Introduction
Leukemia is a malignant hematologic neoplasm that
occurs at the level of hematopoietic stem cells, accounts for ~5%
of all malignancies, and is one of the most common types of cancer
among children and adolescents (1).
Acute promyelocytic leukemia (APL), a high mortality-associated
subtype of acute leukemia, predominantly manifests as disseminated
intravascular coagulation or hyperfibrinolysis-induced severe
bleeding (2). Chemotherapy with
all-trans retinoic acid, anthracycline antibiotics and arsenic
trioxide is a typical treatment for leukemia, and has been widely
used with satisfactory effect (3).
However, due to different genetic phenotypes and repeated drug
exposure, drug resistance has begun to decrease the effectiveness
of chemotherapy and the overall survival time of patients with
leukemia, including those with APL (4). Thus, developing chemotherapy drugs that
may aid in the treatment of patients with APL despite the presence
of drug resistance is crucial.
A previous study demonstrated that, via the
regulation of downstream nuclear transcription factors, the
recombinant activated factor (RAF)/mitogen-activated protein kinase
(MEK)/extracellular signal-regulated kinase (ERK) signaling pathway
is associated with the increased expression of the
multidrug-resistant permeability glycoprotein, and may thereby
induce drug resistance in leukemia cells (5). Sorafenib is an oral RAF kinase
inhibitor, and has been used for the treatment of liver and kidney
cancer (6). Sorafenib inhibits the
RAF/MEK/ERK signaling pathway thereby suppressing tumor cell
proliferation and angiogenesis (7).
Multiple studies have demonstrated that sorafenib induces tumor
cell apoptosis in numerous types of cancer, including
hepatocellular carcinoma (8),
melanoma (9) and human glioblastoma
cells (10), and acute myelocytic
leukemia (11). Numerous studies have
focused on the clinical application of sorafenib in acute leukemia
and have demonstrated that sorafenib may exert a satisfactory
chemotherapeutic effect (12–14). However, the effects and underlying
mechanisms of sorafenib on APL remain to be fully understood.
Therefore, in the present study, sorafenib was used
to treat the APL cell line NB4. The effect of sorafenib on the cell
cycle, proliferation and apoptosis were subsequently assessed.
Furthermore, the present study examined the mechanisms underlying
the effect of sorafenib on the proliferation and apoptosis of APL
cells.
Materials and methods
Cell culture
The APL cell line NB4 was purchased from the
Shanghai Institute of Biochemistry and Cell Biology (Shanghai,
China) and subsequently maintained in RPMI-1640 medium supplemented
with 10% fetal bovine serum (10%; both from Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) in a 37°C humidified incubator with
5% CO2. The cells in logarithmic phase were used in
subsequent experiments.
MTT assay
NB4 cells with a density of 1×104
cells/ml were cultured on a 96-well plate (180 µl/well). On day
two, multiple concentrations of sorafenib (0, 3, 6 or 12 µM; 20
µl/well; Bayer AG, Leverkusen, Germany) were added onto wells and
cultured for 24, 48 or 72 h. Cells were subsequently incubated with
10 µl of MTT (5 mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) for 4 h at the same time each day. Subsequently, 100 µl of
DMSO (KeyGen Biotech Co. Ltd., Nanjing, China) was added to
solubilize the formazan crystals at room temperature. Zero (medium,
MTT, DMSO) and blank holes were set up. The absorbance was measured
as the optical density (OD) value at a wavelength of 492 nm on a
microplate reader. The inhibition rate of cell proliferation was
calculated as follows: 1-(Experimental OD-Blank OD)/(Negative
control OD-Blank OD)×100%.
Flow cytometry
NB4 cells were treated with sorafenib at a final
concentration of 0, 3, 6 or 12 µM. Following treatment for 24 and
48 h, cells were collected via centrifugation (180 × g for 5 min)
at room temperature. For subsequent cell cycle analysis, cells were
fixed with precooled 75% ethanol at 4°C for 1 h. Cells were then
centrifuged (350 × g for 6 min) at room temperature and the ethanol
was removed by washing with PBS. Cells were resuspended in 300 µl
of PBS and treated with 50 µg/ml ribonuclease A (Shanghai Sangong
Pharmaceutical Co., Ltd., Shanghai, China) at 37°C for 30 min.
Subsequently, cells were maintained in propidium iodide (PI; KeyGen
Biotech Co. Ltd.) in the dark at 4°C for 15 min. A flow cytometer
(BD Biosciences, Franklin Lakes, NJ) was used to detect the cell
cycle. The Annexin V-fluorescein isothiocyanate (FITC) Apoptosis
Detection kit was used to detect cell apoptosis (KeyGen Biotech Co.
Ltd.). The collected cells were incubated with Annexin V-FITC and
PI at 25°C for 15 min in the dark. Following incubation with
Binding buffer (provided by the Annexin V-FITC Apoptosis Detection
kit), cells were detected using the flow cytometer. The experiment
was repeated three times.
