Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies globally and the second-leading cause of
mortality in China (1). There are ~62
million newly diagnosed cases of HCC annually worldwide (2). Numerous factors, including hepatitis B
virus infection and alfatoxin induction, may contribute to the
development of HCC. These factors may lead to abnormal gene
expression, resulting in increased cancer cell proliferation and
escape from immune surveillance (3–5). The
majority of patients with HCC are diagnosed at advanced stages of
the disease. Currently, there is no effective surgical or
pharmaceutical treatment for HCC. Therefore, investigation of the
molecular mechanisms underlying HCC development is necessary to
develop therapeutic strategies for HCC.
With the development of sequencing technology and
the in-depth studies of oncological diseases in previous years,
long non-coding RNAs (lncRNAs), once considered to be the ‘noise’
in genomic transcription, have come to the attention of scholars
(6–8).
lncRNAs are non-protein-coding transcripts that are >200
nucleotides, which may interact with various biomolecules,
including DNA, RNA and proteins, to regulate gene expression at
transcriptional, post-transcriptional and epigenetic levels
(9), serving important functions in a
wide range of biological processes, including cell proliferation,
survival, differentiation and chromatin remodeling (10,11). To
date, the encyclopedia of DNA elements project (GENCODE v26) has
conservatively annotated in humans close to 16,000 lncRNA genes
that give rise to >28,000 distinct transcripts (12). In previous years, researchers have
made notable advances in the study of lncRNA and diseases,
particularly in the area of tumors (13–15). It
has been suggested that the abnormal expression of lncRNA may be
the major inducer and accelerant of tumorigenesis (16). However, the functions of lncRNA and
their underlying molecular mechanisms are not well understood,
meaning that further studies are required.
Increasingly, evidence demonstrates that a number of
abnormally expressed lncRNAs are closely associated with the
occurrence, invasion, metastasis and recurrence of HCC, and have
great potential in its prediction, diagnosis and treatment
(17). The regulatory mechanism of
lncRNA is complex, and its role in HCC is diverse (18,19). The
present study utilized bioinformatic analysis to investigate the
regulatory network of HCC-associated lncRNAs and their target genes
in order further understanding on the functions of lncRNA in the
pathogenesis of HCC, which is also conducive to follow-up
studies.
Materials and methods
Identification of HCC-associated
lncRNAs
The lncRNA disease database (http://www.cuilab.cn/lncrnadisease) has identified
>200 types of lncRNA-associated diseases and can be utilized as
a bioinformatic tool to predict human lncRNA-associated diseases
(20). In the present study,
HCC-associated lncRNAs were screened and candidates lncRNAs were
selected using the lncRNA disease database.
lncRNA-miRNA interaction analysis
The interaction between miRNAs (6–8mer sites) and
lncRNAs was predicted from the crosslinking immunoprecipitation RNA
sequencing (CLIP-Seq) data using the starBase platform (http://starbase.sysu.edu.cn/) (21).
Identification of HCC-associated
miRNAs
HCC-associated miRNAs were mapped into the HMDD
database (http://202.38.126.151/hmdd/mirna/md/) (22).
Target gene prediction
ChIPBase (http://deepbase.sysu.edu.cn/chipbase/) is a database
for decoding the transcriptional regulation of lncRNAs and miRNAs
(23). The candidate transcription
factors for lncRNAs and miRNAs were predicted using ChIPBase and
analyzed using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/).
Identification of the target genes for
the candidate miRNAs
miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/) is
a comprehensive atlas of microRNA-target interactions, which
provides the possible miRNA binding sites within the complete
sequence of a gene (24). The target
genes for the candidate miRNAs were identified using the miRWalk
database.
Identification of the HCC-associated
target genes
OncoDB.HCC (http://oncodb.hcc.ibms.sinica.edu.tw/index.htm) is the
first integrated oncogenomic database of hepatocellular carcinoma,
which may aid identification aberrant cancer target genes and loci
(25). Target genes that were not
HCC-associated were further screened using the OncoDB.HCC
database.
Functional enrichment analysis
DAVID (https://david.ncifcrf.gov/home.jsp) is a database that
provides functional interpretation of a large list of genes derived
from genomic studies. DAVID accelerates the analysis of
genome-scale datasets by facilitating the transition from data
collection to biological meaning (26). In the present study, miRNA target
genes were further analyzed using DAVID, including Gene Ontology
(GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
analysis, and it was considered to be significant if the
Benjamini-corrected P-value was <0.05.
Identification of transcription
factors for the target lncRNAs and miRNAs
ConSite is a web-based tool for identifying
cis-regulatory elements in genomic sequences (http://consite.genereg.net). Predictions are made
based on the integration of binding site prediction generated with
high-quality transcription factor models and cross-species
comparison filtering (27). The
transcription factors for each target gene were predicted using
ConSite. The transcription factors that were associated with HCC
were further screened using PubMed.
Construction of the regulatory network
of lncRNAs in HCC
On the basis of the genomic information that was
collected, the possible regulatory network of the selected lcnRNAs
in HCC was constructed by Cytoscape software version 3.5.0
(http://www.cytoscape.org/index.html).
Results
HCC-associated lncRNAs
Of the 12 lncRNAs that were associated with HCC
obtained from the lncRNA disease database (Table I), HOX antisense intergenic RNA
(HOTAIR), metastasis-associated lung adenocarcinoma transcript 1
(MALAT1) and highly upregulated in liver cancer (HULC) were
selected for further analysis.
 | Table I.Hepatocellular carcinoma-associated
lncRNAs obtained from the lncRNA disease database. |
Table I.
