Introduction
Transplantation of mesenchymal stem cells (MSCs) is
a promising treatment for many diseases (1). However, the effect of transplantation of
MSCs on the development of cancer remains controversial. The
influence of MSCs on tumour development was studied using in
vitro co-cultivation (2) and
xenotransplantation models, where human-derived stem cells were
transplanted into study animals (3).
Controversial effects of MSCs on cancer might be related to
differences in approaches towards cancer modelling, e.g., chemical
induction (4) or xenotransplantation
of cancer cell lines (5).
Colorectal cancer (CC) is the second major type of
cancer in Europe and the third major type in the USA (6). Symptoms of the disease appear only at
later stages of the tumour, hence only 39% of the cases are
diagnosed at early stages, and prognosis of the disease development
remains unfavourable for the remaining cases, with the five-year
survival rate of 11–69% (7).
Dimethylhydrazine (DMH)-induced CC rat model follows
the initiation and development processes of human spontaneous colon
tumours closely compared to those followed by the
xenotransplantation models of human CC cells and mouse cell lines
exhibiting spontaneous carcinogenesis (8). The DMH model follows the morphological
and molecular stages of CC in humans at every stage of tumour
development (9). Under the influence
of DMH, mutations occur in the proto-oncogenes, Apc, Kras,
and Trp53, in the colonocytes of rats. Similar mutations
also cause CС in humans. Additionally, like human CC cells,
DMH-induced cancer cells show microsatellite instability and
chromosomal instability (9).
Chemically induced CC allows one to study other aspects including
natural invasiveness of tumours into the intestine wall, and the
engraftment of intravenously administered MSCs in spontaneous
tumours, which cannot be observed when models of
xenotransplantation with CC cell lines are used.
Placenta-derived multipotent cells (PDMCs) satisfy
the criteria of International Society for Cellular Therapy
(10), but exhibit some features that
distinguish them from other MSCs (11,12). The
placenta, as source of MSCs, has a number of advantages; no
invasive procedures are required to obtain the cells, no moral and
ethical issues arise, and there is no lack of cell material. The
safety and efficiency of using PDMCs to treat intestinal
inflammation have already been established in clinical trials
(13). Despite these positive
results, the effect of PDMCs on CC that often occurs together with
inflammatory processes has not been sufficiently investigated.
Previous study showed that intravenous infusion of
human and rat PDMCs did not affect the number and dimensions of
DMH-induced colon tumours in rats (14). Intravenous administration was chosen
because it is the most widely used technique of MSC transplantation
in clinical practice (15,16). Therefore, the aim of this study was to
investigate the effects of allogeneic intravenous transplantation
of PDMCs in the middle phase of colon carcinogenesis on the
progression of different types of colon tumours and rat
survival.
Materials and methods
Isolation and culture of rat
PDMCs
All animal experiments were performed in accordance
with the international guidelines of the European Convention for
the Protection of Vertebrate Animals used for experimental and
other scientific purposes (European Convention, Strasburg, 1986)
and Article 26 of the Law of Ukraine ‘On protection of animals from
cruelty’ (no. 3447-IV, 21.02.2006), in addition to following all
norms of bioethics and biosafety. The protocol was approved by The
Bioethics Committee of The Educational and Scientific Centre
‘Institute of Biology and Medicine’ of Taras Shevchenko National
University of Kyiv (Protocol no. 8, 03.04.14). Female albino rats
(n=5) were euthanized on the 21st day of pregnancy through carbon
dioxide asphyxiation, and the placentas were immediately collected
and processed. Briefly, the CO2 flow rate was started
with 8 l/min in a 30-litre chamber. As unconsciousness occurred as
indicated by the loss of the righting reflex, the flow of the gas
was increased to 12 l/min to fill the chamber and decrease the time
to death. CO2 flow was maintained for at least 1 min
after respiratory arrest. Death was confirmed when the animal was
unresponsive to a toe pinch. Placentas from male foetuses were
selected (n=22).
PDMCs were isolated from the subculture of placental
explants outgrowth cells as described previously (14). Placental tissue fragments were placed
in cell culture dishes (Sarstedt, Nümbrecht, Germany) containing
high-glucose Dulbecco's modified Eagle's medium (DMEM; Life
Technologies Co., Paisley, UK) supplemented with 7% foetal bovine
serum (FBS; HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
and 3% allogeneic rat serum (BioWest, Nuaillé, France). Primary
cultures were cultivated in standard conditions, i.e., in a
humidified atmosphere at 37°C and 5% CO2, and the
culture medium was changed twice a week.
Generation of PDMC lines expressing
green fluorescent protein (GFP) using lentiviruses
Lentiviral transfer vector pCDH-CMV-MCS-EF1-copGFP
(System Biosciences, Mountain View, CA, USA) and psPAX2, pMD2. G
packaging plasmids were kindly provided by Dr. D. Trono (Ecole
Polytechnique Federale de Lausanne, Lausanne, Switzerland)
(17). EGFP sequence was
inserted into transfer vector pCDH-CMV-MCS-EF1-copGFP after PCR
amplification of the corresponding DNA fragment (786 bp) using
pEGFP-C1 plasmid as a template and specific primers (forward,
5′-TCCGCTAGCGCTACCGGTCGCCACC-3′ and reverse,
5′-GAGAATTCATCAGTTATCTAGAAGCTTGAGCTCGA-3′) containing NheI
and EcoRI endonuclease restriction sites respectively.
293T cells were seeded on a 100-cm tissue culture
dish and incubated until they reached ~70% confluence. 2 h prior to
transfection cell medium was replaced with 10 ml of fresh DMEM
supplemented with 10% FBS. 5 µg of EGFP-containing transfer
vector same as 3,5 and 1,5 µg of packaging plasmids (psPAX2 and
pMD2.G, respectively) were diluted in 1 ml of serum-free DMEM with
subsequent addition of 25 µl of briefly vortexed TurboFect™
Transfection Reagent (Thermo Fisher Scientific, Inc., Vilnius,
Lithuania). After pipetting and 20 min incubation, transfection
reagent/DNA mixture was dropwise added to 293T cells. 5 h later
cell growth media was changed and incubation was continued for 2
days. Post the 48-h incubation, cell culture supernatant was
transferred to 15 ml tube and centrifuged at 3,000 × g for 10 min
and then passed through a 0.22 µM ultra-low protein-binding
Millex-GV Syringe Filter (Durapore®; Merck Millipore,
Cork, Ireland).
The PDMCs were transduced twice with
virus-containing supernatant plus polybrene at the final
concentration of 8 µg/ml (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) overnight and media were changed between
transductions.
Flow cytometry
The number of GFP-expressing PDMCs (n=4) was
determined 24 h after transduction. Cells were harvested by
trypsinization, resuspended in DMEM supplemented with 1% FBS,
passed through 70 µm cell strainer (BD Biosciences, Franklin Lakes,
NJ, USA) and analysed on a BD FACSAria cell sorter using FACS Diva
6.1.2 software (both from BD Biosciences). To measure the viability
of PDMCs, cells were incubated with 5 µl of 7-AAD (BD Pharmingen,
San Diego, CA, USA) for 10 min at 4°C in the dark. Using forward
and side scatter parameters we eliminated debris from the analysis.
GFP and 7-AAD were excited by argon blue laser (488 nm) and
fluorescence was detected in the FITC channel using a 530/30 nm
bandpass filter and in PerCP-Cy5.5 channel using 695/40 nm bandpass
filter respectively. PDMCs without transduction were used as
negative control. Single-stained samples (7-AAD single-stained
cells without transduction and GFP-expressing cells without 7-AAD)
were used to adjust the compensation of fluorochrome overlap. At
least 10.000 cells were analyzed for each sample.
