonstrate abnormal
enhancement. A round long T2 signal (size, 10×7 mm) was observed in
the S2 horizontal sacral area, which appeared to indicate the
presence of a small sacral cyst (Fig.
3C). Electrocardiogram, chest radiograph, liver and kidney
function, and coagulation profile all appeared normal. Following
preoperative preparation, partial resection of the right kidney was
planned, but the patient and his family requested to be
discharged.
Follow-up of the patient indicated that the patient
underwent partial resection of the right kidney at the West China
Hospital Affiliated to Sichuan University (Chengdu, China).
Postoperative pathological analysis confirmed that the tumor was a
renal carcinoma (clear-cell carcinoma of the right kidney; Fuhrman
Grade II) (6,7). The tumor was close to the renal capsule
and did not demonstrate significant invasion. The incision area was
not invaded by the tumor. Following recovery, the patient returned
to the Affiliated Hospital of Zunyi Medical College on 21 September
2015. Bilateral renal CT indicated that the lower part of the right
kidney was altered following surgery, and multiple small cystic
lesions were detected in the left and right kidneys. The diameter
of the largest lesion was ~6 mm. Contrast-enhanced scanning did not
reveal marked enhancement. Small cystic lesions were also detected
in the pancreas (Fig. 3D). The
pedigree of the patient's family was analyzed and established
(Fig. 3E). Genetic tests were
performed on several relatives with informed consent. The patient
and 6 other family members, volunteered to take the VHL genetic
test. The tests revealed that the same missense mutation in the VHL
gene [c.500G>A (p.Arg167Gln)] was found in the patient and his
two sons, which led to a substitution of arginine with glutamine at
position 167 of the VHL-encoded protein, and this was confirmed by
Sanger sequencing (Fig. 3F).
Abdominal ultrasound examination of the patient's two sons revealed
no abnormality. MRI of the brain was subsequently planned, but the
patient and his two sons declined. Follow-up examinations conducted
until October 2015 indicated that the patient was in good condition
in general, and no new tumors were identified elsewhere. Follow-up
examinations of the other family members remain underway.
Discussion
VHL syndrome is a rare autosomal dominant genetic
disease. The offspring of patients with VHL syndrome have a 50%
chance of inheriting the disease. The gene responsible for this
disease is located on chromosome 3p25-26. At the age of 60, the
penetrance may exceed 98% (8). The
main clinical manifestations include retinal and CNS
hemangioblastoma, which are frequently accompanied by renal clear
cell carcinoma, renal cysts, pancreatic tumors, pancreatic cysts
and internal lymphoma. In 1859, von Hippel, a German
ophthalmologist, for the first time reported two cases of retinal
hemangioblastoma with familial inheritance (2,9–11). In 1926, Swiss pathologist Lindau
performed a systemic clinical observation of the disease and
reported that cerebellar and retinal hemangioblastoma were only
parts of the CNS hemangioblastomas, and that the manifestations of
these types of hemangioblastoma were associated (2,9–11).
In 1964, Hayden et al (12) first named the disease VHL syndrome,
and this nomenclature has been widely recognized. In 1993, Latif
et al (3) located the VHL gene
on chromosome 3p25-26 and for the first time cloned this gene
successfully. A previous study (13)
confirmed that the VHL gene is a tumor suppressor that exhibits the
classic features of tumor suppressor genes. Mutations and deletions
in the VHL gene are the fundamental causes of VHL syndrome
(4). Currently, a common diagnostic
criterion for VHL syndrome in patients who do not have a family
history is that they should exhibit ≥2 of the following
characteristic lesions: i) ≥2 retinal, spinal cord or brain
hemangioblastomas, or a single hemangioblastoma associated with
visceral organ lesions (multiple renal or pancreatic cysts); ii)
renal clear cell carcinoma; iii) adrenal or extra-adrenal
pheochromocytoma and iv) rare lesions including, internal lymphoma,
papillary cystadenoma of the epididymis and broad ligament, and
neuroendocrine tumors of the pancreas (14). Patients with a family history of VHL
syndrome should have ≥1 of the following lesions: i) Retinal
hemangioblastoma, ii) spinal cord or cerebellar hemangioblastoma,
iii) adrenal or extra-adrenal pheochromocytoma, iv) renal clear
cell carcinoma and v) multiple renal or pancreatic cysts. In
patients with atypical clinical manifestations, the VHL genetic
test is recommended to exclude the diagnosis of VHL syndrome
(14).