Hepatocellular carcinoma-associated
lncRNAs obtained from the lncRNA disease database.
| lncRNA | Dysfunction
type | Chromosome | Genbank |
|---|
| H19 | Expression | 11 | NR_002196.1 |
|
| Expression | 11 | NR_002196.48 |
|
| N/A | 11 | NR_002196.17 |
|
| Expression | 11 | NR_002196.1 |
|
| Regulation | 11 | NR_002196.1 |
| HEIH | Expression | 5 | NR_045680.1 |
| HOTAIR | Expression | 12 | NR_047517.1 |
|
| Expression | 12 | NR_047517.10 |
|
| Expression | 12 | NR_047517.13 |
|
| Expression | 12 | NR_047517.3 |
|
| Expression | 12 | NR_047517.1 |
|
| Expression | 12 | NR_047517.15 |
|
| Regulation | 12 | NR_047517.1 |
| HOTTIP | Expression | 7 | NR_037843.3 |
|
| Expression | 7 | NR_037843.3 |
|
| Expression | 7 | NR_037843.2 |
| HULC | Expression | 6 | NR_004855.2 |
|
| Regulation | 6 | NR_004855.2 |
|
| Regulation | 6 | NR_004855.2 |
|
| Expression | 6 | NR_004855.3 |
|
| Expression | 6 | NR_004855.4 |
|
| Expression | 6 | NR_004855.6 |
|
| Expression | 6 | NR_004855.2 |
|
| Regulation | 6 | NR_004855.2 |
| IGF2-AS | N/A | 11 | NR_028044.1 |
| KCNQ1OT1 | Mutation | 11 | NR_002728.3 |
| MALAT1 | Expression | 11 | NR_002819.2 |
|
| Expression | 11 | NR_002819.2 |
|
| Expression | 11 | NR_002819.7 |
|
| Expression | 11 | NR_002819.12 |
|
| Expression | 11 | NR_002819.17 |
|
| Mutation | 11 | NR_002819.5 |
|
| Expression | 11 | NR_002819.2 |
| MEG3 | Expression | 14 | NR_002766.2 |
|
| Expression | 14 | NR_002766.15 |
|
| Regulation | 14 | NR_002766.2 |
| MINA | Expression | 3 | XR_241516.2 |
| MIR7-3HG | Expression | 19 | NR_027148.1 |
| NPTN-IT1 | Expression | 15 | AK055007.1 |
|
| Expression | 15 | AK055007.1 |
Analyses of the target lncRNAs
The three target lncRNAs, HOTAIR, MALAT1 and HULC,
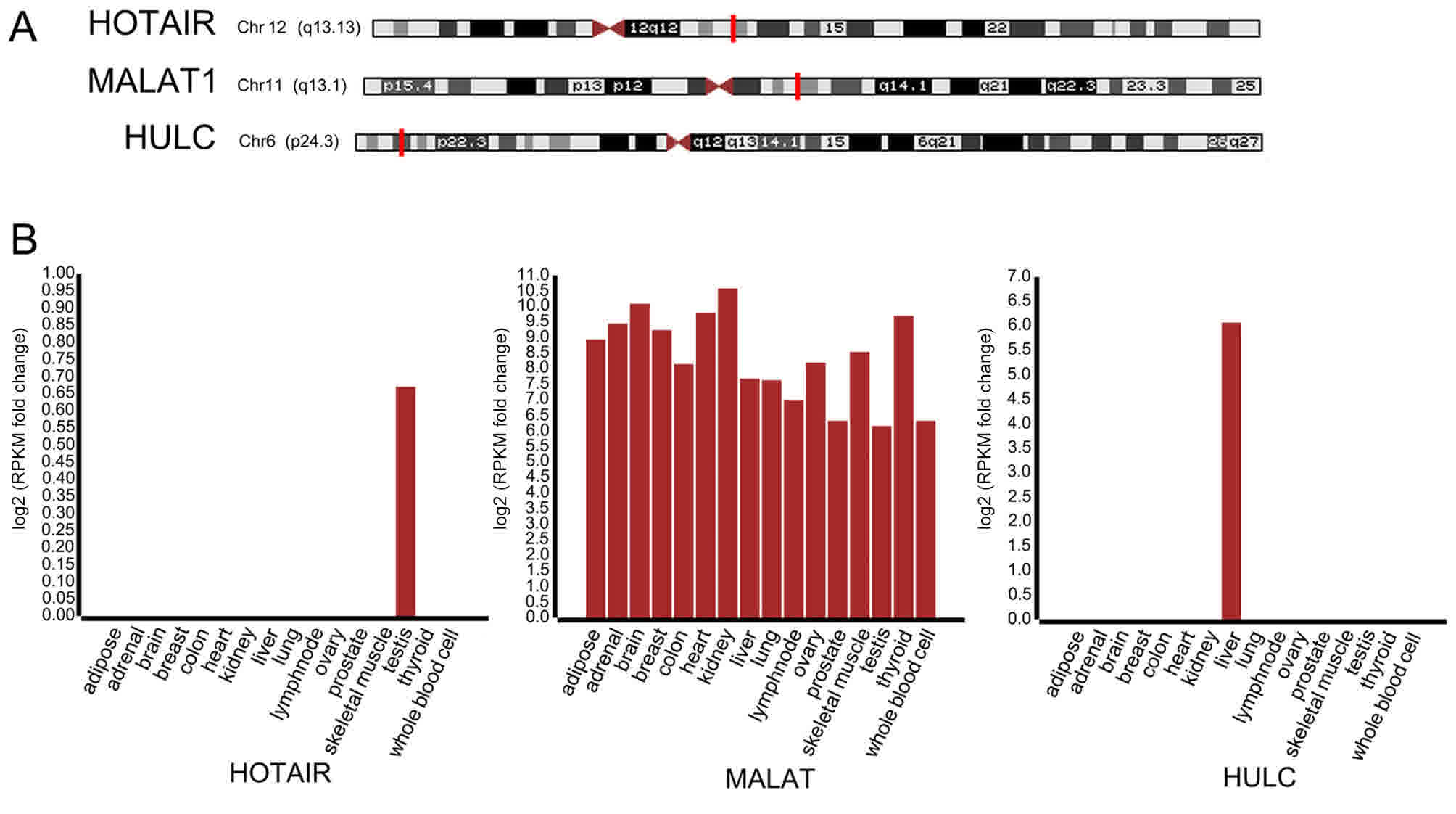
were analyzed using UCSC Genome Browser. As presented in Fig. 1A, HOTAIR, MALAT1 and HULC were located
on chromosome 12, 11 and 6, respectively. Additionally, MALAT1 and
HULC were highly expressed in the liver, whereas HOTAIR was
expressed in the liver at a lower level (Fig. 1B).
