Introduction
Regulated cell death (RCD) is essential for the
development of living organisms and the maintenance of homeostasis.
As a novel form of RCD, ferroptosis has begun to attract increased
attention (1–4). Ferroptosis is an iron-dependent and
peroxidation-driven form of cell death that is distinct from
apoptosis, necrosis and other types of cell death in terms of
morphology, genetics, metabolism and molecular biology (2,5–10).
Apoptosis is characterized by morphological changes
that include chromosome shrinkage, chromatin condensation and
cytoplasmic fragment formation (2,11).
Positive regulators of apoptosis include pro-apoptotic B-cell
lymphoma-2 (Bcl-2) family proteins, for example BCL2 associated X,
apoptosis regulator and BCL2 antagonist/killer 1. Negative
regulators of apoptosis include Bcl-2 and other anti-apoptotic
Bcl-2 family proteins. Concerning the process of necrosis, loss of
plasma membrane integrity occurs instead of the mitochondrial
shrinkage and increased mitochondrial membrane density that occurs
during ferroptosis. Regulators involved in necrosis include
receptor interacting serine/threonine kinase 1 (RIPK1), receptor
interacting serine/threonine kinase 3 and mixed lineage kinase
domain like pseudokinase (12,13).
The regulators involved in apoptosis and necrosis
are not required for ferroptosis. Ferroptosis is regulated by
glutathione peroxidase 4 (GPX4), a lipid repair enzyme (14,15), and
is associated with the following group of genes: Iron responsive
element binding protein 2, citrate synthase, ribosomal protein L8
and ATP synthase, H+ transporting, mitochondrial Fo complex subunit
C3 (subunit 9) (2). Ferroptosis is a
type of cellular sabotage that results in cell death, whereas
apoptosis, pyroptosis and necroptosis are considered types of
programmed cell death (16).
Ferroptosis is not blocked by
carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)-fluoromethylketone,
an inhibitor of caspase-dependent apoptosis and pyroptosis, or by
necrostatin-1, an inhibitor of RIPK1-dependent necroptosis
(17,18).
Ferroptosis is driven by inhibition of extracellular
cysteine uptake and inactivation of the function of GPX4 (5,15). These
processes lead to the depletion of polyunsaturated fatty acids
(PUFAs) in lipid bilayers and the accumulation of lipid-based
reactive oxygen species (L-ROS) (2,3,7,9,15). Furthermore, iron metabolism and
cellular iron abundance simultaneously impact the onset of
ferroptosis. The inducers and inhibitors of ferroptosis are
categorized and summarized in Table I
(2,5,7,10,15,18–24).
 | Table I.Inducers and inhibitors of
ferroptosis. |
Table I.
Inducers and inhibitors of
ferroptosis.
| A, Inducers |
|---|
|
|---|
| Author, year | Type | Molecules and
drugs | Mechanism | (Refs.) |
|---|
| Hayano et
al, 2016; Yang and Stockwell, 2008 | I | Erastin Glutamate
Buthioninesulfoximine Acetaminophen Sorafenib Sulfasalazine | Inhibit the
glutamate/cysteine antiporter system and block glutathione
synthesis, indirectly inhibiting GPX4 | (10) (18) |
| Hayano et
al, 2016; Yang et al, 2014 | II | (1S,3R)-RSL3,
DPI19, DPI18, DPI17, DPI13, DPI12, DPI10, DPI7 Altretamine | Directly inactivate
GPX4 without glutathione decrease | (10) (15) |
|
| B,
Inhibitors |
|
| Author,
year | Type | Molecules and
drugs |
Mechanism | (Refs.) |
|
| Dixon et al,
2012 | I | Cycloheximide | Suppress protein
synthesis and lipid | (2) |
| Magtanong et
al, 2016 |
|
β-mercaptoethanol | peroxidation | (5) |
| Skouta et
al, 2014 |
| Trolox |
| (7) |
| Hayano et
al, 2016 |
| Baicalein |
| (10) |
| Yang et al,
2014 |
| Zileuton |
| (15) |
|
|
| Liproxstatin-1 |
|
|
|
|
| Ferrostatin-1 |
|
|
| Dixon et al,
2012 | II | Deferoxamine | Chelate lysosomal
iron or the labile iron | (2) |
| Hayano et
al, 2016 |
| Ciclopirox
olamine | pool in the
cytoplasm to protect against | (10) |
| Yang et al,
2008 |
| 2,2-BP | lipid
peroxidation | (18) |
| Louandre et
al, 2013 |
|
|
| (19) |
| Dixon et al,
2014 |
|
|
| (20) |
| Xie et al,
2016 |
|
|
| (21) |
| Kurz et al,
2006 |
|
|
| (22) |
| Barradas et
al, 1989 |
|
|
| (23) |
| Soupene and
Kuypers, 2008 |
|
|
| (24) |
Ferroptosis is associated with multiple
physiopathological processes and human diseases, particularly the
occurrence and progression of multiple types of cancer. Previous
studies have revealed that hepatocellular carcinoma (HCC), renal
cell carcinoma, diffuse large B-cell lymphoma, pancreatic carcinoma
and ovarian cancer cells are susceptible to ferroptosis (15,25,26). The
metabolic peculiarities of ferroptosis vary among different types
of cancer cell.