Reverse transcription-semi
quantitative polymerase chain reaction (RT-sqPCR)
Cells were treated with sorafenib at a final
concentration of 0, 3, 6 or 12 µM for 48 h. Subsequently, total RNA
was extracted from the collected cells using TRIzol reagent (KeyGen
Biotech Co. Ltd.) at room temperature for 15 min and measured using
a UV spectrophotometer (BD Biosciences) at A260; cDNA
was reverse transcribed from 1 µg of total RNA with
A260/A280 of 1.8–2.0 using a PrimeScript RT
reagent kit with a genomic DNA eraser (Takara Biotechnology Co.,
Ltd., Dalian, China) at 37°C for 15 min, followed by 65°C for 15
sec. Briefly, 10 µl of reaction system included: 2 µl 5×
PrimeScript RT master mix, 7 µl RNase-free water and 1 µl RNA.
Reverse cDNA was then used in sqPCR. Primer sequences corresponding
to caspase-3, caspase-8, myeloid cell leukemia (MCL)1 and GAPDH are
provided in Table I. The sqPCR
reaction system was comprised of 1 µl of cDNA, 0.25 µl of each
primer (Sangon Biotech Co., Ltd., Shanghai, China), 12.5 µl of Taq
PCR mix (Takara Biotechnology Co., Ltd.) and double distilled
H2O to adjust the total volume to 25 µl. Initial
denaturation occurred at 94°C for 3 min, followed by 40 30-sec
cycles at 94°C, 58°C and 74°C, respectively. Elongation followed at
74°C for 10 min. The PCR product was visualized using agarose gel
(1.5%), and analyzed on a UV trans-illuminator (UVitec Ltd.,
Cambridge, UK). Subsequently, sqPCR analysis was performed using
the ratio of grey density associated with target genes to that
associated with the internal control. mRNA was quantified using
Labworks™ Analysis Software (version 4.5; Upland, CA,
USA).
 | Table I.Primer sequences of MCL1, caspase-3,
caspase-8 and GAPDH. |
Table I.
Primer sequences of MCL1, caspase-3,
caspase-8 and GAPDH.
| Gene | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| MCL1 | F:
GCGACTTTTGGCTACGGAGA | 246 |
|
| R:
ATGAGGTGAAAGCCGCGAAA |
|
| Caspase-3 | F:
AGGAGCAGTTTTGTTTGTGTGC | 123 |
|
| R:
TCGTGGACCAATAATAAGAACCG |
|
| Caspase-8 | F:
GGGGCTTTGACCACGACCT | 368 |
|
| R:
GTTTGCTCTATATAGGGCCTACTC |
|
| GAPDH | F:
ACGGGAAACCCATCACCATC | 129 |
|
| R: CTACCACTACCCAAA
GGGCA |
|
Western blot analysis
Cells were treated with sorafenib at a final
concentration of 0, 3, 6, or 12 µM for 48 h. Subsequently, cells
were collected and lysed using radioimmunoprecipitation assay
buffer (Beyotime Institute of Biotechnology, Haimen, China). A
bicinchoninic acid protein assay kit based on the standard curve
was used to determine the protein concentration. An equal amount of
protein (30 µg) per lane was separated using SDS-PAGE (10–15% gel).
Following electrophoresis, the separated proteins were transferred
onto a polyvinylidene fluoride membrane. The membranes were
subsequently blocked with 5% skim milk for 2 h at room temperature,
and incubated with rabbit anti-rat MCL1 (cat. no. SAB4501843),
caspase-3 (cat. no. C8487), caspase-8 (cat. no. C2976), cyclin D1
(cat. no. SAB4503501), MEK (cat. no. SAB4501863), phosphorylated
(P)-MEK (cat. no. M7683), ERK (cat. no. M7556) or P-ERK (cat. no.
M7933) polyclonal antibodies (1:500; Sigma-Aldrich; Merck KGaA), or
rabbit anti-rat β-actin monoclonal antibody (cat. no. SAB5500001;
1:1,000, Sigma-Aldrich; Merck KGaA) at 4°C overnight. β-actin was
used as an internal control. Following washing with PBS, the
membrane was incubated with horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (cat. no. A16110; 1:10,000; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) for 2 h at room
temperature. The membrane was subsequently washed with PBS and
developed using 3,3′-diaminobenzidine (Shanghai Sangong
Pharmaceutical Co., Ltd.). Image Pro Plus software (version 6.3,
Media Cybernetics, Inc., Rockville, MD, USA) was used to detect
target protein expression.
Statistical analysis
SPSS software (Version 16.0; SPSS, Inc., Chicago,
IL, USA) was used to analyze data. All experiments were repeated
three times. All data were expressed as mean ± standard deviation.