Prediction of the interaction between
target lncRNAs and miRNAs
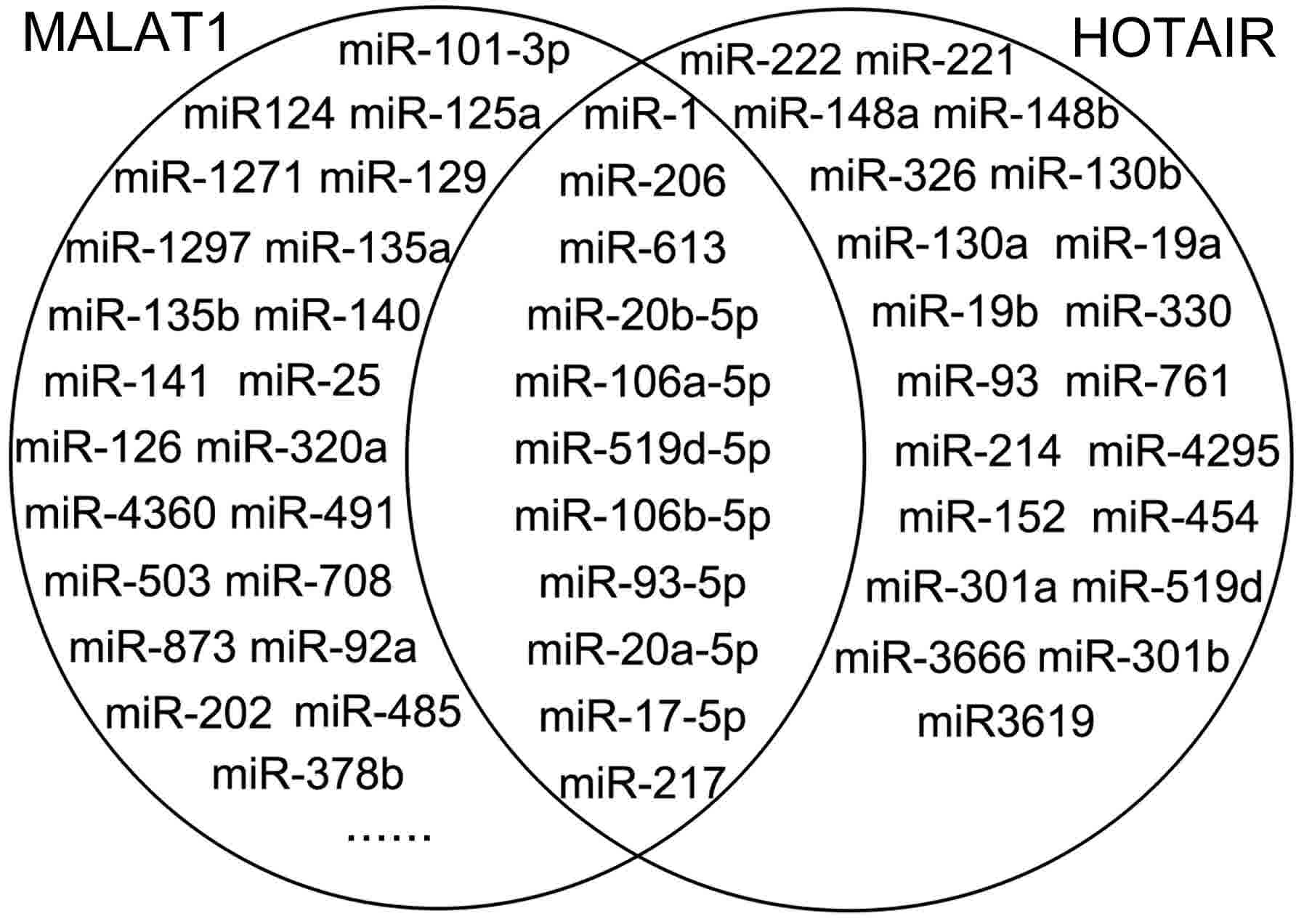
The miRNAs that were associated with HOTAIR, MALAT1
and HULC, were predicted using starBase2.0. A total of 32 miRNAs
were predicted to interact with HOTAIR, and 113 miRNAs were
predicted to interact with MALAT1. Among these miRNAs, miR-206,
miR-1, miR-17-5p, miR-20a-5p, miR-93-5p, miR-106a-5p, miR-106b-5p,
miR-613, miR-519d-5p, miR-217 and miR-20b-5p were overlapping
(Fig. 2). However, no miRNA was
identified to interact with HULC. Therefore, HULC was excluded from
further analysis.