The present review provides a comprehensive overview
of studies concerning the metabolic networks involved in
ferroptosis in cancer cells.
Metabolism and ferroptosis
Cysteine is critical for
ferroptosis
Cellular cysteine is primarily obtained by
extracellular cysteine uptake through the glutamate/cysteine
antiporter (Xc−) (6,27,28). In addition to extracellular uptake,
certain mammalian cells are able to use methionine as a sulfur
donor to synthesize de novo cysteine through the
trans-sulfuration pathway (6,28–30).
However, mammalian cells normally depend on only one of these
patterns as the major source of cysteine. The trans-sulfuration
pathway provides a compensatory source of cysteine when the uptake
pattern is inhibited.
The Xc− system consists of a 12-pass
transmembrane protein transporter solute carrier family 7 member 11
(SLC7A11) and a single-pass transmembrane regulatory protein solute
carrier family 3 member 2. Acting as a glutamate-cystine
antiporter, inhibition of the Xc− system may lead to
depletion of the intracellular cysteine pool, one of the molecular
events that induces ferroptosis (2,7,21,28). As a
classic inducer of ferroptosis, erastin suppresses the
glutamate-cystine antiporter (21).
Upregulation of SLC7A11 prevents cells from erastin-induced
ferroptosis, while downregulation of SLC7A11 inhibits the growth of
cancer cells during erastin treatment (2,21).
As another source of cysteine, the trans-sulfuration
pathway is catalyzed and regulated by cystathionine-β-synthase
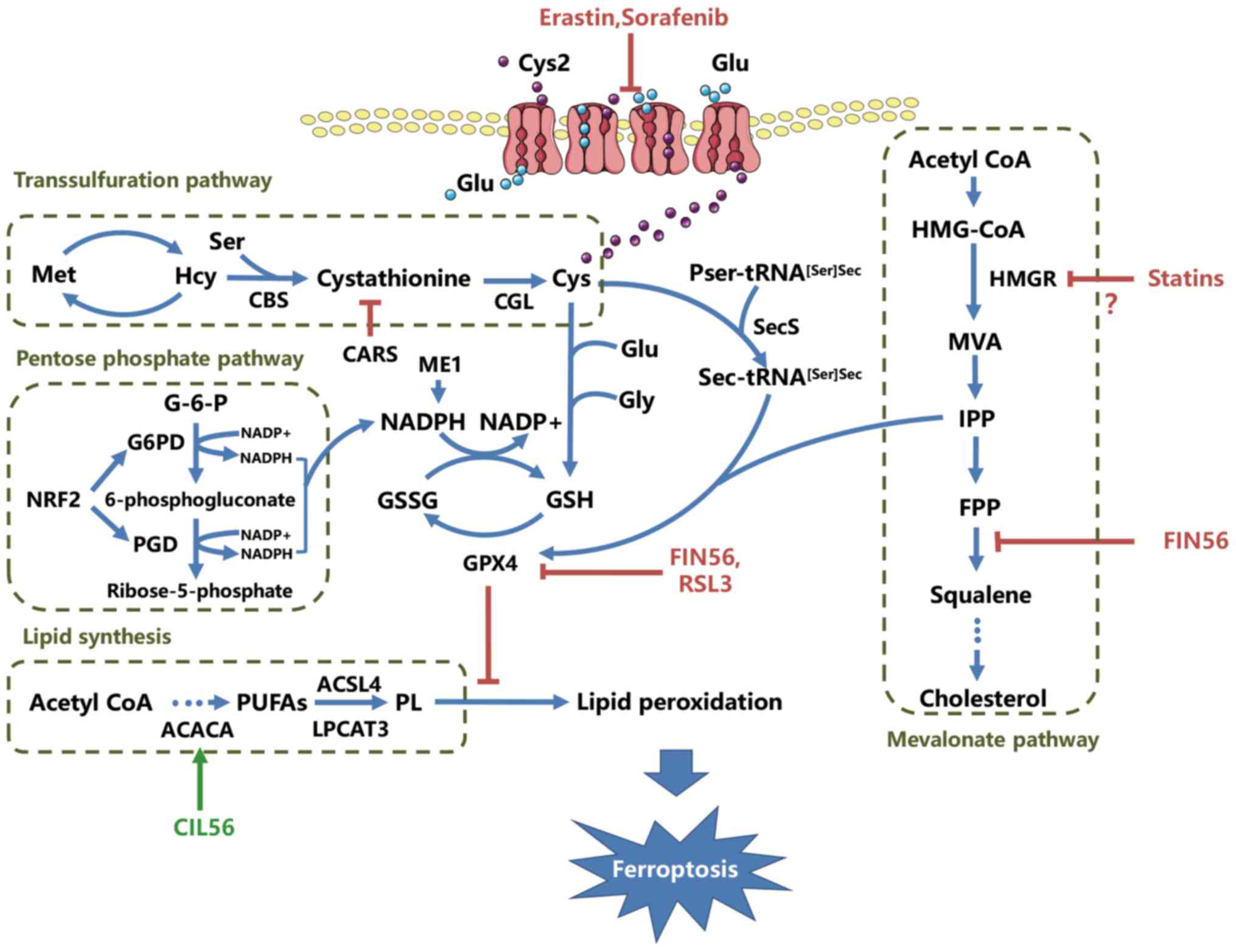
(CBS) and cystathionine-γ-lyase (CGL) (31) (Fig. 1).