CC modelling and PDMC administration
In vivo experimental design
Experiments were performed using 4-month-old male
albino Wistar rats (n=137), weighing 180–200 g, obtained from the
Central Animal House of the Institute of Pharmacology and
Toxicology of NAMS of Ukraine. The animals were maintained under
12-h light/dark cycle, 60% humidity at 20–22°C and fed on standard
diet and tap water ad libitum. DMH (Sigma-Aldrich; Merck
KGaA) was dissolved in physiological saline (PS) and adjusted to pH
6.5 with 2 M sodium hydroxide immediately prior to use. To induce
tumour development, rats were subcutaneously injected with 20 mg/kg
DMH in 0.1 ml PS once weekly for 20 weeks (n=122) as described
previously (9). Intact control
animals (n=15) received PS subcutaneously once a week for 20
weeks.
At the 20th week of modelling, rats with DMH-induced
CC were sacrificed (group base, n=11) to confirm the initiation of
tumour development equivalent to stage
Т1-2N0-1M0 of CC in humans. To
confirm this, a thorough histological analysis of the developing
tumours was conducted.
Further, at the start of the 20th week, rats with
DMH-induced cancer were intravenously administered with 0.5 ml PS
(group PS, n=13), or PDMCs (group PDMCs, n=13) from four rat
fetuses (4 placenta tissue samples) at a dose of
2.2×106/kg body mass of rat in 0.5 ml PS (Fig. 1). The rats were sacrificed at the 25th
week.
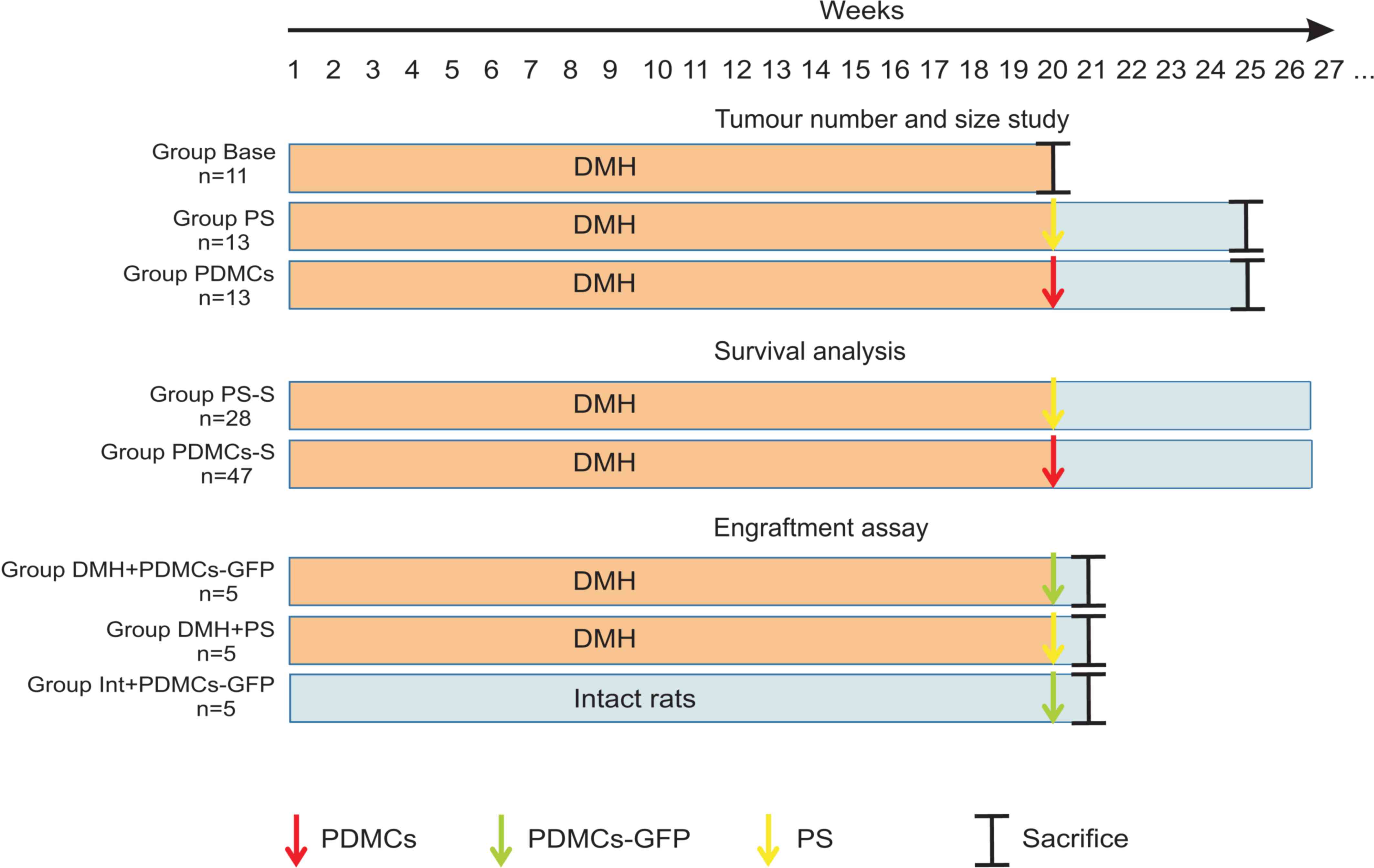 | Figure 1.Treatment protocols for the different
study groups. Group Base received DMH subcutaneously at a dose of
20 mg/kg once a week for 20 weeks. At the start of 21th week, the
animals were sacrificed to confirm tumour formation; Group PS
received DMH subcutaneously at a dose of 20 mg/kg once a week for
20 weeks. At the start of 20th week, the animals were injected with
0.5 ml PS and then maintained without supplements for 5 weeks of
the experimental period. At the end of 25th week, rats were
sacrificed. Group PDMCs was treated similarly to Group PS; however,
at the start of week 20, PDMCs were intravenously injected at a
dose of 2.2×106/kg body mass of rat in 0.5 ml PS. Group
PS-S was treated similarly to Group PS; however, all animals were
used for survival analysis; Group PDMCs-S was treated similarly to
Group PDMCs; however, all animals were used for survival analysis.
Group DMH + PDMCs-GFP received DMH subcutaneously at a dose of 20
mg/kg once a week for 20 weeks. At the start of week 20, PDMCs-GFP
were intravenously injected at a dose of 2.2×106/kg body
mass of rat in 0.5 ml PS. At the end of 20th week, rats were
sacrificed. Group DMH-PS was treated similarly to Group DMH +
PDMCs-GFP; however, at the start of week 20, 0.5 ml PS was
intravenously injected. Group Int + PDMCs-GFP received PS
subcutaneously once a week for 20 weeks. At the start of week 20,
PDMCs-GFP were intravenously injected at a dose of
2.2×106/kg body mass of rat in 0.5 ml PS. At the end of
20th week, rats were sacrificed. PDMCs, placenta-derived
multipotent cells; DMH, dimethylhydrazine; PS, physiological
saline; GFP, green fluorescent protein. |
Evaluation of colon lesions
The tumour nodule sizes were calculated and
histological analysis was performed as described by Svitina et
al (14). Briefly, the rats were
euthanized through carbon dioxide asphyxia, after which their
abdomens were opened, and their entire gastrointestinal tracts were
removed and cut longitudinally. The colon lesions were defined
macroscopically and colon was photographed with the ruler at least
three times. The 10 mm bar was set in each photo, lesions were
counted, and their area was calculated using ImageJ 1.46r software
(National Institutes of Health, Bethesda, MD, USA). The mean from
no less than three photos was calculated for each lesion. The total
tumour area per animal was calculated as the sum of areas of all
lesions in the colon. The average tumour area was calculated by
dividing of total tumour area on number of lesions per rat. Tissue
lesions were excised for standard H&E histological analysis
(14).