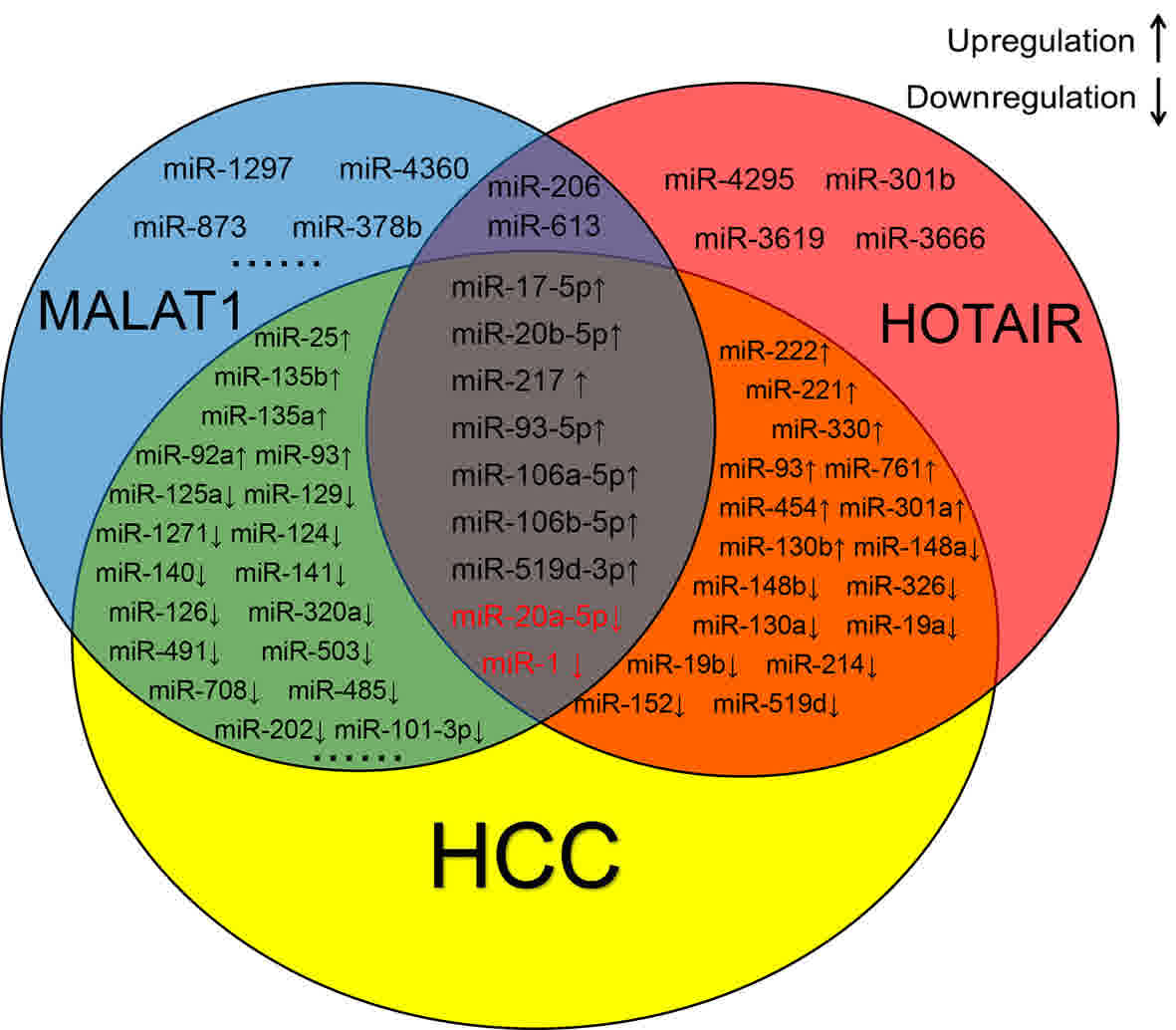
Identification of HCC-associated
miRNAs
The HCC-associated miRNAs were obtained from HMDD
and compared with the previously identified miRNAs. As presented in
Fig. 3, miR-1, miR-17-5p, miR-20a-5p,
miR-93-5p, miR-106a-5p, miR-106b-5p, miR-519d-5p, miR-217 and
miR-20b-5p were involved in the progression of HCC and were
identified to interact with HOTAIR and MALAT1. A previous study
demonstrated the negative association between miRNA and lncRNA
(28). Therefore, in the present
study, miR-1 and miR-20a-5p were selected for further analysis,
owing to their low expression in HCC.