One-way analysis of variance (ANOVA) was used to analyze data among
different time points. Data among different concentrations of
sorafenib were analyzed using one-way ANOVA. The Bonferroni method
was used for pairwise comparison. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of sorafenib on NB4 cell
proliferation
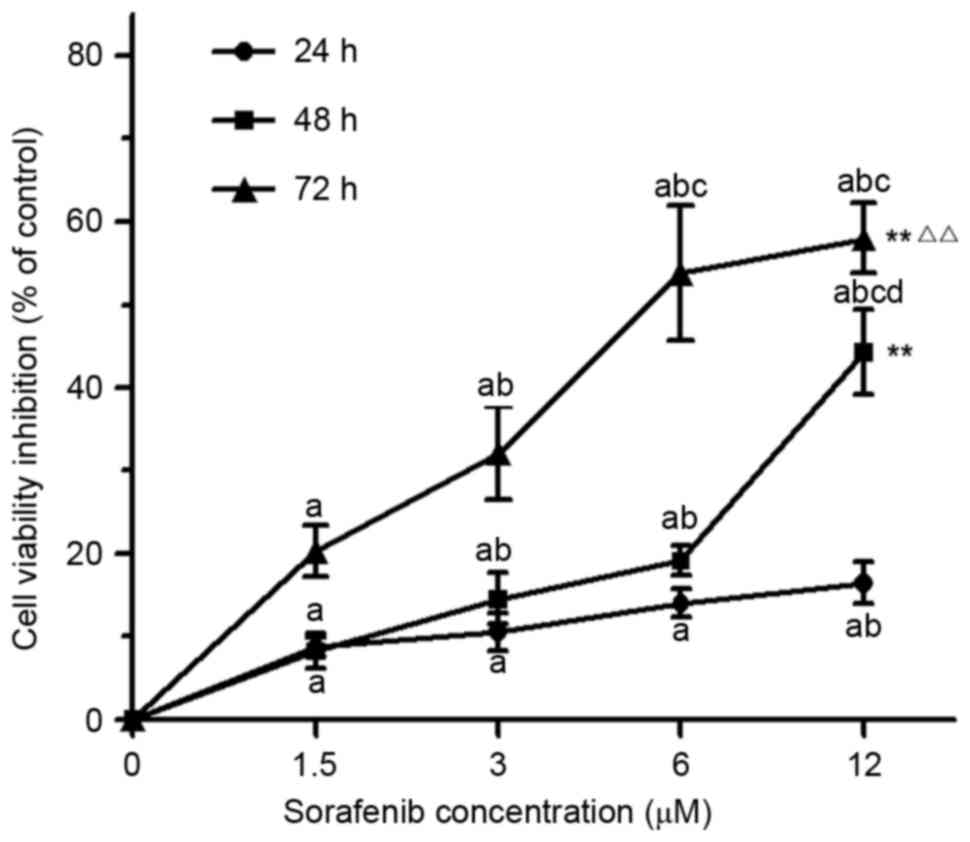
MTT analysis demonstrated that, compared with
untreated cells, 1.5–12 µM of sorafenib significantly inhibited the
proliferation of NB4 cells following treatment for 24, 48, or 72 h
(P<0.05). Furthermore, the inhibiting effect of sorafenib on NB4
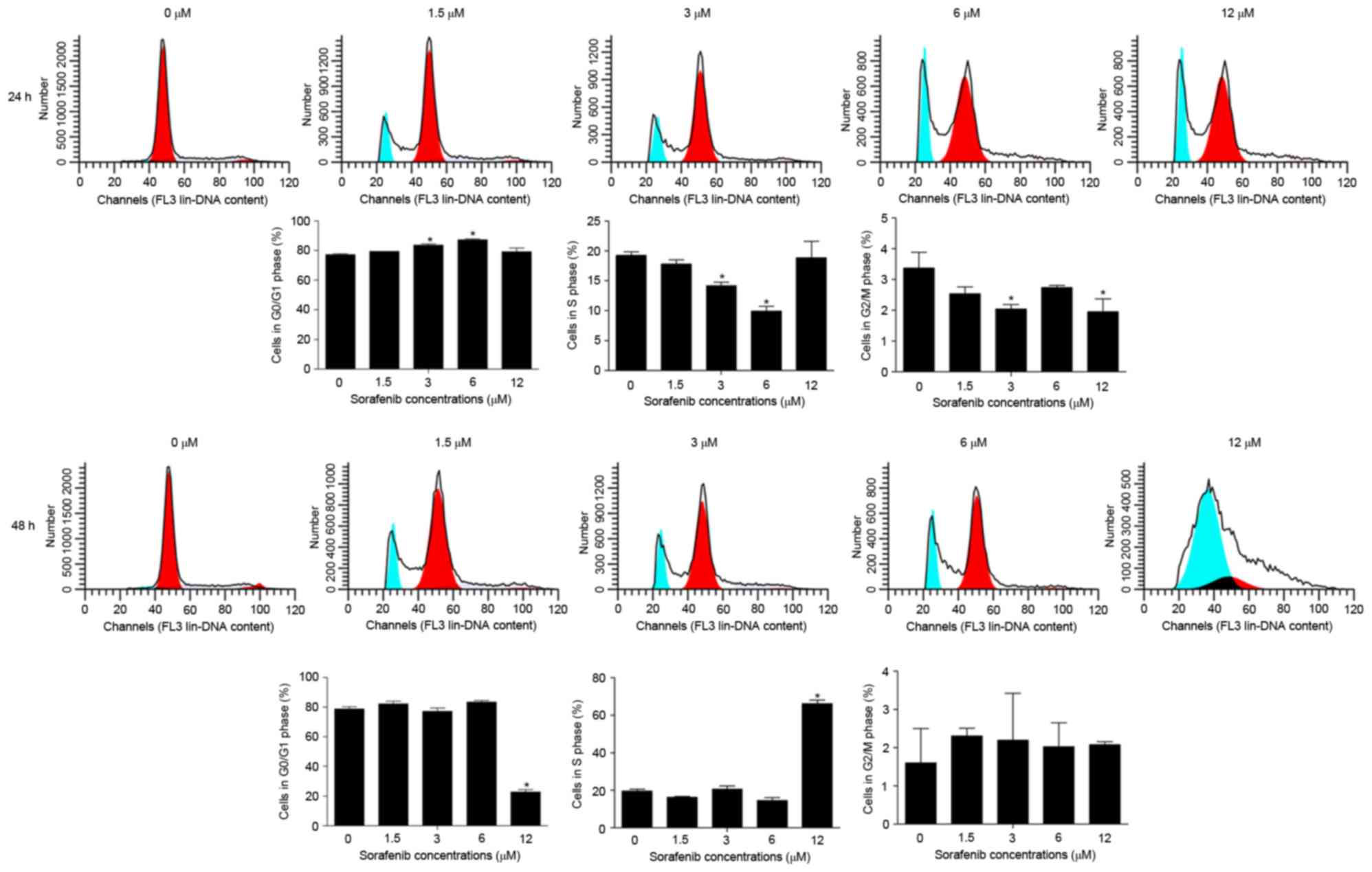
cells was dose- and time-dependent (P<0.01; Fig. 1). The results of cell cycle analysis