From the perspective of clinical practice, diagnosis
rates of VHL syndrome in the early stages remain low. This may be
due to physicians lacking a thorough understanding of VHL syndrome
and therefore overlooking the comprehensive examination of other
body systems, family history of the patient and genetic tests.
Additionally, VHL syndrome is characterized by the presence of
multiple tumors in multiple organs; however, the majority of
patients with VHL syndrome present with only a single lesion at the
time of admission. In the present report, brain hemangioblastoma
was detected in cases 1 and 2 when the patients visited the
hospital for the first time. According to Glasker et al
(15), 19% of patients with VHL
syndrome who are admitted to the hospital for the first time due to
neurological symptoms are observed to have multiple
hemangioblastomas, and 36% of patients possess tumor-associated
symptoms that are indicative of VHL syndrome. Since the severity of
clinical symptoms and the number of tumors affected organs vary,
early diagnosis of VHL syndrome is relatively difficult (1,2). In cases
1 and 2, VHL syndrome was considered when lesions in the visceral
organs were detected following resection of the brain
hemangioblastomas. The diagnosis could not be confirmed until the
results of the genetic tests with the clinical symptoms were
combined. According to Conway et al (16), the sensitivity of the VHL genetic test
is 86%, and all patients with positive results were confirmed by
clinical diagnostic criteria. Therefore, the accuracy of genetic
test-based diagnosis is markedly improved compared with clinical
manifestation-based diagnosis.
Conway et al (16) found that 67% of patients with VHL
syndrome developed a new CNS tumor within a mean of 2.1 years. In
cases 1 and 2, the mean time for developing a new tumor was 3.5
years. The patient in case 3 remains under close follow-up, and no
new tumor has been detected to date. Surgical resection of the
tumor remains the main treatment method. In the case of CNS
hemangioblastomas, resection is preferred by certain clinicians,
while others prefer regular follow-up and decide on surgery when
clinical symptoms occur (16).
Retinal hemangioblastomas should be removed as soon as possible to
avoid loss of vision and other complications. Additionally, VHL
syndrome accompanied by pheochromocytoma requires surgery. Partial
resection of the adrenal gland may be considered. In patients
without hypertension, an α-receptor blocker still should be
administered (17).
Notably, there are no standard criteria for surgery
in cases of VHL syndrome accompanied by renal clear cell carcinoma
(17). Duffey et al (18) analyzed 108 VHL syndrome patients with
a renal tumor (diameter, <3 cm) and reported that following a
mean of 28 months (range, 0–244 months), no tumor metastasis was
observed. Additionally, in VHL syndrome patients (n=73) with a
renal tumor with diameter, >3 cm, following an average of 72.9
months (range, 0–321 months), metastasis occurred in 20 cases
(27.4%). Therefore, VHL syndrome patients with a tumor diameter
<3 cm require close follow-up and monitoring. Abdominal CT and
serum creatinine should be examined at least every 6 months. When
the diameter of the largest tumor is >3 cm, surgery should be
considered (19–22). Linehan et al (23) suggest that if the size of the tumor is
<3 cm and it is present on the renal surface, tumor enucleation
may be performed. If the tumor size is relatively large (>4 cm)
and is located on one side of the kidney, partial resection should
be considered. If the tumor is present in the center of the kidney
and is the kidney will be difficult to preserve, radical
nephrectomy should be considered (23). The patient in case 1 underwent radical
nephrectomy, because the tumor in the right kidney was large and
located in the center. Potential invasion into the renal pelvis
could not be excluded. In addition, Roupret et al (24) suggested that VHL syndrome-induced
renal clear cell carcinoma tends to present as multiple lesions in
the left and right kidneys and has a high relapse rate following
nephron-sparing surgery. Therefore, thorough elimination of all
potential relapses and metastasis may be possible only with
bilateral nephrectomy. Following surgery, the patient requires
renal replacement therapy, including dialysis and renal
transplantation. However, there is evidence that the use of
immunosuppressants in patients who require dialysis or renal
transplantation following nephrectomy would increase the risk of
tumors developing elsewhere (25).