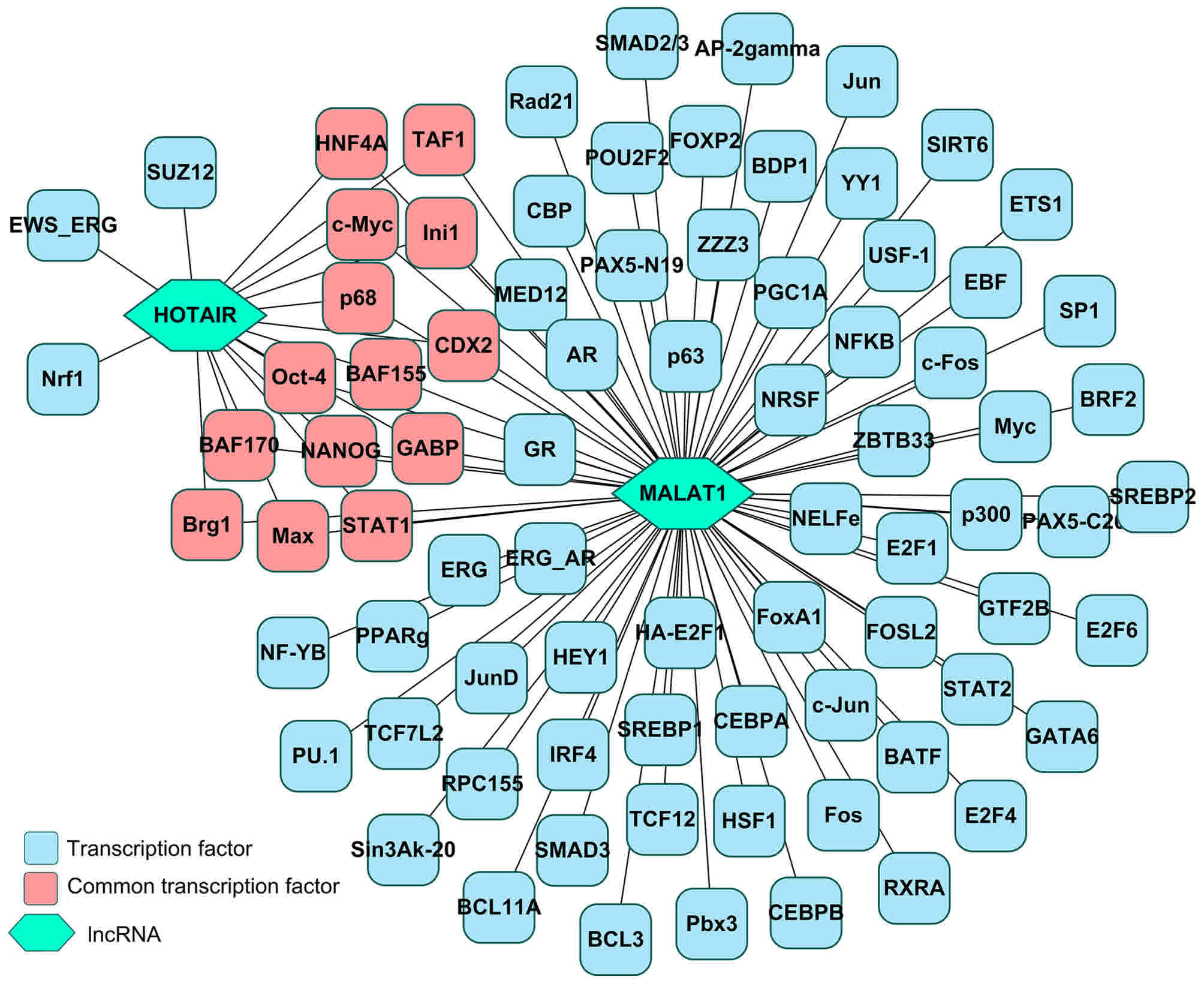
Prediction of the regulatory
association between transcription factors and target
lncRNAs/miRNAs
The transcription factors for lncRNAs (MALAT1 and
HOTAIR) and miRNAs (hsa-miR-1 and hsa-miR-20a-5p) were separately
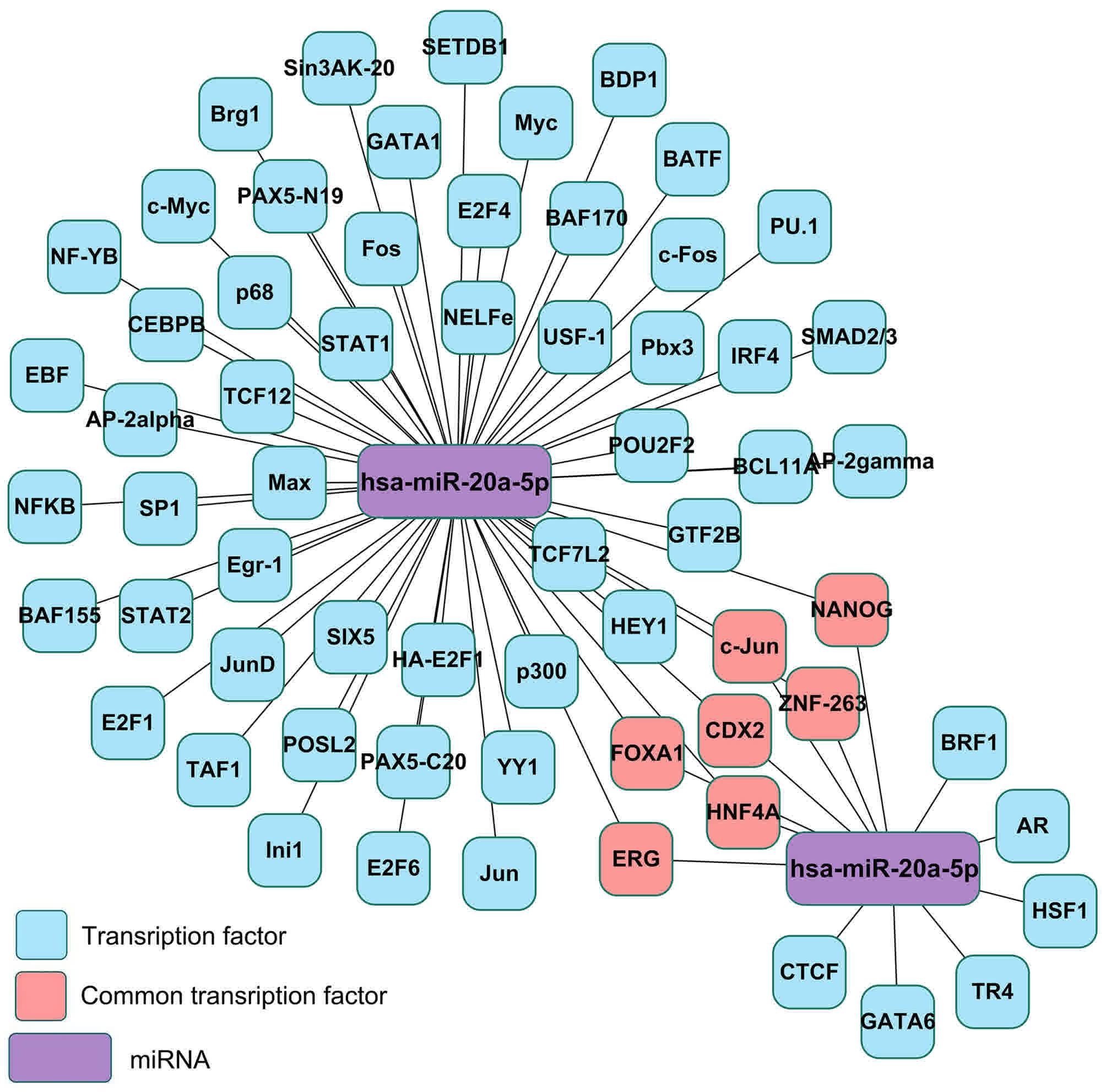
predicted using ChIPBase. As presented in Fig. 4, a total of 78 and 17 transcription
factors were identified for MALAT1 and HOTAIR, respectively. As
presented in Fig. 5, a total of 13
and 59 transcription factors were identified for hsa-miR-1 and
has-miR-20a-5p, respectively. A total of 7 overlaps were identified
between hsa-miR-1 and has-miR-20a-5p (Fig. 5).
Identification of HCC-associated
transcription factors
HCC-associated transcription factors were identified