Genome-wide siRNA screening has revealed that silencing of
cysteinyl tRNA synthetase (CARS) suppresses erastin-induced
ferroptosis (10,28). CBS and CGL are upregulated in
CARS-deprieved cells, and metabolites accumulate in the
trans-sulfuration pathway following erastin treatment (6,10,32). These results support the hypothesis
that the transsulfuration pathway is a regulator of ferroptosis
resistance, compensating for cysteine depletion caused by the
inhibition of cysteine uptake.
GSH biosynthesis is connected to
cysteine and GPX4
In the 1970s, deprivation of Cys2 was revealed to
lead to marked depletion of GSH and the promotion of cell death
(3,33), suggesting that cysteine uptake may be
the limiting factor for GSH biosynthesis. Several subsequent
pharmacological studies of glutamate- or erastin-induced
ferroptosis further demonstrated that decreased GSH levels
triggered by cysteine deprivation may induce the initiation of
oxidative stress and ferroptotic cell death (2,3,7,15,20,28). GSH
biosynthesis is critical for protecting cells from oxidative
damage, and the cysteine-GSH pathway is one of the most pivotal
upstream mechanisms for the execution of ferroptosis.
GSH biosynthesis is critical for the functional
activity of GSH-dependent enzymes, including selenium glutathione
peroxidase (GPX). GPX uses the thiol group in GSH as an electron
donor and affects the cellular antioxidant reaction (15,28).
Inactivation of GPX4 induced by GSH depletion increases
intracellular lipid peroxides, resulting in ferroptosis (10,15).
The mevalonate pathway is crucial for
GPX4 maturation in ferroptosis
In addition to its dependence on GSH, GPX also
relies on cysteine metabolism for maturation. GPX, a typical
selenoprotein, uses its catalytic center selenocysteine (Sec)
during defense against antioxidants. During the process of GPX
maturation, Sec transfer RNA (Sec-tRNA[Ser]Sec) is one
of the key regulatory elements modulated by isopentenyl
pyrophosphate (IPP), a product of the mevalonate pathway (Fig. 1) (34–36).
Serving as a primary source of IPP, the mevalonate pathway is a
crucial signaling network for GPX4 maturation and ferroptosis
induction. FIN56 is a novel inducer of ferroptosis discovered
during the study of nonapoptotic cell death (37). Unlike erastin, FIN56 treatment does
not result in GSH depletion, but causes GPX4 loss at the
post-translational level and the decrease of mevalonate-derived
lipophilic antioxidants, indicating that FIN56-induced ferroptosis
is modulated through the mevalonate pathway (37). Thus, GPX4 maturation may link the
mevalonate pathway and ferroptosis.
Previous studies investigating the functions of
statins in the prevention of obesity-associated cardiovascular
diseases have demonstrated that 3-hydroxy-3-methylglutaryl-coenzyme
A reductase serves as a target of statins in the mevalonate pathway
(38). Preclinical studies have
demonstrated the pro-apoptotic effects of statins (39,40).
Furthermore, human prostate cancer PC3 cells treated with
atorvastatin undergo autophagy, whereas simvastatin leads to the
induction of apoptosis in HCT116 colorectal cancer cells and renal
cell carcinoma cells (41). Statins
also downregulate the mevalonate pathway and block the biosynthesis
of cellular isoprenoids, including IPP, which are responsible for
the post-translational modification of Sec-tRNA[Ser]Sec
and the synthesis of GPX4 (6,35,42).
Although there is no experimental evidence demonstrating the link
between statins and ferroptosis, statins downregulate the
mevalonate pathway, which is a crucial signaling event for GPX4
maturation. Thus, ferroptosis may be a form of statin-induced cell
death. Further research is required to investigate the association
between statins and ferroptosis.