To analyse the distribution of tumours by the degree
of invasion, every tumour was assigned a certain value from 0 to 4
according to the international tumour classification system, TNM:
T0, (Тis) carcinoma in situ, the tumour is located in the
epithelial layer; T1, the tumour reaches submucosa; T2, the tumour
grows to the muscular layer; T3, the tumour infiltrates tela
subserosa; T4, the tumour grows into the visceral peritoneum or
other organs. The grade of tumour was evaluated depending on the
state of epithelium differentiation: well-differentiated tumour:
the epithelium contains all types of colon epithelium
(epitheliocytes and goblet cells) and reflects native colon crypt
structure; moderatory: crypt get deformed, epithelium get
multilayer structure; poorly: there are no native structure and
gland forming.
Survival analysis
Colon cancer was induced as described above. At the
start of the 20th week the rats with DMH-induced CC were
intravenously administered 0.5 ml PS (group PS-S, n=28), or PDMCs
(group PDMCs-S, n=47) from three pregnant rats (14 placenta tissue
samples) at a dose of 2.2×106/kg body mass of rat in 0.5
ml PS. Survival of all the rats was noted daily until death. None
of the rats had to be sacrificed because of excessive bleeding,
open wound infection, or moribund status.
Cell engraftment assay
Spreading of PDMCs-GFP was studied using healthy
rats and those with cancer. Аt the start of the 20th week, rats
with DMH-induced CC were intravenously administered with PDMCs-GFP
(Group DMH + PDMCs-GFP, n=5) from two pregnant rats (3 placenta
tissue samples), or 0.5 ml PS (Group DMH + PS, n=5), and intact
rats received PDMCs-GFP (Group Int + PDMCs-GFP, n=5) from two
pregnant rats (3 placenta tissue samples) (Fig. 1).
The cells were administered intravenously at a dose
of 2.2×106/kg body mass of rat in 0.5 ml PS. The rats
were euthanized at the 21st week through carbon dioxide
asphyxiation and the tumour and colon tissue (from healthy rats)
fragments, liver, lungs, spleen, and kidneys were extracted. PDMCs
engraftment was studied by PCR to contain EGFP DNA.
Immunohistochemistry was used to analyse localisation of engrafted
cells in colon tumour tissue.
DNA isolation and polymerase chain
reaction (PCR)
For PCR analysis, tissue samples were frozen in
liquid nitrogen and stored at −70°C. Genomic DNA was extracted
according to the standard phenol-chloroform extraction procedure
with modifications as described previously (14). Extracted DNA was stored at −20°C once
the concentration evaluation was performed using NanoDrop 2000
(Thermo Fisher Scientific, Inc., Wilmington, DE USA). The quality
of the isolated DNA was verified by PCR for the β-actin gene
(forward, 5′-ACCAACTGGGACGATATGGAGAAGA-3′ and reverse,
5′-TACGACCAGAGGCATACAGGGACAA-3′), and the PCR products were
examined by electrophoresis. After amplification, two products were
obtained: 680 bp (corresponding to the Actb gene) and 214 bp
(low specific of Acta1 gene). A 533 bp region of the
eGFP transgene was amplified using the following primers:
Forward, 5′-CCGCTAGCGCTACCGGTCGCCACC-3′ and reverse,
5′-GGCGGATCTTGAAGTTCACC-3′.
PCR reactions were performed on an Applied
Biosystems 2720 thermal cycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.) to final volumes of 20 µl containing 1X Hot Start
PCR buffer (2.0 mM Mg2+), 0.2 mM dNTPs, 0.3 µM of each
primer and 1.25 units of Maxima Hot Start Taq DNA polymerase
(Thermo Fisher Scientific, Inc., Vilnius, Lithuania). Each sample
was assayed in triplicate, and each run included water blanks. PCR
conditions were as follows: 95°C for 4 min, then 40 cycles of 95°C
for 40 sec, 59°C for 20 sec and 72°C for 40 sec, with a final
extension for 7 min at 72°C. PCR products were analysed using
ethidium bromide stained 1% agarose gels electrophoresis. Weight
Marker ‘GeneRuler DNA Ladder Mix’ (Thermo Fisher Scientific, Inc.)
and the ladder were supplied with 6X DNA Loading Dye. Detection of
the gel images was performed using a ChemiDoc™ XRS + System
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Immunohistochemistry
Following PDMCs administration, colon tumours from
DMH-treated rats (Group DMH + PDMCs-GFP; n=4) were fixed in 10%
buffered formalin at 4°C for 24 h, embedded in paraffin and
sectioned on 5-µm thick slices using Microm HM325 microtome (Carl
Zeiss AG, Oberkochen, Germany). Paraffin-embedded tissues were
deparaffinised in 100% xylene two times for 5 min each and
rehydrated in decreasing concentrations of ethanol: 96, 70 and 50%.
Slides were boiled in Citrate buffer (10 mM sodium citrate, pH 6.0)
for 30 min to retrieve the antigen. Nonspecific reactivity was
reduced by incubating tissue sections in a blocking solution (0.1 M
PBS with 0.5% BSA) at room temperature for 30 min. Immunostaining
was completed following an overnight incubation at 4°C with
polyclonal goat anti-GFP primary antibody (ab6673; 1:100; Abcam,
Cambridge, UK). Following washing three times for 10 min each with
0.1 M PBS, samples were incubated with donkey anti-goat IgG (H + L)
secondary antibody with Alexa Fluor 488 conjugate (ab150129;
1:1,000; Abcam) for 1 h at room temperature in the dark. Following
nuclear staining with Hoechst 33342 (H3570; 1:5,000; Thermo Fisher
Scientific, Inc.), slides were mounted using Mowiol 4–88
(Sigma-Aldrich; Merck KGaA). Confocal analysis was performed using
a Zeiss LSM 510 Meta microscope (magnification, ×630) and images
were captured with Zeiss LSM Image Browser Version 4.2.0.121
software (all from Carl Zeiss AG).