using PubMed. The results demonstrated that there were nine lncRNA
transcription factors associated with HCC: Signal transducer and
activator of transcription 1 (STAT1), hepatocyte nuclear factor 4α
(HNF4A), octamer-binding transcription factor 4 (Oct-4), Nanog
homeobox (NANOG), caudal type homeobox 2 (CDX2), DEAD-box helicase
5 (p68), brahma-related gene 1 (Brg1), MYC-associated factor X
(Max) and MYC proto-oncogene, BHLH transcription factor (c-Myc)
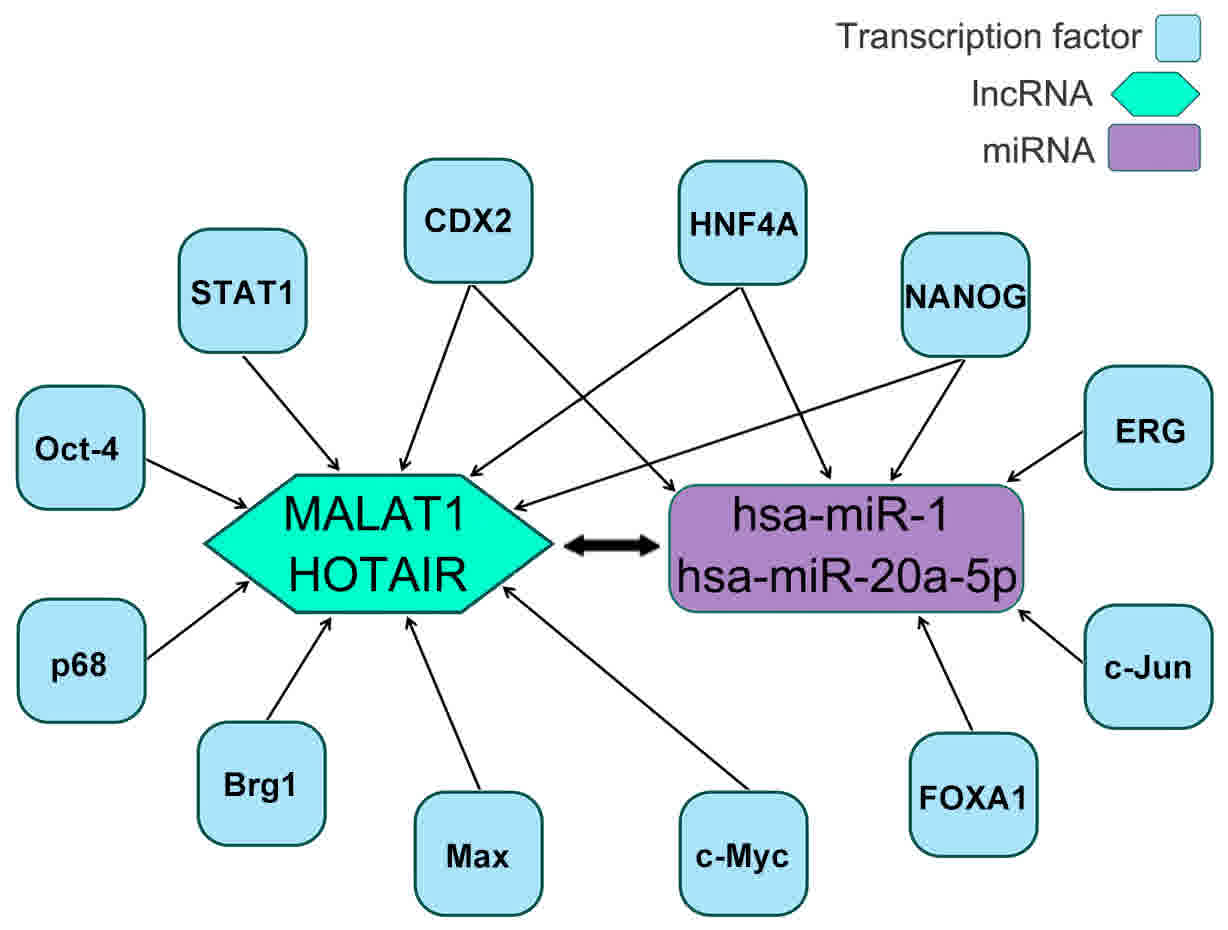
(29–36). Additionally, CDX2, erythroblast
transformation-specific-related gene (ERG), forkhead box protein A1
(FOXA1), HNF4A, c-Jun and NANOG were identified as the
transcription factors for hsa-miR-1 and hsa-miR-20a-5p (30–32,37–39).