Therefore, this treatment plan should only be selected when the
left and right kidneys cannot be preserved, and a matched kidney is
available for transplantation.
The aim of surgical treatment of VHL syndrome
accompanied by renal clear cell carcinoma should be thorough
elimination of the tumor and preservation of as much renal tissue
as possible. The optimal surgical plan should be selected based on
tumor size and site, and the physical conditions of the patient.
During postoperative follow-up, relapse in the same kidney or new
tumors in the other kidney should be closely monitored (23–25). Based
on the diagnosis and treatment in case 2, where VHL syndrome was
accompanied by tumors in multiple organs, follow-up surgery for the
complete resection of other tumors should be performed as soon as
possible following the primary surgery. Even if follow-up surgery
is not performed, follow-up should be performed every 3 months. The
patient in case 2 was diagnosed with bilateral adrenal
pheochromocytoma. Resection of the left adrenal tumor was
performed, and postoperative pathological analysis confirmed that
it was an adrenal pheochromocytoma. However, the right adrenal
tumor was not removed rapidly enough, and the patient did not
return for routine follow-up. A total of 8 months later, the
patient returned for resection of the right adrenal tumor. However,
preoperative chest CT indicated the presence of multiple metastases
in the left and right lungs, which was indicative of malignant
pheochromocytoma. No surgery could be performed at that time.
Currently, there are certain limitations associated
with the surgical treatment of VHL syndrome, as surgical treatment
does not prevent tumor relapse and metastasis elsewhere (26). Therefore, postoperative follow-up of
the patients and their family members is essential. Emerging
investigations of the VHL gene and novel therapeutics, including
gene therapy and targeted therapy may be beneficial for patients
with VHL syndrome. Early diagnosis, treatment and regular
follow-ups are critical for good prognosis of VHL syndrome. Missed
diagnosis or misdiagnosis often results from an insufficient
understanding of VHL syndrome among physicians (26). Therefore, patients with CNS
hemangioblastoma and multiple lesions in visceral organs should be
examined carefully, and VHL syndrome should be considered as a
potential diagnosis. Genetic tests for VHL should also be conducted
to exclude the possibility of VHL syndrome when necessary. Since
there is a risk of tumorigenesis with VHL syndrome, lifelong
follow-up of the patient is essential. The follow-up should include
(14,26) the following measurements: i) urinary
catecholamine levels every 6 months; ii) fundus examination
annually; iii) CNS MRI every 6 months; and iv) abdominal CT and
ultrasound examination annually.
Since VHL syndrome is an autosomal dominant genetic
disorder, pedigree analysis and genetic diagnosis may provide
important insights into treatment and prognosis, as well as
prenatal and postnatal care. In case 1, a total of 7 family members
volunteered for the genetic test. The proband (II-6) carried a
deletion in the VHL gene [c.227_229delTCT (p.Phe76del)] that led to
the deletion of phenylalanine at position 76 of the VHL-encoded
protein. p.Phe76del blocked the binding of the VHL protein with
hypoxia-induced factor-α (HIF-α), and this affected the stability
of HIF-α and induced VHL syndrome (27). The p.Phe76del mutation has been
reported to be associated with type I VHL syndrome (27). The remaining family members, including
the father of the patient (I-1), did not carry mutations in the VHL
gene. However, since the mother of the patient was previously
deceased, the authors of the present study hypothesize that this
may have been associated with VHL syndrome, and that the patient
may have inherited the mutation from his mother.