demonstrated that, compared with untreated cells, the
sorafenib-treated NB4 cells exhibited a significantly increased
percentage of cells in the G0/G1 phase, but a significantly
decreased percentage of cells in S phase (P<0.05; Fig. 2), suggesting sorafenib may induce cell
cycle arrest in the G0/G1 phase of NB4 cells.
Effect of sorafenib on NB4 cell
apoptosis
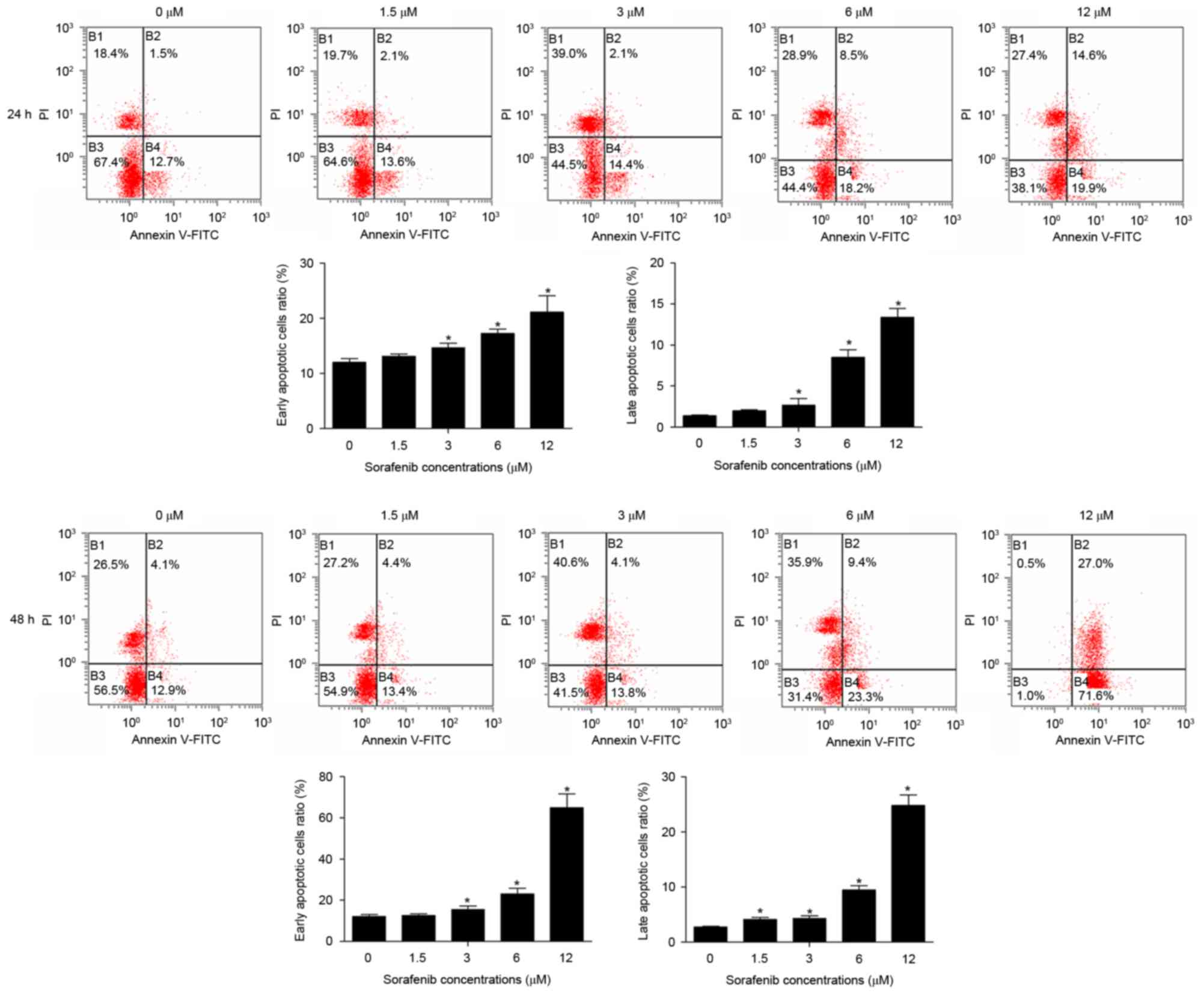
Compared with untreated cells, the early and late
apoptotic cells ratio was significantly increased in NB4 cells
treated with 1.5–12 µM of sorafenib for 24 or 48 h (Fig. 3), and sorafenib induced an increase in
NB4 cell apoptosis dose-dependently (P<0.05). These results