CDX2, HNF4A and NANOG were identified to be common between the
target lncRNAs and miRNAs (Fig.
6).
Identification of target genes for
hsa-miR-1 and hsa-miR-20a-5p
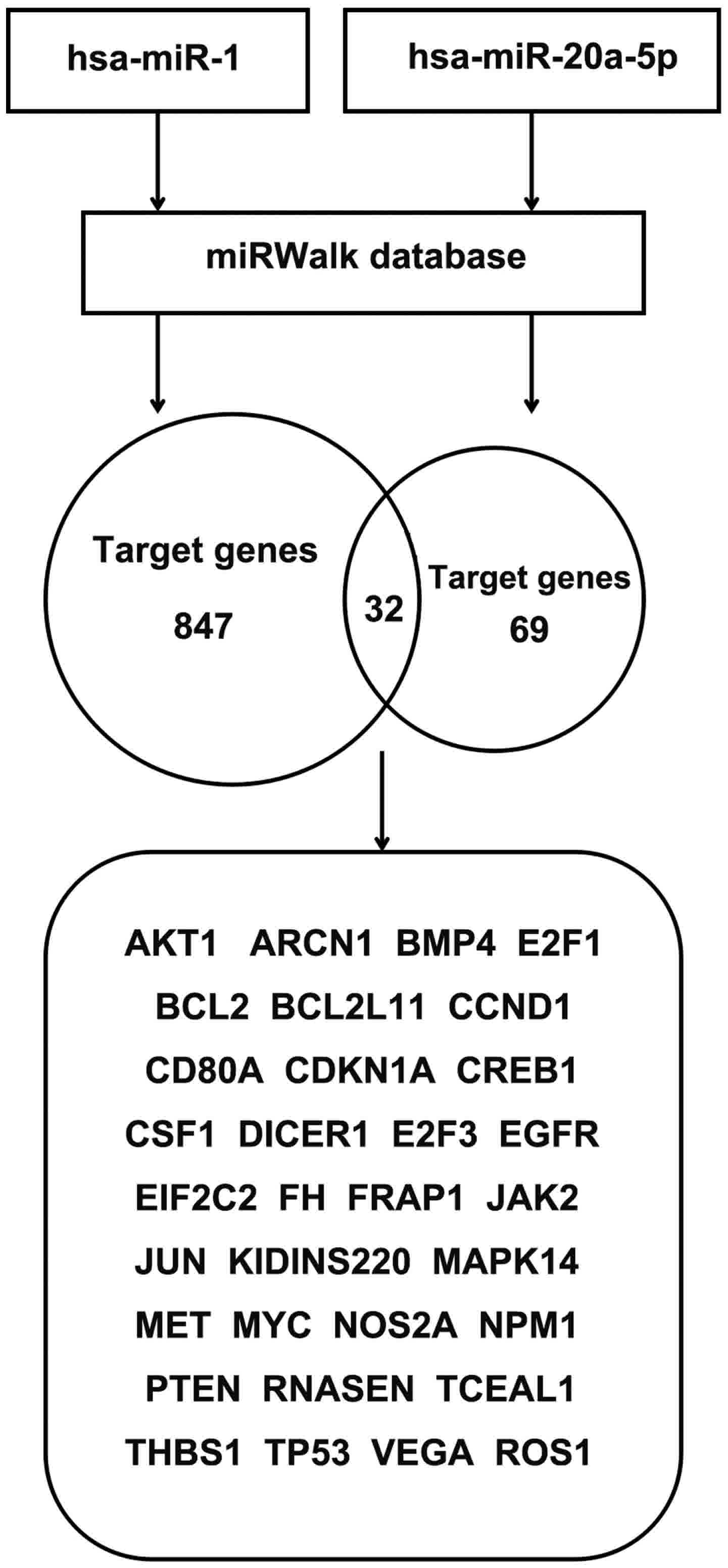
The target genes for hsa-miR-1 and hsa-miR-20a-5p
were predicted using the miRWalk database. A total of 879 and 101
target genes were identified for hsa-miR-1 and has-miR-20a-5p,
respectively. A total of 32 overlaps were identified between these
target miRNAs (Fig. 7).