Dual effects of nicotinamide adenine
dinucleotide phosphate (NADPH) on sensitivity to ferroptosis
NADPH, the predominant reducing agent in organisms,
participates in a number of metabolic reactions. GSH is
dehydrogenated to form glutathione disulfide, which is in turn
reduced to GSH by glutathione reductase in the presence of NADPH
(43). Given the functions of GSH,
the synthesis of NADPH is important in resistance to
peroxidation-induced damage during ferroptosis.
NADPH is produced by the pentose phosphate pathway,
which is catalyzed by glucose-6-phosphate dehydrogenase (G6PD) and
6-phosphogluconate dehydrogenase (PGD; Fig. 1). Several studies have demonstrated
that nuclear factor E2-related factor 2 (NRF2) targets the genes
encoding G6PD and PGD (44–46). Silencing of NRF2 and these
enzyme-associated genes causes HCC cells to be sensitized to the
ferroptosis inducers erastin and sorafenib (47). Consequently, NRF2 functions as a
negative regulator of ferroptosis in liver cancer cells,
participates in NADPH production and subsequently affects GSH
function, which is essential for the initiation of ferroptosis.
Notably, NADPH depletion sensitizes fibrosarcoma
HT-1080 cells to ferroptosis inducers, indicating that NADPH is
negatively associated with ferroptosis sensitivity (48). However, well-established studies
concerning the NADPH oxidase (NOX) protein family have demonstrated
that NADPH provides electrons for NOX to generate superoxide from
oxygen (49), which may promote the
ferroptosis process. Furthermore, inhibition of the pentose
phosphate pathway partially rescues Calu-1 cells from ferroptosis
(2). The results of these studies
support the contradictory function of NADPH in ferroptosis. Further
investigations are required to determine the function of NADPH, as
an inducer or an inhibitor of ferroptosis.
Ferroptosis is induced by lipid
peroxidation
Cell lines selected for Xc− system
inhibition resistance have been demonstrated to overexpress
aldo-keto reductase family members, which detoxify oxidative lipid
fragments (6,20). Thus, the lipid fragments may be
downstream products generated from cysteine depletion. This
discovery provides insights into the potential associations between
cysteine and lipid metabolism and the mechanisms of lipid
peroxidation in ferroptosis (Fig.
1).
Two lipid metabolism-associated genes, acyl-CoA
synthetase long-chain family member 4 (ACSL4) and
lysophosphatidylcholine acyltransferase 3 (LPCAT3), encode
enzymes required for the acylation and insertion of PUFAs into
membrane phospholipids, respectively (24,50). A
previous study demonstrated that deletion of ACSL4 and
LPCAT3 prevents RSL3 and ML162-induced ferroptosis in KBM7
cells (51). Thus, inhibition of
phospholipid synthesis may suppress ferroptotic cell death.
Furthermore, initiation of ferroptosis results in the depletion of
PUFAs in lipid bilayers and the accumulation of L-ROS and
lysophospholipids (7,9,15).
Lysophospholipids and oxidized PUFAs are products of
glycerophospholipids, and are formed via a degradation reaction
catalyzed by phospholipase A2 (9).
These results suggest that the PUFAs provided by
glycerophospholipids are required as substrates for lipid
peroxidation during ferroptosis.
PUFAs oxidized and cleaved from glycerophospholipid
backbones are subsequently degraded, producing a series of toxic
metabolites in ferroptosis. The peroxidation of PUFAs in membranes
generates toxic lipid hydroperoxides, resulting in the formation of
lethal lipid radicals in the presence of ferrous iron, while
inhibiting GPX4 (7,15,52). Lipid
radicals react with adjacent PUFAs in lipid membranes and induce
lipid peroxidation in ferroptosis. However, the precise pathways
through which lipid peroxidation directly or indirectly leads to
ferroptosis remain unclear.
Ferroptosis is associated with
‘ferritinophagy’
Iron serves a pivotal function in various
fundamental metabolic processes due to its role as an auxiliary
factor of proteins (53). As
ferroptosis is inhibited by an iron chelator, desferrioxamine
B-methane sulfonate (DFO), the association between intracellular
iron and ferroptosis has become a topic of interest.
Although the mechanisms through which cellular iron
facilitates ferroptosis remain unclear, cellular iron homeostasis
is recognized as a key factor in ferroptotic cell death (Fig. 2). An excess of iron is stored in
ferritin heavy chain 1 (FTH1) and ferritin light chain, and genetic
inhibition of FTH1 promotes erastin and sorafenib-induced
ferroptosis in HCC cells (47).
Furthermore, increased transferrin receptor 1 and decreased
ferritin expression occur in ferroptosis-sensitive cells with Ras
mutations (18). These results
suggest that the abundance of free iron contributes to the
induction of ferroptosis via increased iron intake and decreased
iron storage.