Statistical analysis
The data were analysed using GraphPad Prism 7.0
(GraphPad Software, Inc., La Jolla, CA, USA) using one- or two-way
ANOVA followed by Tukey's post-hoc test for multiple comparisons
and presented as mean ± standard deviation. The survival rates were
calculated by the Kaplan-Meier method in SPSS 10.0. (SPSS, Inc.,
Chicago, IL, USA), the analysis of survival between two groups was
carried out using log-rank test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of transplantation of PDMCs on
the body mass, number and area of colon tumours
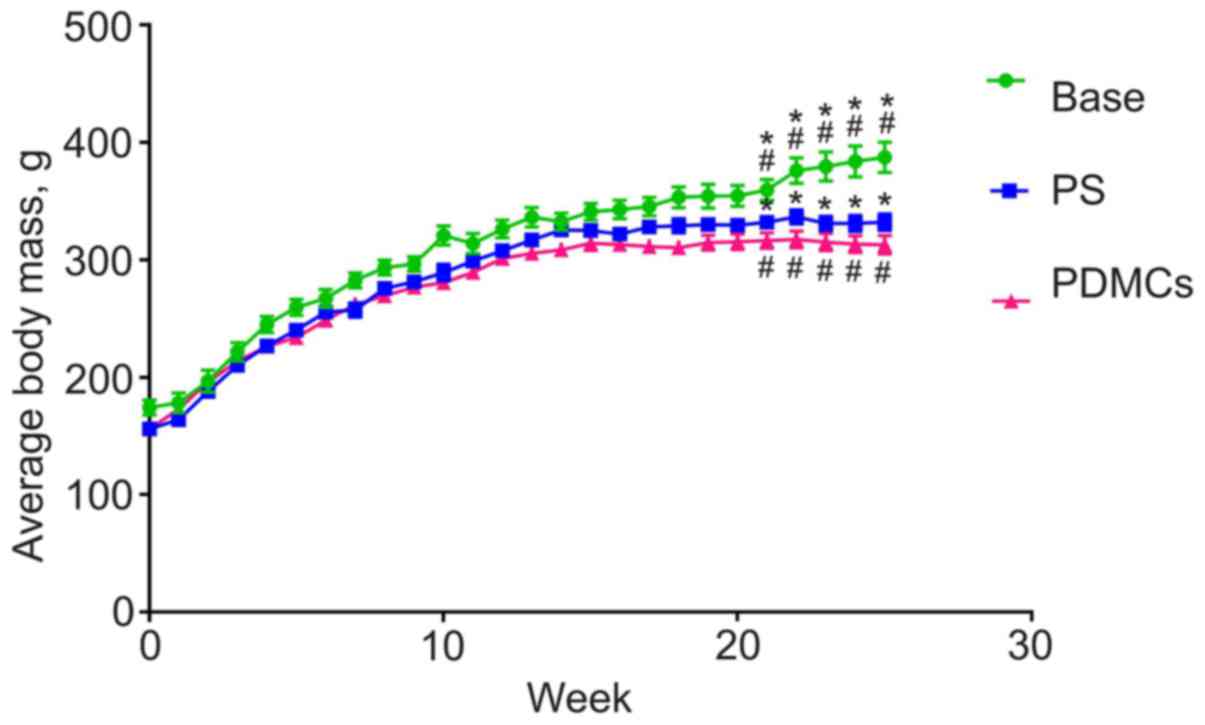
Body mass of intact control rats increased during
the 25 weeks of observation, while the masses of animals with
DMH-induced cancer began decreasing 16 weeks after the start of CC
induction. At the day of PDMC administration, mean mass of rats in
the control and the experimental groups differed significantly
(Fig. 2). It is important to note
that transplantation of PDMCs did not lead to normalisation of body
mass.
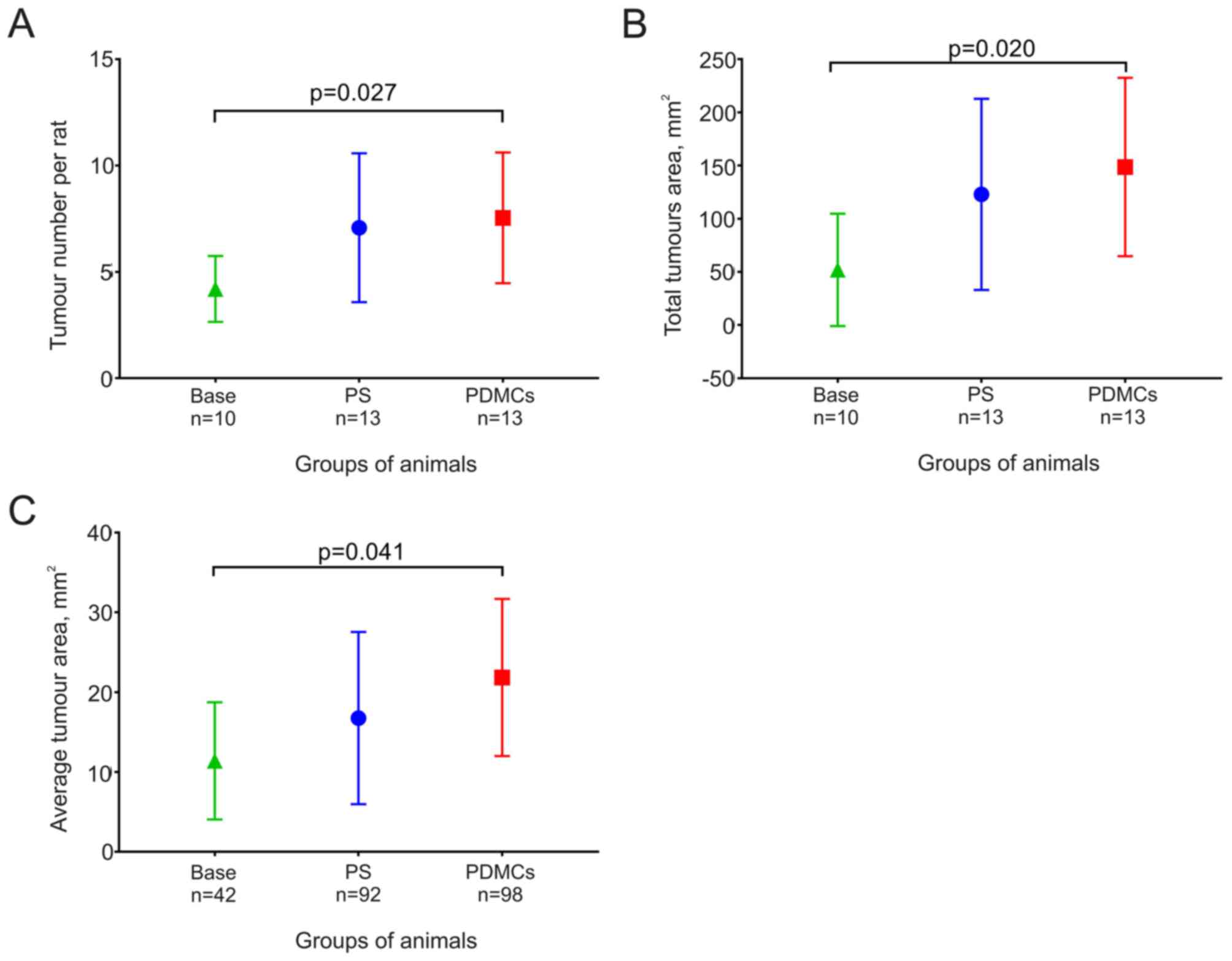
The mean number of tumours per rat did not change
significantly in animals from the PDMCs and the PS groups, although
this number increased significantly during five weeks after PDMCs
were transplanted (Fig. 3А).
The total and mean areas of tumours from the PDMCs
group were higher than those of the tumours from the PS group,
although the difference did not reach statistical significance. The
values of these parameters were higher in rats in the PS and PDMCs
groups than in the Base group, although a significant increase was
noted in animals after PDMC transplantation (Fig. 3B and C).
After DMH-induced development of colon cancer,
tumours with different degree of invasion and differentiation were
observed, depending on the depth of tumour growth into bowel wall
and state of epithelium (Fig. 4A and
B). Hence, our next task was to study the effect of intravenous
administration of PDMCs on size and number of different colon
tumour types (Fig. 4C-F).