suggest that sorafenib may induce apoptosis in NB4 cells.
Effect of sorafenib on the expression
of MCL1, caspase-8, caspase-3 and cyclin D1 in NB4 cells
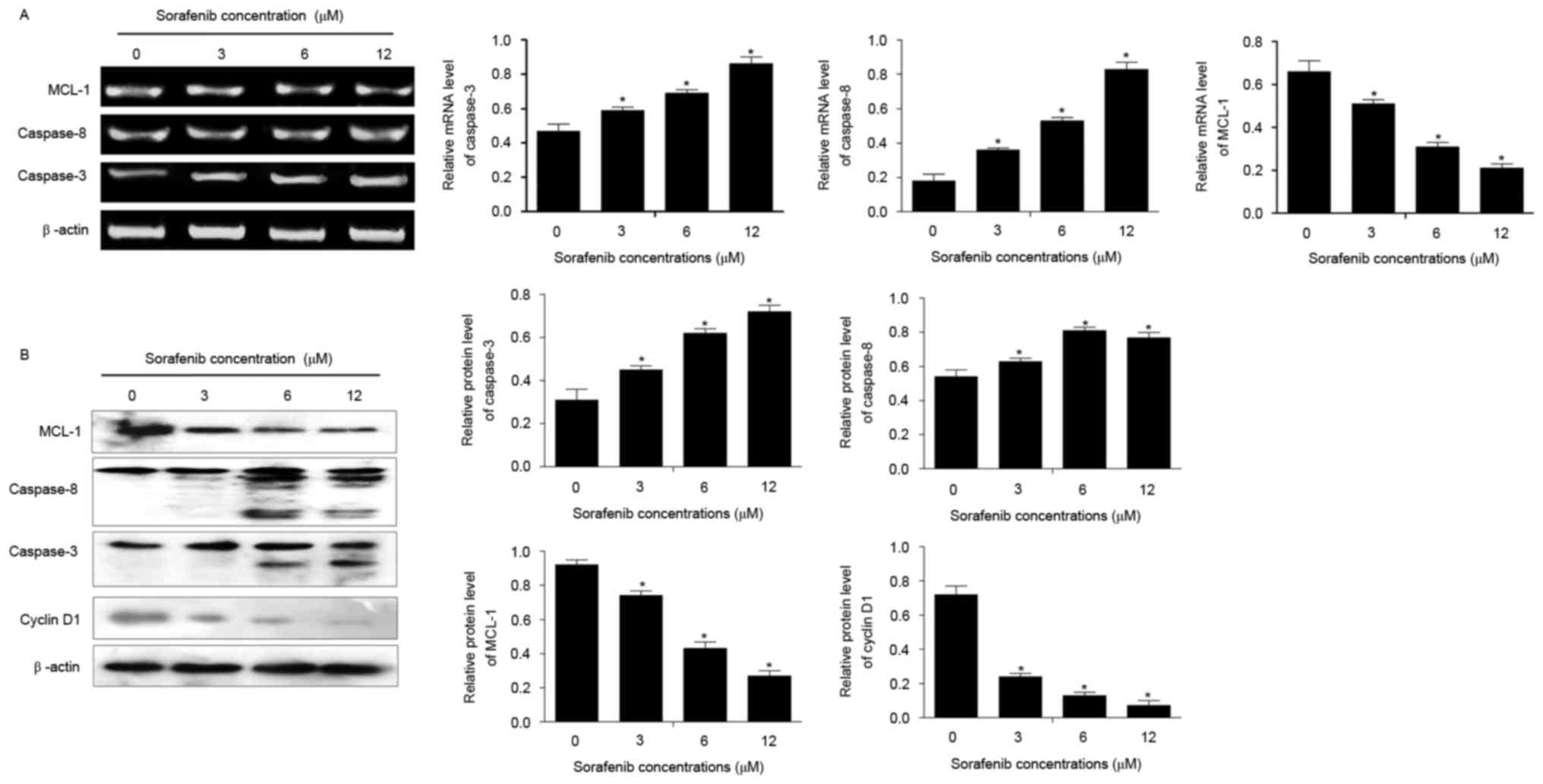
Compared with untreated cells, sorafenib
significantly increased the mRNA and protein expression of
caspase-3 and caspase-8, and inhibited the expression of MCL1,
following treatment for 48 h (P<0.05; Fig. 4). In addition, the expression of
cyclin D1 was decreased in NB4 cells treated with 1.5–12 µM of
sorafenib for 24 and 48 h compared with that in untreated cells
(P<0.05; Fig. 4B). Sorafenib
induced alterations to the expression of each of these genes
dose-dependently (P<0.05).