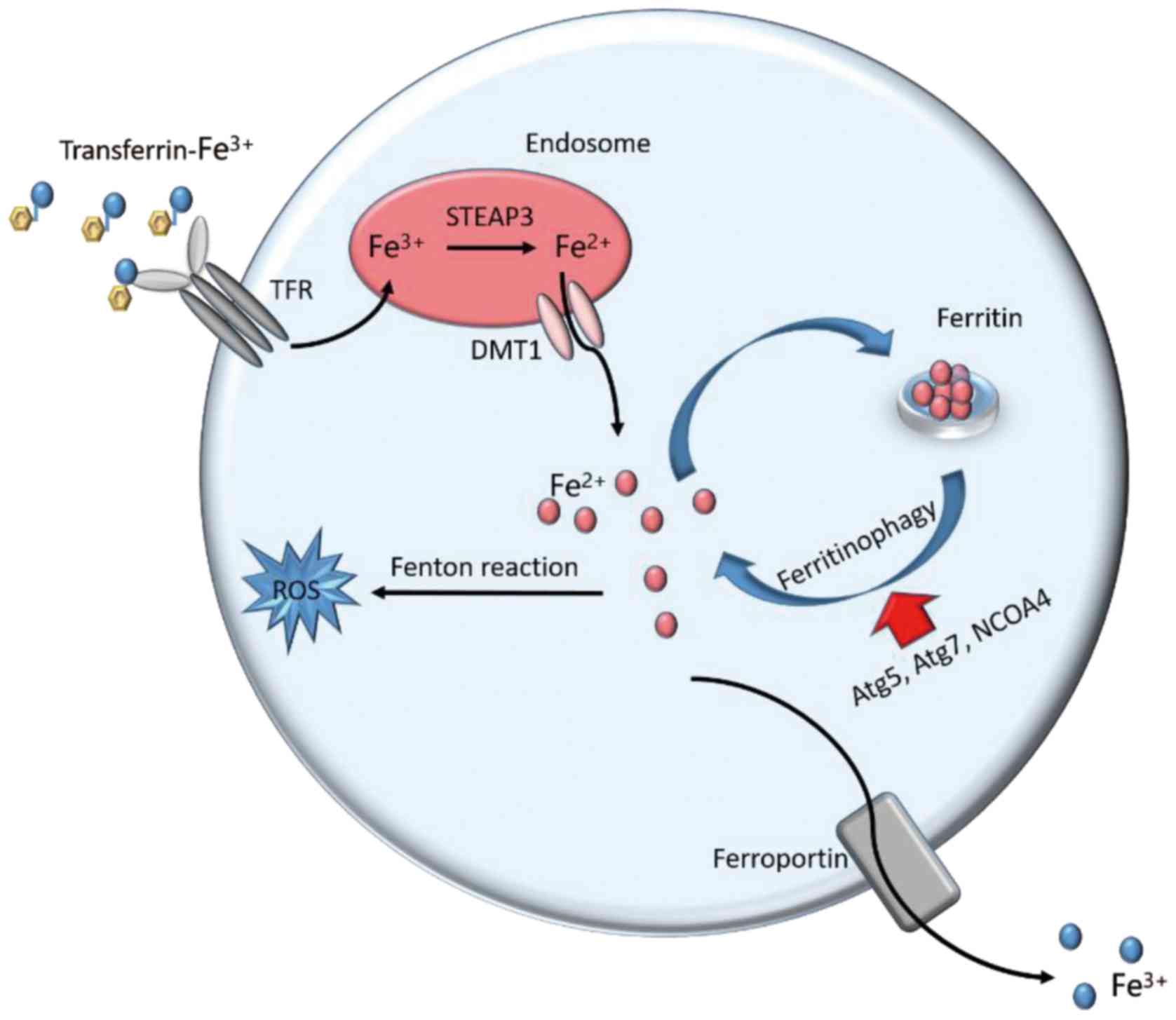 | Figure 2.Iron metabolism in ferroptosis.
Cellular iron homeostasis is dependent on the coordination of iron
uptake, export, utilization, and storage. Extracellular
Fe3+ binds to transferrin and is taken up into cells
through TFR1. The freed Fe3+ is reduced to
Fe2+ by STEAP3 metalloreductases in the endosome.