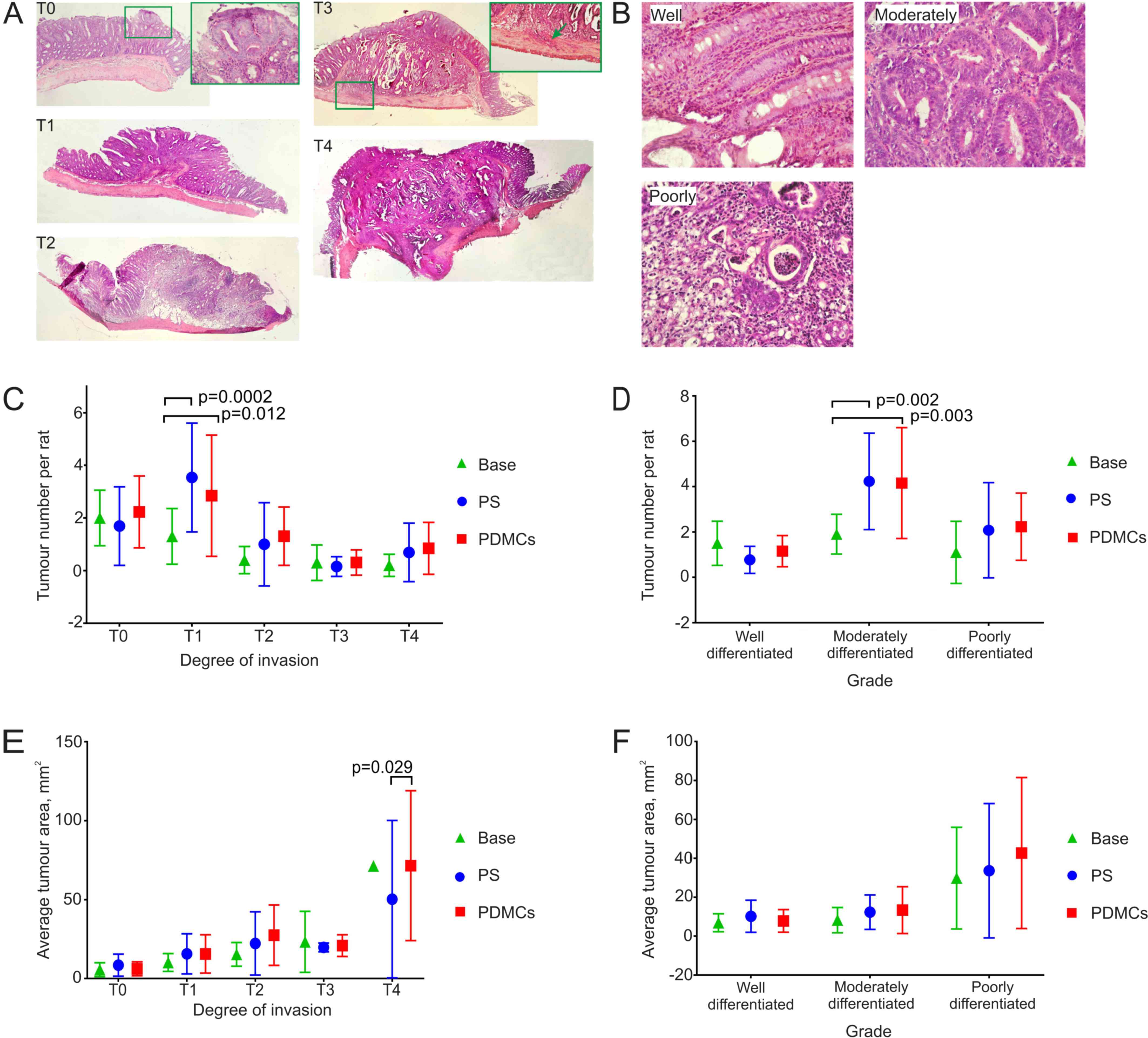 | Figure 4.The heterogeneity of colon tumour
types and their morphometric parameters in experimental groups. (A)
Histological analysis of the DMH induced colon tumours with
different degree of invasion into the intestine wall. T0, a tumour
is located in the epithelial layer; T1, a tumour reaches tela
submucosa; T2, a tumour grows to the muscular layer; T3, a tumour
infiltrates tela subserosa (green arrow); T4, a tumour grows into
the visceral peritoneum or other organs. H&E stain,
magnification ×40, magnification in the green frame T0 ×400,
magnification in the green frame T3 ×200. (B) Grade of tumours was
assigned according to epithelium state: well-differentiated tumour
(the epithelium contains all types of colon epithelial cells and
reflects native colon crypt structure); moderately differentiated
(crypt gets deformed, epithelium gets multilayer and/or
multicellular instead of unicellular single row); poorly
differentiated (there is no native structure and gland forming),
H&E stain, magnification ×400. The number of both tumours at
the 1st degree of invasion into the intestinal wall (C) and tumours
with moderately differentiated grade (D) increased in PS and PDMCs
groups compared to those in Base group. (E) The average area of
tumours at the 4th degree of invasion into intestinal wall
increased in PDMCs group as compared to PS and Base groups. (F) The
average area of tumours at different degrees of differentiation did
not change in all experimental groups. Two-way ANOVA followed by
Tukey's post-hoc test for multiple comparisons was employed for
statistical analysis. The results are presented as mean ± standard
deviation. PDMCs, placenta-derived multipotent cells; DMH,
dimethylhydrazine; PS, physiological saline. |
The distribution of tumours by the degree of
invasion almost did not differ between groups; a significant
difference was observed only in rats from the PDMCs and PS groups
compared to those in the Base group for the stage T1 tumours
(Fig. 4A, invasion into submucosa of
the intestine), which is expected, because during 5 weeks the
tumours invaded deeper compared to the time of treatment (Fig. 4C).
Measuring the differentiation degree has important
prognostic significance and depends on the structure of the
colonocytes in the epithelium. Considering the tumour grade
distribution by the degree of differentiation of the tumours, a
significant difference was observed in moderately differentiated
tumours (Fig. 4B) between the PDMCs
and Base groups and PS and Base groups (Fig. 4D). Additionally, we observed a
tendency towards an increase in the number of less differentiated
tumours in the PDMCs and PS groups as compared to those in the Base
group (Fig. 4D).
Interestingly, the mean size of the T4 tumours that
had invaded the deepest (Fig. 4A)
differed between the PDMCs and the PS groups (Fig. 4E), although we did not see a
difference in the mean size of tumours with different degrees of
differentiation (Fig. 4F).
Distribution of PDMCs in the body
To study the distribution of PDMCs in the rats'
bodies, the PDMCs were pre-marked by transduction of a genetic
vector containing EGFP. The level of transduction of PDMCs
was noted to be 7.6 to 57.7% (n=4). EGFP was detected in
different rat organs on the 7th day after transplantation (Fig. 5A and Table
I). Additionally, alive PDMCs-GFP were visualized by
immunohistochemistry in tumour colon tissues (Fig. 5B).