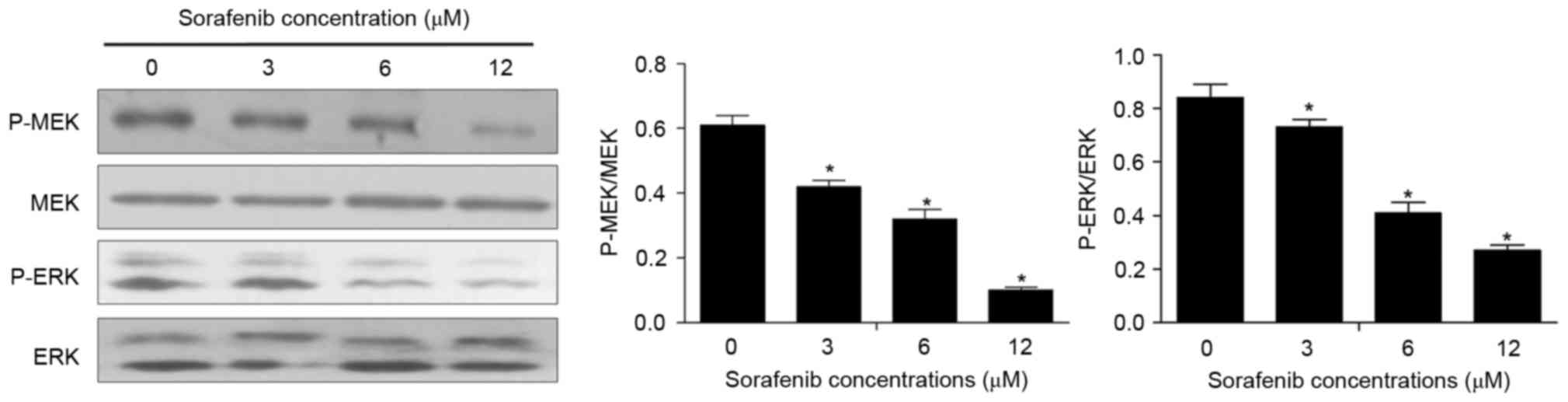
Effect of sorafenib on the MEK/ERK
signaling pathway in NB4 cells
To further assess the mechanisms underlying the
effects of sorafenib, the present study detected the expression of
MEK/ERK signaling pathway-associated proteins in sorafenib-treated
NB4 cells. The results of the present study demonstrated that,
compared with untreated cells, sorafenib significantly inhibited
the expression of P-MEK and P-ERK (P<0.05; Fig. 5), and that the expression of P-MEK and
P-ERK decreased with increasing sorafenib concentration
(P<0.05).
Discussion
Due to the toxicity, side effects, possibility of
APL drug resistance or relapse associated with APL chemotherapy
(15), developing APL chemotherapy
drugs that may overcome drug resistance is crucial. The present
study demonstrated that, in NB4 cells, sorafenib inhibited cell
growth, induced cell cycle arrest in the G0/G1 phase and apoptosis
dose-dependently. Such effects may be induced by the upregulation
of the proapoptotic proteins caspase-3 and caspase-8, and the
downregulation of the antiapoptotic protein MCL1 and the cell
cycle-associated protein cyclin D1. Furthermore, sorafenib
inhibited the phosphorylation of MEK and ERK.