The patient in case 2 exhibited type II VHL
syndrome. Type II VHL syndrome occurs in ~10–20% of patients with
VHL syndrome (1). Sequencing of the
VHL gene of the 7 family members revealed the same missense
mutation in the VHL gene [c.499C>T (p.Arg167Trp)] in the proband
(II-4), his sister (II-2) and brother (II-3). The mutation resulted
in the substitution of arginine at position 167 with tryptophan.
Crossey et al (28) and Brauch
et al (29) reported that the
p.Arg167Trp mutation was closely associated with VHL syndrome.
Mutation of the amino acid at position 167 has been reported to
increase the risk of pheochromocytoma (28,29). In
the present report, the mutation was at position 238. In a study
with 469 pedigrees with VHL syndrome across North America, Europe
and Japan, Zbar et al (30)
observed that p.Arg167Trp mutation was a hotspot, and the detection
rate of this mutation was high in VHL syndrome families with
pheochromocytoma. A total of 21 patients with pheochromocytoma in
33 families carrying the p.Arg167Trp mutation were reported. Siu
et al (31) performed genetic
screening of 9 Chinese pedigrees with VHL syndrome and detected the
p.Arg167Trp mutation in a patient with pheochromocytoma. The mother
of the patient exhibited a brain tumor and coronary heart disease,
and it was likely that she succumbed due to VHL syndrome (31). In the present report, it is possible
that the proband (II-4), sister (II-2) and brother (II-3) inherited
the mutation from their mother.
In case 3, a total of 7 members of the family
volunteered for the genetic test, and the same missense mutation
[c.500G>A (p.Arg167Gln)] in the VHL gene was detected in the
proband (II-4) and his two sons (III-6 and III-7). This mutation
led to the substitution of arginine at position 167 with glutamine.
Hes et al (32) reported that
the p.Arg167Gln mutation was detected in 11 members from 3 typical
VHL syndrome pedigrees. Among the 11 members, 4 had renal carcinoma
and 3 exhibited renal cysts. Additionally, Ciotti et al
(33) found in a controlled study of
43 VHL syndrome patients and 42 healthy controls that the
p.Arg167Gln mutation was detected in 2 VHL syndrome patients, where
one patient exhibited bilateral renal carcinoma and the other had
renal carcinoma, pancreatic cysts and ovarian cysts. The proband of
the pedigree also exhibited pancreatic and bilateral renal cysts.
Therefore, the authors of the present report hypothesize that the
p.Arg167Gln mutation may be associated with cyst formation in
visceral organs. However, this hypothesis requires validation in
additional cases. In the present report, all members from the three
pedigrees who remain alive are under follow-up, particularly those
carrying the VHL mutations.
VHL syndrome is a rare autosomal dominant genetic
disorder with symptoms that may not be evident, meaning early
diagnosis is rare. DNA analysis of VHL genetic mutations is the
primary method used to confirm the diagnosis of the disease. The
main treatment method of VHL syndrome is surgery. A reasonable
surgical strategy should be selected according to the site and
number of tumors as well as the physical condition of the patient.
Patients with VHL syndrome accompanied by tumors in multiple organs
should undergo complete resection of the tumors as soon as
possible. Additionally, pedigree analysis and genetic testing are
essential. Physicians should pay sufficient attention to VHL
syndrome in order to avoid missed diagnosis or misdiagnosis.
Acknowledgements
The present study was supported by the Science and
Technology Fund Project of Guizhou Province [grant no. (2015)
31].
References
|
1
|
Lonser RR, Glenn GM, Walther M, Chew EY,
Libutti SK, Linehan WM and Oldfield EH: von Hippel-Lindau disease.