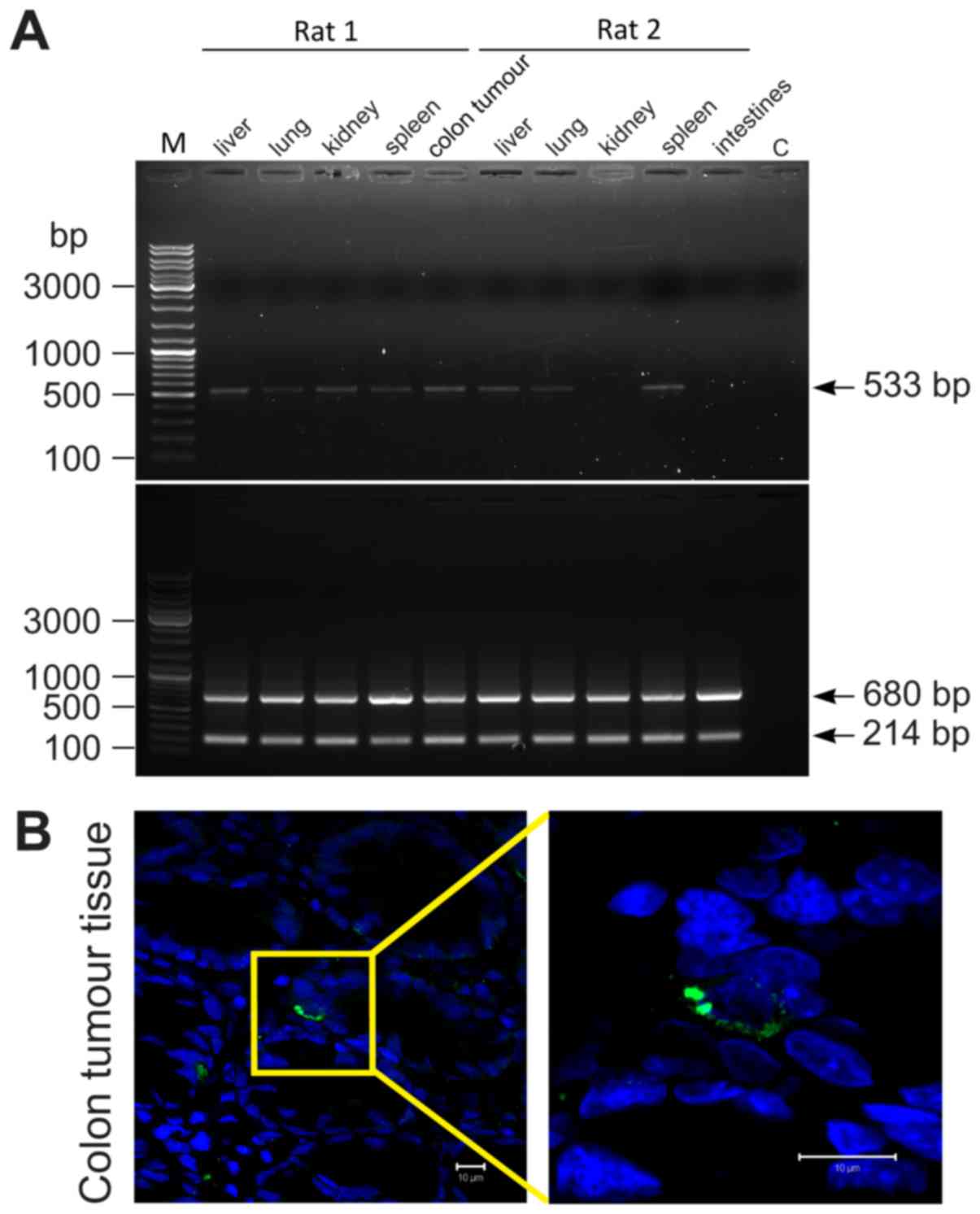 | Figure 5.PDMCs engrafted in different rat's
organs and colon tumours. (A) A representative example of PDMC-GFP
distribution in the rat body one week after administration. A PCR
analysis of EGFP (upper panel) and ACTB (bottom
panel) in rat's different tissues; rats 1 and 2 refer to groups
DMH+PDMCs-GFP and Int+PDMCs-GFP respectively; М, GeneRuler™ DNA
Ladder Mix (Thermo Fisher Scientific, Inc., Waltham, MA, USA), C,
water was used as negative control. (B) PDMCs-GFP were identified
in rat tumour colon tissues by anti-GFP staining.
Immunohistochemistry fluorescence: Anti-GFP are green, nuclei are
blue. Scale bar, 10 µm; magnification, ×630. PDMCs,
placenta-derived multipotent cells; GFP, green fluorescent protein;
EGFP, enhanced green fluorescent protein. |
 | Table I.Measuring EGFP in different
rat organs after transplantation of PDMCs-GFP. |
Table I.
Measuring EGFP in different
rat organs after transplantation of PDMCs-GFP.
|
| Colon |
|
|
|
|
|---|
|
|
|
|
|
|
|
|---|
| Animal group | Tumour | Norm | Liver | Spleen | Lungs | Kidney |
|---|
| DMH +
PDMCs-GFP | 4/5 | NA | 4/5 | 4/5 | 5/5 | 5/5 |
| DMH + PS | 0/5 | NA | NA | NA | NA | NA |
| Int +
PDMCs-GFP | NA | 4/4 | 1/4 | 1/4 | 5/5 | 1/4 |
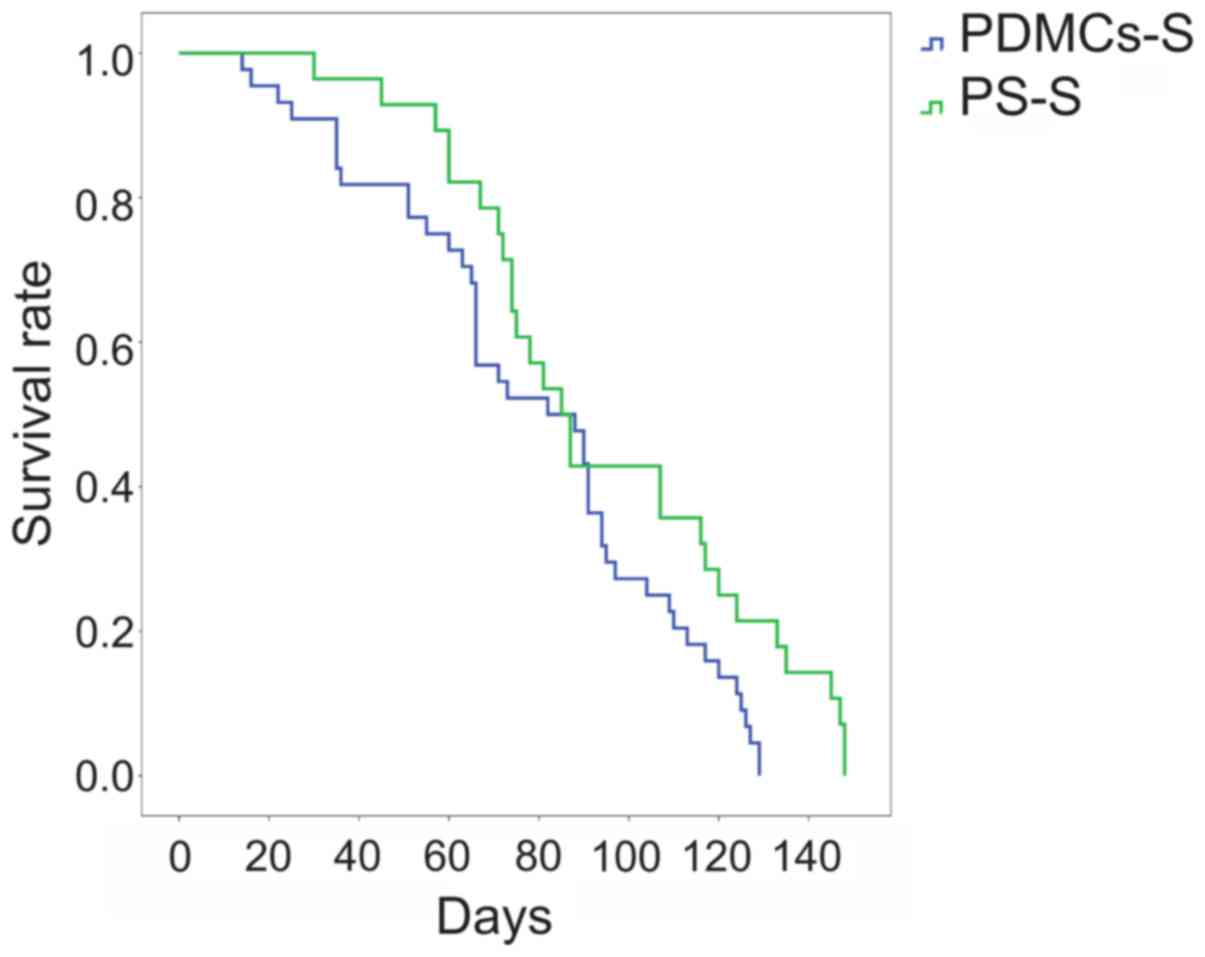
Effect of PDMCs transplantation on the
survival rate of rats with DMH-induced CC
One of the most important criteria when estimating
the effect of anti-tumour pharmacological and cell preparations is
survival analysis. Intravenous allogeneic transplantation of PDMCs
led to a reduction in the survival rate of rats with DMH-induced CC
by 17 days (Fig. 6). The median
survival rate for the untreated control (PS-S) group was 107 days,
and that for the PDMCs-S group was 90 days.