Sorafenib is a novel multi-target anticancer drug
that may simultaneously inhibit multiple cell surface and
intracellular kinases that serve key functions in regulating tumor
growth (16). A previous study
demonstrated that sorafenib inhibited the RAF/MEK/ERK signaling
pathway to directly exert an antitumor effect, and suppressed the
expression of vascular endothelial growth factor receptor to
inhibit tumor angiogenesis and thereby indirectly inhibit tumor
cell growth in hepatocellular carcinoma (16). The present study demonstrated that
sorafenib inhibited the proliferation of NB4 cells by promoting
cell cycle arrest in the G0/G1 phase, a similar result to the
effect of sorafenib on human synovial sarcoma cells, as
demonstrated by Peng et al (17). Cyclin D1 is a key protein in cell
cycle progression (18). The present
study demonstrated that cyclin D1 was downregulated in
sorafenib-treated cells compared with untreated cells, indicating
that cyclin D1 may be associated with sorafenib-inhibited cell
growth. In addition, the present study demonstrated that
sorafenib-induced cell apoptosis may be associated with the
expression of the apoptosis-associated proteins caspase-3,
caspase-8 and MCL1. A previous study suggested a proapoptotic
effect of sorafenib on the human myeloma cell line RPMI8226
(19). Furthermore, Schult et
al (20) demonstrated that
sorafenib induced cell apoptosis by upregulating the expression of
caspase-3 and caspase-7 in acute lymphoblastic leukemia cells. In
addition, Meng et al (21)
suggested that MCL1 is an antiapoptotic B-cell lymphoma (Bcl)-2
homolog that aids the inhibitory apoptosis function of sorafenib in
acute myelogenous leukemia. Combined with the results of the
present study, these results suggest that sorafenib serves an
inhibitory function in the development of APL by modulating cell
proliferation and apoptosis.
To further assess the molecular mechanism underlying
the activity of sorafenib in APL cells, the present study detected
MEK/ERK signaling pathway-associated protein expression. Previous
studies have demonstrated that sorafenib inhibited tumor growth of
certain types of cancer through the regulation of the RAF/MEK/ERK
signaling pathway (22,23). The RAF/MEK/ERK signaling pathway is
associated with cell cycle progression and apoptosis in numerous
cells, and mutations in the pathway may be associated with cancer
(24). Multiple studies have
demonstrated that the mediation of G1 arrest by certain cell
cycle-associated genes, including p15, p16 and p21 depended on the
RAF/MEK/ERK signaling pathway (25–27). In
addition, Boucher et al (28).
suggest that the MEK/ERK signaling pathway may inhibit apoptosis by
regulating the expression of Bcl-2 and MCL1 in human pancreatic
cancer cells. The results of the present study suggest that the
inhibitory effects of sorafenib on APL cells were potentially
achieved by the downregulation of the activity of the MEK/ERK
signaling pathway. Therefore, the present study suggests that the
role of sorafenib in inhibiting cell growth in APL cells may be
regulated by the MEK/ERK signaling pathway.
To conclude, the present study demonstrated that
sorafenib inhibited proliferation and induced apoptosis in human
APL cells, the underlying mechanism of which may involve the
MEK/ERK signaling pathway. Further studies are required in order to
fully understand the mechanism underlying the effect of sorafenib
on APL cells.
Acknowledgements
The authors would like to thank Dr Kai Hu, Mr Fei
Gao, Mr Xiaobo Zhang and Ms Lu Wang (Institute of Hematology, Xi'an
Central Hospital, Xi'an, Shaanxi, China), and Dr Bingcheng Liu and
Ms Yuan Li (Leukemia Center, Institute of Hematology and Blood
Diseases Hospital, Chinese Academy of Medical Sciences, Tianjin,
China) for their assistance. The present study was supported by the
Science and Technology Research and Development Program of Shaanxi
Province (grant no. 2014K11-01-01-16).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tallman MS: Acute promyelocytic leukemia.
Best Pract Res Clin Haematol. 27:12014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lo-Coco F, Avvisati G, Vignetti M, Thiede
C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona
E, et al: Retinoic acid and arsenic trioxide for acute
promyelocytic leukemia. N Engl J Med. 369:111–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bally C, Fadlallah J, Leverger G, Bertrand
Y, Robert A, Baruchel A, Guerci A, Recher C, Raffoux E, Thomas X,
et al: Outcome of acute promyelocytic leukemia (APL) in children
and adolescents: An Analysis in two consecutive trials of the
European APL Group. J Clin Oncol. 30:1641–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takeshita A, Shigeno K, Shinjo K, Naito K,
Ohnishi K, Hayashi H, Tanimoto M and Ohno R: All-trans retinoic
acid (ATRA) differentiates acute promyelocytic leukemia cells
independently of P-glycoprotein (P-gp) related multidrug
resistance. Leuk Lymphoma. 42:739–746. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adnane L, Trail PA, Taylor I and Wilhelm
SM: Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that
targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases
VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 407:597–612.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Miyagi T, Hosui A, Tatsumi T, Ishida H, Noda T, Nagano H, et al:
The let-7 family of microRNAs inhibits Bcl-xL expression and
potentiates sorafenib-induced apoptosis in human hepatocellular
carcinoma. J Hepatol. 52:698–704. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Panka DJ, Wang W, Atkins MB and Mier JW:
The Raf inhibitor BAY 43-9006 (Sorafenib) induces
caspase-independent apoptosis in melanoma cells. Cancer Res.