Divalent metal transporter 1 mediates the transport of
Fe2+ from the endosome into a labile iron pool in the
cytoplasm. The labile iron is exported via the membrane protein
ferroportin to maintain plasma iron levels. Alternatively, or in
parallel, excess iron from the labile iron pool is stored in
ferritin heteropolymers (ferritin heavy chain 1 or ferritin light
chain), a redox-inactive form of iron, to protect cells and tissues
from iron-mediated damage. Notably, the autophagic degradation of
ferritin, a process known as ferritinophagy, increases labile iron
levels and contributes to ferroptosis. Fe3+, ferric
iron; Fe2+, ferrous iron; TFR1, transferrin receptor 1;
STEAP3, six-transmembrane epithelial antigen of prostate 3; DMT1,
doublesex and mab-3 related transcription factor 1; ROS, reactive
oxygen species; Atg5, autophagy related 5; Atg7, autophagy related
7; NCOA4, nuclear receptor coactivator 4. |
The introduction of iron chelators further supports
the involvement of iron in the process of ferroptosis. Iron
chelators are divided into lipophilic iron chelators (including
311, ciclopirox olamine, and 2,2-BP) and membrane impermeable iron
chelators (including DFO), which inhibit ferroptosis via diverse
mechanisms (2,15,18). DFO
chelates lysosomal iron, which should be present at a different
location in the cell, promoting L-ROS production. In addition,
lipophilic iron chelators are able to cross membranes and chelate
the labile iron pool, which is critical for the fragmentation and
peroxidation of PUFAs (22,23,52).
Excess active iron donates electrons to generate ROS based on the
Fenton reaction, promoting lipid peroxidation and the initiation of
ferroptosis (54).
Ferritin, a form of stored labile iron, is an
antioxidant that inhibits iron-mediated lipid peroxidation
(55). Autophagic degradation of
ferritin (a process known as ferritinophagy) contributes to
ferroptosis via increased labile iron levels in fibroblasts and
cancer cells, supporting the association between autophagy and
ferroptosis (56). At the genetic
level, multiple autophagy-related genes have been identified as
positive regulators of ferroptosis. Genetic inhibition of
autophagy-related 5 and autophagy-related 7 abrogates the
accumulation of labile iron and prevents erastin-induced
ferroptosis (56,57). In addition, knockdown of nuclear
receptor coactivator 4 (NCOA4), a ferritinophagy cargo receptor,
also inhibits ferritinophagy and ferroptotic cell death. In
contrast, overexpression of NCOA4 increases ferritinophagy and
promotes ferroptosis (56,57). These results suggest that autophagy
activation leads to ferritinophagy and promotes ferroptosis by
regulation of intracellular iron homeostasis.
Conclusion
Ferroptosis is an aberrant metabolic process
involving amino acids, lipids, NADPH and microelements. Metabolism
of these substances serves a crucial function in cell proliferation
and differentiation. However, cysteine depletion, GPX4
inactivation, and iron overload cause cells to experience metabolic
stress or ferroptotic cell death. Ferroptosis is characterized by a
metabolic imbalance and the perturbation of redox homeostasis. The
abundance of the amino acid cysteine and the existence of NADPH,
which is primarily generated by the pentose phosphate pathway, are
essential for the antioxidant function of GPX4. Furthermore, the
inactivation of GPX4 contributes to lipid peroxidation and results
in the induction of ferroptosis. The metabolic processes in
ferroptosis are not independent, but are instead a part of an
intricate metabolic network.
Multiple physiopathological processes and human
diseases are involved in ferroptosis. Several types of cancer cell
are susceptible to ferroptosis; thus, ferroptosis may represent a
novel anticancer therapy. In acute kidney failure, hemorrhagic
stroke and nephrotoxic folic acid induce acute kidney injury, and
inhibitors of ferroptosis (for example, ferrostatin-1) reduce the
damage caused by cell sabotage. Ferrostatin-1 preserves renal
function and decreases injury, oxidative stress and tubular cell
death in mice with nephrotoxic folic acid-induced acute kidney
injuries (17,58,59).
Therefore, ferrostatin-1 may have a prophylactic effect in these
non-neoplastic diseases.
Ferritinophagy has also been demonstrated to serve
as a bridge between ferroptosis and autophagy. Autophagy exerts
positive effects in the regulation of ferroptosis. The mechanisms
through which autophagy is connected to ferroptosis and through
which this relationship is regulated are important. Thus, further
studies are needed to determine whether there are any other
metabolic processes involved in the association between ferroptosis
and autophagy, and whether there is a link between other forms of
RCD and ferroptosis.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81472317)
and the Science and Technology Planning Project of Guangdong
Province, China (grant no. 2016A020215232).