In this study, it was demonstrated that PDMCs
administered in rats at the middle phase of colon carcinogenesis,
led to an increase in the size of the most invasive tumours and a
rise in mortality.
Discussion
The safety and efficacy of the PDMCs transplantation
for the treatment of inflammatory colon diseases have been
demonstrated by clinical trials (13,18).
Despite the positive results, the effect of PDMCs on the CC, that
often accompanies inflammatory processes in the intestine, has not
been studied sufficiently. Currently, the two-side effect of MSCs,
including placenta-derived MSCs (19), on the growth of transformed cells is a
topic of research and some possible mechanisms for this phenomenon
have been proposed (20,21). Most of the experiments for evaluation
of the MSCs' impact on cancer have been carried out in vitro
(22) while xenotransplantation
models were used in vivo (23,24). In
her review, O'Malley et al (25) indicates that most studies regarding
the effect of MSCs on CC were performed on immunodeficient animals
with allo/xenografted tumours and local administration of high
doses MSCs.
The results of such studies cannot be used to
predict the impact of MSCs on the development of colon cancer in
humans (25). In our study, PDMCs
transplanted at middle phase of CC did not affect either the number
or the area of the forming tumours, but detailed analysis of the
area of tumours depending on the invasion depth revealed an
unexpected result due to the selectivity of stimulation of stage T4
tumours. After all, previously, it was shown that after
administering bone marrow-derived MSCs at early stages of
azoxymethane/dextran sulfate sodium colitis-associated
tumourigenesis modelling, the number of tumours decreased, but the
mean tumour size did not change (26,27).
Katsuno et al observed that after administering bone
marrow-derived MSCs to rats in early phase of DMH-induced
carcinogenesis, both the number and the volume of generated tumours
decreased, while at the late phase it did not have any effect
(4).
However, we did not observe any stimulation of colon
tumours with low and medium degree of invasion after systemic cell
administration that could suggest the selective effect of PDMCs
depending on colon cancer cell properties. According to the ‘Big
Bang’ model of CC (28), the colon
tumour is heterogenic, that is, it gradually accumulates mutations
of different genes. It was recently found that different CC cell
lines are characterized by cell line-specific growth dependency on
MSCs; for example, MSCs stimulate the growth of COLO 320, HCT116,
and LoVo, and inhibit the growth of DLD-1, HCT-15, and HT-29 if
they are xenotransplanted to SCID mice (29). Bearing in mind that the aforementioned
cell lines differ in mutation and gene expression profiles, one can
propose that spontaneous colon tumours at stage T4 contain cells
that would be able to answer the contact-dependent and/or paracrine
effects of PDMCs. A similar contrasting effect was shown after
co-transplantation of Wharton's jelly MSC (WJMSCs) with cancer stem
cell (CSCs) lines from different types of lung tumours (30). The authors suggested that the
stimulatory or inhibitory effect of MSCs on pulmonary tumours
depends on the degree of its differentiation. It was also found
that the co-transplantation of WJMSCs with the LCSC lines in the
NOD/SCID mice led to the elevation in the expression of the CSCs
marker CD133 indicating a reduction in the degree of
differentiation of the tumours, and so an increase of their growth
and metastasis (30).
We showed that PDMCs were able to engraft into
tissues of various organs-the liver, spleen, lungs, kidneys, colon,
and colon tumours. The engraftment of human PDMCs after intravenous
transplantation was detected earlier in different organs (31) and colon cancer (14) of rodents. Also using the
immunohistochemistry method to detect GFP-labelled PDMCs, we showed
the presence of the alive PDMCs in the tissue of spontaneous colon
tumour on day 35 after administration (14). Since the traces of PDMCs were detected
in many different organs, we must take into account not only the
interaction between MSCs and various cell populations of the tumour
(endotheliocytes, epitheliocytes, tumour-associated macrophages,
and cancer-associated fibroblasts), but also the complex effect of
MSCs on the organism as a whole that changes the functioning of
other organ systems, which can reflect on carcinogenesis.
Particularly, along with stimulation of the tumour
growth at stage T4, we found deterioration in animal survival rate
after PDMC transplantation that has not been established until now.
Taking into account the fact that, as mentioned earlier, the total
number and area of tumours does not change at the same time, it
indicates the necessity to study MSCs effect on different
spontaneous colon tumours which are as usual very heterogenic.
Furthermore, the animal survival rate as a critical indicator has
to be evaluated before postulating a conclusion about the effect of
MSC on carcinogenesis. It should be noted that the effect of MSC
transplantation on animal survival with carcinogenesis of the colon
has almost not been investigated. Only Shinagawa et al in
their study showed that co-transplantation of colon cancer cell
line KM12SM and MSCs led to a decrease of mice survival rate as
compared to animals with KM12SM alone (5). Decreased survival of mice after
co-transplantation may be caused by the enhancement of
proliferation, angiogenesis, migration, and invasion along with the
inhibition of apoptosis of tumour cells by MSCs (5).
Further investigations designed to explore the
mechanisms underlying the effects of PDMCs on different colon
tumour subtypes may shed light on the complex effects of MSC
systemic transplantation on cancer progression.
Acknowledgements
The authors thank Mr. S. Martynenko and Dr. M.
Sokolov for their help in organisation and support of this
research. This study was supported by the Institute of Cell Therapy
and The National Academy of Sciences of Ukraine (grant no.
0114U003877).
Glossary
Abbreviations
Abbreviations:
|
MSCs
|
mesenchymal stem cells
|
|
PDMCs
|
placenta-derived multipotent cells
|
|
CC
|
colorectal cancer
|
|
EGFP
|
enhanced green fluorescent protein
|
|
PCR
|
polymerase chain reaction
|
|
SCID
|
severe combined immunodeficiency
|
|
DMH
|
dimethylhydrazine
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
rPDMCs
|
rat PDMCs
|
References
|
1
|
Squillaro T, Peluso G and Galderisi U:
Clinical trials with mesenchymal stem cells: An update. Cell
Transplant. 25:829–848. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mele V, Muraro MG, Calabrese D, Pfaff D,
Amatruda N, Amicarella F, Kvinlaug B, Bocelli-Tyndall C, Martin I,
Resink TJ, et al: Mesenchymal stromal cells induce
epithelial-to-mesenchymal transition in human colorectal cancer
cells through the expression of surface-bound TGF-β. Int J Cancer.