66:1611–1619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang F, Brown C, Buettner R, Hedvat M,
Starr R, Scuto A, Schroeder A, Jensen M and Jove R: Sorafenib
induces growth arrest and apoptosis of human glioblastoma cells
through the dephosphorylation of signal transducers and activators
of transcription 3. Mol Cancer Ther. 9:953–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang W, Konopleva M, Ruvolo VR, McQueen
T, Evans RL, Bornmann WG, McCubrey J, Cortes J and Andreeff M:
Sorafenib induces apoptosis of AML cells via Bim-mediated
activation of the intrinsic apoptotic pathway. Leukemia.
22:808–818. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ravandi F, Cortes JE, Jones D, Faderl S,
Garcia-Manero G, Konopleva MY, O'Brien S, Estrov Z, Borthakur G,
Thomas D, et al: Phase I/II study of combination therapy with
sorafenib, idarubicin, and cytarabine in younger patients with
acute myeloid leukemia. J Clin Oncol. 28:1856–1862. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pratz KW, Cho E, Levis MJ, Karp JE, Gore
SD, McDevitt M, Stine A, Zhao M, Baker SD, Carducci MA, et al: A
pharmacodynamic study of sorafenib in patients with relapsed and
refractory acute leukemias. Leukemia. 24:1437–1444. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ravandi F, Alattar ML, Grunwald MR, Rudek
MA, Rajkhowa T, Richie MA, Pierce S, Daver N, Garcia-Manero G,
Faderl S, et al: Phase 2 study of azacytidine plus sorafenib in
patients with acute myeloid leukemia and FLT-3 internal tandem
duplication mutation. Blood. 121:4655–4662. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gallagher RE, Moser BK, Racevskis J, Poiré
X, Bloomfield CD, Carroll AJ, Ketterling RP, Roulston D,
Schachter-Tokarz E, Zhou DC, et al: Treatment-influenced
associations of PML-RARα mutations, FLT3 mutations, and additional
chromosome abnormalities in relapsed acute promyelocytic leukemia.
Blood. 120:2098–2108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng CL, Guo W, Ji T, Ren T, Yang Y, Li
DS, Qu HY, Li X, Tang S, Yan TQ and Tang XD: Sorafenib induces
growth inhibition and apoptosis in human synovial sarcoma cells via
inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol Ther.
8:1729–1736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnson DG and Walker CL: Cyclins and cell
cycle checkpoints. Annu Rev Pharmacol Toxicol. 39:295–312. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou NC, Liu BL, Qi MY, Xu B and Liu X:
Effects of sorafenib on proliferation and apoptosis of human
multiple myeloma cell RPMI 8226. Zhongguo Shi Yan Xue Ye Xue Za
Zhi. 22:1331–1335. 2014.(In Chinese). PubMed/NCBI
|
|
20
|
Schult C, Dahlhaus M, Ruck S, Sawitzky M,
Amoroso F, Lange S, Etro D, Glass A, Fuellen G, Boldt S, et al: The
multikinase inhibitor Sorafenib displays significant
antiproliferative effects and induces apoptosis via caspase 3, 7
and PARP in B- and T-lymphoblastic cells. BMC cancer. 10:5602010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meng XW, Lee SH, Dai H, Loegering D, Yu C,
Flatten K, Schneider P, Dai NT, Kumar SK, Smith BD, et al: Mcl-1 as
a buffer for proapoptotic Bcl-2 family members during TRAIL-induced
apoptosis: A mechanistic basis for sorafenib (bay 43-9006)-induced
TRAIL sensitization. J Biol Chem. 282:29831–29846. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang K, Luo C, Li Y, Lu C, Zhou W, Huang H
and Chen X: The study of a novel sorafenib derivative HLC-080 as an
antitumor agent. PLoS One. 9:e1018892014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Locatelli SL, Giacomini A, Guidetti A,
Cleris L, Mortarini R, Anichini A, Gianni AM and Carlo-Stella C:
Perifosine and sorafenib combination induces mitochondrial cell
death and antitumor effects in NOD/SCID mice with Hodgkin lymphoma
cell line xenografts. Leukemia. 27:1677–1687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chang F, Steelman LS, Shelton JG, Lee JT,
Navolanic PM, Blalock WL, Franklin R and McCubrey JA: Regulation of
cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway
(Review). Int J Oncol. 22:469–480. 2003.PubMed/NCBI
|
|
25
|
Blalock WL, Weinstein-Oppenheimer C, Chang
F, Hoyle PE, Wang XY, Algate PA, Franklin RA, Oberhaus SM, Steelman
LS and McCubrey JA: Signal transduction, cell cycle regulatory, and
anti-apoptotic pathways regulated by IL-3 in hematopoietic cells:
Possible sites for intervention with anti-neoplastic drugs.
Leukemia. 13:1109–1166. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang F and McCubrey JA: P21(Cip1) induced
by Raf is associated with increased Cdk4 activity in hematopoietic
cells. Oncogene. 20:4354–4364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Malumbres M, Pérez De Castro I, Hernández
MI, Jiménez M, Corral T and Pellicer A: Cellular response to
oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor
p15(INK4b). Mol Cell Biol. 20:2915–2925. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|