Glossary
Abbreviations
Abbreviations:
|
ACSL4
|
acyl-CoA synthetase long-chain family
member 4
|
|
CARS
|
cysteinyl tRNA synthetase
|
|
CBS
|
cystathionine-β-synthase
|
|
CGL
|
cystathionine-γ-lyase
|
|
FTH1
|
ferritin heavy chain 1
|
|
GPX
|
glutathione peroxidase
|
|
GPX4
|
glutathione peroxidase 4
|
|
GSH
|
glutathione
|
|
G6PD
|
glucose-6-phosphate dehydrogenase
|
|
IPP
|
isopentenyl pyrophosphate
|
|
L-ROS
|
lipid-based reactive oxygen
species
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
NCOA4
|
nuclear receptor coactivator 4
|
|
NOX
|
NADPH oxidase
|
|
NRF2
|
nuclear factor E2-related factor 2
|
|
PGD
|
6-phosphogluconate dehydrogenase
|
|
PUFAs
|
polyunsaturated fatty acids
|
|
RCD
|
regulated cell death
|
|
Sec
|
selenocysteine
|
|
Sec-tRNA[Ser]Sec
|
selenocysteine transfer RNA
|
|
Xc-
|
glutamate/cysteine antiporter
|
References
|
1
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Magtanong L, Ko PJ and Dixon SJ: Emerging
roles for lipids in non-apoptotic cell death. Cell Death Differ.
23:1099–1109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayano M, Yang WS, Corn CK, Pagano NC and
Stockwell BR: Loss of cysteinyl-tRNA synthetase (CARS) induces the
transsulfuration pathway and inhibits ferroptosis induced by
cystine deprivation. Cell Death Differ. 23:270–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al Nomenclature Committee on Cell Death
2009, : Classification of cell death: Recommendations of the
nomenclature committee on cell death 2009. Cell Death Differ.
16:3–31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the nomenclature committee on cell
death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Conrad M and Friedmann Angeli JP:
Glutathione peroxidase 4 (Gpx4) and ferroptosis: What's so special
about it? Mol Cell Oncol. 30:e9950472015. View Article : Google Scholar
|
|
15
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dixon SJ: Ferroptosis: Bug or feature?
Immunol Rev. 277:150–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Muller T, Dewitz C, Schmitz J, Schröder
AS, Bräsen JH, Stockwell BR, Murphy JM, Kunzendorf U and Krautwald
S: Necroptosis and ferroptosis are alternative cell death pathways
that operate in acute kidney failure. Cell Mol Life Sci.
27:2017.(Epub ahead of print).
|
|
18
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem. Biol. 15:234–245. 2008.
|
|
19
|
Louandre C, Ezzoukhry Z, Godin C, Barbare
JC, Mazière JC, Chauffert B and Galmiche A: Iron-dependent cell
death of hepatocellular carcinoma cells exposed to sorafenib. Int J
Cancer. 133:1732–1742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kurz T, Gustafsson B and Brunk UT:
Intralysosomal iron chelation protects against oxidative
stress-induced cellular damage. FEBS J. 273:3106–3117. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barradas MA, Jeremy JY, Kontoghiorghes GJ,
Mikhailidis DP, Hoffbrand AV and Dandona P: Iron chelators inhibit
human platelet aggregation, thromboxane A2 synthesis and
lipoxygenase activity. FEBS Lett. 245:105–109. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soupene E and Kuypers FA: Mammalian
long-chain acyl-CoA synthetases. Exp Biol Med (Maywood).
233:507–521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eling N, Reuter L, Hazin J, Hamacher-Brady
A and Brady NR: Identification of artesunate as a specific
activator of ferroptosis in pancreatic cancer cells. Oncoscience.
2:517–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Louandre C, Marcq I, Bouhlal H, Lachaier
E, Godin C, Saidak Z, François C, Chatelain D, Debuysscher V,
Barbare JC, et al: The retinoblastoma (Rb) protein regulates
ferroptosis induced by sorafenib in human hepatocellular carcinoma
cells. Cancer Lett. 356:971–977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McBean GJ: Cerebral cystine uptake: A tale
of two transporters. Trends Pharmacol Sci. 23:299–302. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shimada K and Stockwell BR: tRNA synthase
suppression activates de novo cysteine synthesis to compensate for
cystine and glutathione deprivation during ferroptosis. Mol Cell
Oncol. 3:e10910592015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stipanuk MH, Dominy JE Jr, Lee JI and
Coloso RM: Mammalian cysteine metabolism: New insights into
regulation of cysteine metabolism. J Nutr. 136:1652S–1659S. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McBean GJ: The transsulfuration pathway: A
source of cysteine for glutathione in astrocytes. Amino Acids.
42:199–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kabil O, Vitvitsky V, Xie P and Banerjee
R: The quantitative significance of the transsulfuration enzymes
for H2S production in murine tissues. Antioxid Redox Signal.