Lancet. 361:2059–2067. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shuin T, Yamazaki I, Tamura K, Kamada M
and Ashida S: Recent advances in ideas on the molecular pathology
and clinical aspects of Von Hippel-Lindau disease. Int J Clin
Oncol. 9:283–287. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Latif F, Tory K, Gnarra J, Yao M, Duh FM,
Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim W and Kaelin WG Jr: The von
Hippel-Lindau tumor suppressor protein: New insights into oxygen
sensing and cancer. Curr Opin Genet Dev. 13:55–60. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McNeill A, Rattenberry E, Barber R,
Killick P, MacDonald F and Maher ER: Genotype-phenotype
correlations in VHL exon deletions. Am J Med Genet A.
149A:2147–2151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patard JJ, Leray E, Rioux-Leclercq N,
Cindolo L, Ficarra V, Zisman A, De La Taille A, Tostain J, Artibani
W, Abbou CC, et al: Prognostic value of histologic subtypes in
renal cell carcinoma: A multicenter experience. J Clin Oncol.
23:2763–2771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leibovich BC, Cheville JC, Lohse CM,
Zincke H, Frank I, Kwon ED, Merchan JR and Blute ML: A scoring
algorithm to predict survival for patients with metastatic clear
cell renal cell carcinoma: A stratification tool for prospective
clinical trials. J Urol. 174:1759–1763. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vortmeyer AO, Choo D, Pack S, Oldfield E
and Zhuang Z: VHL gene inactivation in an endolymphatic sac tumor
associated with von Hippel-Lindau disease. Neurology. 55:4602000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neumann HP and Wiestler OD: Clustering of
features of von Hippel-Lindau syndrome: Evidence for a complex
genetic locus. Lancet. 337:1052–1054. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weil RJ, Vortmeyer AO, Zhuang Z, Pack SD,
Theodore N, Erickson RK and Oldfield EH: Clinical and molecular
analysis of disseminated hemangioblastomatosis of the central
nervous system in patients without von Hippel-Lindau disease.
Report of four cases. J Neurosurg. 96:775–787. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanno H, Yamamoto I, Yoshida M and
Kitamura H: Meningioma showing VHL gene inactivation in a patient
with von Hippel-Lindau disease. Neurology. 60:1197–1199. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hayden MG, Gephart R, Kalanithi P and Chou
D: Von Hippel-Lindau disease in pregnancy: A brief review. J Clin
Neurosci. 16:611–613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bader HL and Hsu T: Systemic VHL gene
functions and the VHL disease. FEBS Lett. 586:1562–1569. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maher ER, Neumann HP and Richard S: von
Hippel-Lindau disease: A clinical and scientific review. Eur J Hum
Genet. 19:617–623. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Glasker S, Bender BU, Apel TW, Natt E, van
Velthoven V, Scheremet R, Zentner J and Neumann HP: The impact of
molecular genetic analysis of the VHL gene in patients with
haemangioblastomas of the central nervous system. J Neurol
Neurosurg Psychiatry. 67:758–762. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Conway JE, Chou D, Clatterbuck RE, Brem H,
Long DM and Rigamonti D: Hemangioblastomas of the central nervous
system in von Hippel-Lindau syndrome and sporadic disease.
Neurosurgery. 48:55–63. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Benhammou JN, Boris RS, Pacak K, Pinto PA,
Linehan WM and Bratslavsky G: Functional and oncologic outcomes of
partial adrenalectomy for pheochromocytoma in patients with von
Hippel-Lindau syndrome after at least 5 years of followup. J Urol.