134:2583–2594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Boeck A, Pauwels P, Hensen K, Rummens
JL, Westbroek W, Hendrix A, Maynard D, Denys H, Lambein K, Braems
G, et al: Bone marrow-derived mesenchymal stem cells promote
colorectal cancer progression through paracrine neuregulin 1/HER3
signalling. Gut. 62:550–560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katsuno T, Ochi M, Tominaga K, Tanaka F,
Sogawa M, Tanigawa T, Yamagami H, Shiba M, Watanabe K, Watanabe T,
et al: Mesenchymal stem cells administered in the early phase of
tumorigenesis inhibit colorectal tumor development in rats. J Clin
Biochen Nutr. 53:170–175. 2013. View Article : Google Scholar
|
|
5
|
Shinagawa K, Kitadai Y, Tanaka M, Sumida
T, Kodama M, Higashi Y, Tanaka S, Yasui W and Chayama K:
Mesenchymal stem cells enhance growth and metastasis of colon
cancer. Int J Cancer. 127:2323–2333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Altobelli E, D'Aloisio F and Angeletti PM:
Colorectal cancer screening in countries of european council
outside of the EU-28. World J Gastroenterol. 22:4946–4957. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
American Cancer Society, . Colorectal
Cancer Facts & Figures 2014–2016. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2014-2016.pdf
|
|
8
|
Jackstadt R and Sansom OJ: Mouse models of
intestinal cancer. J Pathol. 238:141–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Perše M and Cerar A: Morphological and
molecular alterations in 1,2 dimethylhydrazine and azoxymethane
induced colon carcinogenesis in rats. J Biomed Biotechnol.
2011:4739642011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dominici M, Le Blanc K, Mueller I,
Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A,
Prockop Dj and Horwitz E: Minimal criteria for defining multipotent
mesenchymal stromal cells. The international society for cellular
therapy position statement. Cytotherapy. 8:315–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mariotti E, Mirabelli P, Abate G,
Schiattarella M, Martinelli P, Fortunato G, Di Noto R and Del
Vecchio L: Comparative characteristics of mesenchymal stem cells
from human bone marrow and placenta: CD10, CD49d and CD56 make a
difference. Stem Cells Dev. 17:1039–1041. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miao Z, Jin J, Chen L, Zhu J, Huang W,
Zhao J, Qian H and Zhang X: Isolation of mesenchymal stem cells
from human placenta: Comparison with human bone marrow mesenchymal
stem cells. Cell Biol Int. 30:681–687. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu J, Zhao G, Zhang L, Qiao C, Di A, Gao H
and Xu H: Safety and therapeutic effect of mesenchymal stem cell
infusion on moderate to severe ulcerative colitis. Exp Ther Med.
12:2983–2989. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Svitina H, Kyryk V, Skrypkina І, Kuchma M,
Bukreieva T, Areshkov P, Shablii Y, Denis Y, Klymenko P, Garmanchuk
L, et al: Placenta-derived multipotent cells have no effect on the
size and number of DMH-induced colon tumors in rats. Exp Ther Med.
14:2135–2147. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kean TJ, Lin P, Caplan AI and Dennis JE:
MSCs: Delivery routes and engraftment, cell-targeting strategies
and immune modulation. Stem Cells Int. 2013:7327422013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kurtz A: Mesenchymal stem cell delivery
routes and fate. Int J Stem Cells. 1:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zufferey R, Nagy D, Mandel RJ, Naldini L
and Trono D: Multiply attenuated lentiviral vector achieves
efficient gene delivery in vivo. Nat Biotechnol. 15:871–875. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang J, Lv S, Liu X, Song B and Shi L:
Umbilical cord mesenchymal stem cell treatment for crohn's disease:
A randomized controlled clinical trial. Gut Liver. 12:73–78. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Silini AR, Cancelli S, Signoroni PB,
Cargnoni A, Magatti M and Parolini O: The dichotomy of
placenta-derived cells in cancer growth. Placenta. 59:154–162.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rhee KJ, Lee JI and Eom YW: Mesenchymal
stem cell-mediated effects of tumor support or suppression. Int J
Mol Sci. 16:30015–30033. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klopp AH, Gupta A, Spaeth E, Andreeff M
and Marini F III: Concise review: Dissecting a discrepancy in the
literature: Do mesenchymal stem cells support or suppress tumor
growth? Stem Cells. 29:11–19. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen D, Liu S, Ma H, Liang X, Ma H, Yan X,
Yang B, Wei J and Liu X: Paracrine factors from adipose-mesenchymal
stem cells enhance metastatic capacity through Wnt signaling
pathway in a colon cancer cell co-culture model. Cancer Cell Int.
15:422015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohta N, Ishiguro S, Kawabata A, Uppalapati
D, Pyle M, Troyer D, De S, Zhang Y, Becker KG and Tamura M: Human
umbilical cord matrix mesenchymal stem cells suppress the growth of
breast cancer by expression of tumor suppressor genes. PLoS One.
10:e01237562015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng H, Zou W, Shen J, Xu L, Wang S, Fu
YX and Fan W: Opposite effects of coinjection and distant injection
of mesenchymal stem cells on breast tumor cell growth. Stem Cells
Transl Med. 4:1216–1228. 2016. View Article : Google Scholar
|
|
25
|
O'Malley G, Heijltjes M, Houston AM, Rani
S, Ritter T, Egan LJ and Ryan AE: Mesenchymal stromal cells (MSCs)
and colorectal cancer: A troublesome twosome for the anti-tumour
immune response? Oncotarget. 7:60752–60774. 2016.PubMed/NCBI
|
|
26
|
Nasuno M, Arimura Y, Nagaishi K, Isshiki
H, Onodera K, Nakagaki S, Watanabe S, Idogawa M, Yamashita K,
Naishiro Y, et al: Mesenchymal stem cells cancel
azoxymethane-induced tumor initiation. Stem Cells. 32:913–925.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Z, He X, He X, Chen X, Lin X, Zou Y,
Wu X and Lan P: Bone marrow mesenchymal stem cells ameliorate
colitis-Associated tumorigenesis in mice. Biochem Biophys Res
Commun. 450:1402–1408. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sottoriva A, Kang H, Ma Z, Graham TA,
Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D and
Curtis C: A Big Bang model of human colorectal tumor growth HHS
Public Access new model provides a quantitative framework to
interpret tumor growth dynamics and the origins of ITH with
significant clinical implications. Nat Genet. 47:209–216. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakagaki S, Arimura Y, Nagaishi K, Isshiki
H, Nasuno M, Watanabe S, Idogawa M, Yamashita K, Naishiro Y, Adachi
Y, et al: Contextual niche signals towards colorectal tumor
progression by mesenchymal stem cell in the mouse xenograft model.
J Gastroenterol. 50:962–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vulcano F, Milazzo L, Ciccarelli C, Eramo
A, Sette G, Mauro A, Macioce G, Martinelli A, La Torre R, Casalbore
P, et al: Wharton's jelly mesenchymal stromal cells have
contrasting effects on proliferation and phenotype of cancer stem
cells from different subtypes of lung cancer. Exp Cell Res.
345:190–198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu CG, Zhang JC, Xie CQ, Parolini O,
Silini A, Huang YZ, Lian B, Zhang M, Huang YC and Deng L: In vivo
tracking of human placenta derived mesenchymal stem cells in nude
mice via 14C-TdR labeling. BMC Biotechnol. 15:552015.
View Article : Google Scholar : PubMed/NCBI
|