15:363–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stipanuk MH and Ueki I: Dealing with
methionine/homocysteine sulfur: cysteine metabolism to taurine and
inorganic sulfur. J Inherit Metab Dis. 34:17–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bannai S, Tsukeda H and Okumura H: Effect
of antioxidants on cultured human diploid fibroblasts exposed to
cystine-free medium. Biochem Biophys Res Commun. 74:1582–1588.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kryukov GV, Castellano S, Novoselov SV,
Lobanov AV, Zehtab O, Guigó R and Gladyshev VN: Characterization of
mammalian selenoproteomes. Science. 300:1439–1443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Warner GJ, Berry MJ, Moustafa ME, Carlson
BA, Hatfield DL and Faust JR: Inhibition of selenoprotein synthesis
by selenocysteine tRNA [Ser]Sec lacking isopentenyladenosine. J
Biol Chem. 275:28110–28119. 2000.PubMed/NCBI
|
|
36
|
do Nascimento NC, Menguer PK, Henriques AT
and Fett-Neto AG: Accumulation of brachycerine, an antioxidant
glucosidic indole alkaloid, is induced by abscisic acid, heavy
metal and osmotic stress in leaves of Psychotria brachyceras. Plant
Physiol Biochem. 73:33–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. 12:1–503. 2016.
|
|
38
|
Taylor J: Joint societies CVD prevention
guidelines launched in, May 2012. Eur Heart J.
33:15392012.PubMed/NCBI
|
|
39
|
Gazzerro P, Proto MC, Gangemi G, Malfitano
AM, Ciaglia E, Pisanti S, Santoro A, Laezza C and Bifulco M:
Pharmacological actions of statins: A critical appraisal in the
management of cancer. Pharmacol Rev. 64:102–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ciofu C: The statins as anticancer agents.
Maedica (Buchar). 7:3772012.PubMed/NCBI
|
|
41
|
Altwairgi AK: Statins are potential
anticancerous agents (review). Oncol Rep. 33:1019–1039. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kromer A and Moosmann B: Statin-induced
liver injury involves cross-talk between cholesterol and
selenoprotein biosynthetic pathways. Mol Pharmacol. 75:1421–1429.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W,
Zhang S, Huang Q and Shi M: ROS signaling under metabolic stress:
Cross-talk between AMPK and AKT pathway. Mol Cancer. 16:792017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reisman SA, Yeager RL, Yamamoto M and
Klaassen CD: Increased Nrf2 activation in livers from
keap1-knockdown mice increases expression of cytoprotective genes
that detoxify electrophiles more than those that detoxify reactive
oxygen species. Toxicol Sci. 108:35–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Goven D, Boutten A, Lecon-Malas V,
Marchal-Sommé J, Soler P, Boczkowski J and Bonay M: Induction of
heme oxygenase-1, biliverdin reductase and H-ferritin in lung
macrophage in smokers with primary spontaneous pneumothorax: Role
of HIF-1alpha. PLoS One. 5:e108862010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kirby J, Halligan E, Baptista MJ, Allen S,
Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J
and Shaw PJ: Mutant SOD1 alters the motor neuronal transcriptome:
Implications for familial ALS. Brain. 128:1686–1706. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shimada K, Hayano M, Pagano NC and
Stockwell BR: Cell-Line selectivity improves the predictive power
of pharmacogenomic analyses and helps identify NADPH as biomarker
for ferroptosis sensitivity. Cell Chem Biol. 23:225–235. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: Physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shindou H and Shimizu T: Acyl-CoA:
Lysophospholipid acyltransferases. J Biol Chem. 284:1–5. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dixon SJ, Winter GE, Musavi LS, Lee ED,
Snijder B, Rebsamen M, Superti-Furga G and Stockwell BR: Human
haploid cell genetics reveals roles for lipid metabolism genes in
nonapoptotic cell death. ACS Chem Biol. 10:1604–1609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cheng Z and Li Y: What is responsible for
the initiating chemistry of iron-mediated lipid peroxidation: An
update. Chem Rev. 107:748–766. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bogdan AR, Miyazawa M, Hashimoto K and
Tsuji Y: Regulators of iron homeostasis: New players in metabolism,
cell death and disease. Trends Biochem Sci. 41:274–286. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dixon SJ and Stockwell BR: The role of
iron and reactive oxygen species in cell death. Nat Chem Biol.
10:9–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao G, Arosio P and Chasteen ND: Iron
(II) and hydrogen peroxide detoxification by human H-chain
ferritin. An EPR spin-trapping study. Biochemistry. 45:3429–3436.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Martin-Sanchez D, Ruiz-Andres O, Poveda J,
Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, Ruiz Ortega M, Egido
J, Linkermann A, Ortiz A and Sanz AB: Ferroptosis, but not
necroptosis, is important in nephrotoxic folic acid-induced AKI. J
Am Soc Nephrol. 28:218–229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zille M, Karuppagounder SS, Chen Y, Gough
PJ, Bertin J, Finger J, Milner TA, Jonas EA and Ratan RR: Neuronal
death after hemorrhagic stroke in vitro and in vivo shares features
of ferroptosis and necroptosis. Stroke. 48:1033–1043. 2017.
View Article : Google Scholar : PubMed/NCBI
|