184:1855–1859. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duffey BG, Choyke PL, Glenn G, Grubb RL,
Venzon D, Linehan WM and Walther MM: The relationship between renal
tumor size and metastases in patients with von Hippel-Lindau
disease. J Urol. 172:63–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steinbach F, Novick AC, Zincke H, Miller
DP, Williams RD, Lund G, Skinner DG, Esrig D, Richie JP, deKernion
JB, et al: Treatment of renal cell carcinoma in von Hippel-Lindau
disease: A multicenter study. J Urol. 153:1812–1816. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ghavamian R, Cheville JC, Lohse CM, Weaver
AL, Zincke H and Blute ML: Renal cell carcinoma in the solitary
kidney: An analysis of complications and outcome after nephron
sparing surgery. J Urol. 168:454–459. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Albers P: Treatment approaches in renal
cell carcinoma: past, present and future perspectives. Eur Urol
Suppl. 7:36–45. 2008. View Article : Google Scholar
|
|
22
|
Ploussard G, Droupy S, Ferlicot S, Ples R,
Rocher L, Richard S and Benoit G: Local recurrence after
nephron-sparing surgery in von Hippel-Lindau disease. Urology.
70:435–439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Linehan WM, Rubin JS and Bottaro DP: VHL
loss of function and its impact on oncogenic signaling networks in
clear cell renal cell carcinoma. Int J Biochem Cell Biol.
41:753–756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roupret M, Hopirtean V, Mejean A, Thiounn
N, Dufour B, Chretien Y, Chauveau D and Richard S: Nephron sparing
surgery for renal cell carcinoma and von Hippel-Lindau's disease: A
single center experience. J Urol. 170:1752–1755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marcos HB, Libutti SK, Alexander HR,
Lubensky IA, Bartlett DL, Walther MM, Linehan WM, Glenn GM and
Choyke PL: Neuroendocrine tumors of the pancreas in von
Hippel-Lindau disease: Spectrum of appearances at CT and MR imaging
with histopathologic comparison. Radiology. 225:751–758. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frantzen C, Links T and Giles R: Von
Hippel-Lindau Disease. Seattle (WA); University of Washington,
Seattle: 2000
|
|
27
|
Jia D, Tang B, Shi Y, Wang J, Sun Z, Chen
Z, Zhang L, Xia K and Jiang H: A deletion mutation of the VHL gene
associated with a patient with sporadic von Hippel-Lindau disease.
J Clin Neurosci. 20:842–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crossey PA, Richards FM, Foster K, Green
JS, Prowse A, Latif F, Lerman MI, Zbar B, Affara NA, Ferguson-Smith
MA, et al: Identification of intragenic mutations in the von
Hippel-Lindau disease tumour suppressor gene and correlation with
disease phenotype. Hum Mol Genet. 3:1303–1308. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brauch H, Kishida T, Glavac D, Chen F,
Pausch F, Höfler H, Latif F, Lerman MI, Zbar B and Neumann HP: Von
Hippel-Lindau (VHL) disease with pheochromocytoma in the Black
Forest region of Germany: Evidence for a founder effect. Hum Genet.
95:551–556. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zbar B, Kishida T, Chen F, Schmidt L,
Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA,
Ferguson-Smith MA, et al: Germline mutations in the Von
Hippel-Lindau disease (VHL) gene in families from North America,
Europe, and Japan. Hum Mutat. 8:348–357. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siu WK, Ma RC, Lam CW, Mak CM, Yuen YP, Lo
FM, Chan KW, Lam SF, Ling SC, Tong SF, et al: Molecular basis of
von Hippel-Lindau syndrome in Chinese patients. Chin Med J (Engl).
124:237–241. 2011.PubMed/NCBI
|
|
32
|
Hes FJ, van der Luijt RB, Janssen AL,
Zewald RA, de Jong GJ, Lenders JW, Links TP, Luyten GP, Sijmons RH,
Eussen HJ, et al: Frequency of Von Hippel-Lindau germline mutations
in classic and non-classic Von Hippel-Lindau disease identified by
DNA sequencing, Southern blot analysis and multiplex
ligation-dependent probe amplification. Clin Genet. 72:122–129.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ciotti P, Garuti A, Gulli R, Ballestrero
A, Bellone E and Mandich P: Germline mutations in the von
Hippel-Lindau gene in Italian patients. Eur J Med Genet.
52:311–314. 2009. View Article : Google Scholar : PubMed/NCBI
|