Identification of the HCC-associated
target genes for hsa-miR-1 and hsa-miR-20a-5p
The OncoDB.HCC database was used for the
identification of the HCC-associated target genes for hsa-miR-1 and
hsa-miR-20a-5p. As presented in Table
II, among the 32 target genes, 11 of them were involved in the
progression of HCC. As miRNAs are single-stranded RNA molecules
that bind to targets in a base pair-mediated manner, resulting in
the degradation or inhibition of the expression and function of
target genes, the expression of the target genes were inverse to
that of the miRNAs (40), Then, seven
target genes [cyclin D1, E2F transcription factor 1 (E2F1),
epithelial growth factor receptor (EGFR), MYC, MET proto-oncogene,
receptor tyrosine kinase (MET), nitric oxide synthase 2A (NOS2A)
and vascular endothelial growth factor (VEGF)] with increased
expression in HCC were selected for the further analysis.
| Table II.HCC-associated miRNA target genes
obtained from the OncoDB.HCC database. |
Table II.
HCC-associated miRNA target genes
obtained from the OncoDB.HCC database.
| Gene | Description | Evidence |
|---|
| CCND1 | G1/S-specific
cyclin-D1 (PRAD1 oncogene) | Arrays: C.R.01a,
PNAS01, MBC 02 |
|
| (BCL-1 oncogene).
[Source: Uniprot/SWISSPROT; Acc: P24385] | Experiments:
upregulated (RT-PCR, IHC and western blotting) |
| E2F1 | Transcription
factor E2F1 (E2F-1) (Retinoblastoma-binding protein 3) (RBBP-3)
(PRB-binding protein E2F-1) (PBR3) (Retinoblastoma-associated
protein 1) (RBAP-1). | Experiments:
upregulated (RT-PCR) |
|
| [Source:
Uniprot/SWISSPROT; Acc: Q01094] |
|
| EGFR | Epidermal growth
factor receptor precursor (EC 2.7.10.1) | Experiments:
upregulated (IHC) |
|
| (Receptor
tyrosine-protein kinase ErbB-1). [Source: Uniprot/SWISSPROT; Acc:
P00533] |
|
| MYC | Myc proto-oncogene
protein (c-Myc) (Transcription factor p64). [Source:
Uniprot/SWISSPROT; Acc: P01106] | Arrays: PNAS01,
Onc. 02, MBC 02 Experiments: upregulated (RT-PCR and IHC) |
| NOS2A | Nitric oxide
synthase, inducible (EC 1.14.13.39) (NOS type II) (Inducible NO
synthase) (Inducible NOS) (iNOS) (Hepatocyte NOS) (HEP-NOS). | Experiments:
upregulated (RT-PCR) |
|
| [Source:
Uniprot/SWISSPROT; Acc: P35228] |
|
| VEGFA | Vascular
endothelial growth factor A precursor (VEGF-A) (Vascular
permeability factor) (VPF). [Source: Uniprot/SWISSPROT; Acc:
P15692] | Experiments:
upregulated (Northern blotting and IHC) |
| MET | Hepatocyte growth
factor receptor precursor (EC 2.7.10.1) | Experiments:
upregulated (RT-PCR and mutation) |
|
| (HGF receptor)
(Scatter factor receptor) (SF receptor) (HGF/SF receptor) (Met
proto-oncogene tyrosine kinase) (c-Met). |
|
|
| [Source:
Uniprot/SWISSPROT; Acc: P08581] |
|
| CDKN1A | Cyclin-dependent
kinase inhibitor 1 (p21) (CDK-interacting protein 1) (Melanoma
differentiation-associated protein 6) | Arrays: Gast01,
Onc.02, J.H.03 Experiments: downregulated (IHC and RT-PCR) |
|
| (MDA-6). [Source:
Uniprot/SWISSPROT; Acc: P38936] |
|
| NPM1 | Nucleophosmin (NPM)
(Nucleolar phosphoprotein B23) | Arrays: PNAS01,
MBC02, CCR03, Prot05a, Prot05c |
|
| (Numatrin)
(Nucleolar protein NO38). [Source: Uniprot/SWISSPROT; Acc:
P06748] |
|
| THBS1 | Thrombospondin-1
precursor. [Source: Uniprot/SWISSPROT; Acc: P07996] | Experiments:
downregulated (qPCR) |
| TP53 | Cellular tumor
antigen p53 (Tumor suppressor p53) (Phosphoprotein p53) (Antigen
NY-CO-13). | Experiments: other
(IHC, western blotting, mutation and methylation) |
|
| [Source:
Uniprot/SWISSPROT; Acc: P04637] |
|
GO and KEGG pathway enrichment
analysis of the target genes
GO and KEGG pathway enrichment analysis of
identified target genes was performed using DAVID. GO analysis
revealed that six genes were involved in cellular functions such as
cell growth, cycle and mitosis (Table
III). Additionally, KEGG analysis revealed that six genes were
involved in the cell cycle and focal adhesion, and in disease
progression (Table IV). NOS2A was
excluded from further analysis.
| Table III.GO enrichment analysis for the seven
HCC-associated miRNA target genes. |
Table III.
GO enrichment analysis for the seven
HCC-associated miRNA target genes.
| GO ID | Term | Genes |
Percentagea | P-value |
Benjaminib |
|---|
| GO:0008283 | Cell
proliferation | E2F1, EGFR, MET,
MYC, VEGFA | 83.3 |
5.20×10−6 |
2.40×10−3 |
| GO:0051726 | Regulation of cell
cycle | E2F1, CCND1, EGFR,
MYC | 66.7 |
1.40×10−4 |
3.20×10−2 |
| GO:0008284 | Positive regulation
of cell proliferation | CCND1, EGFR, MYC,
VEFA | 66.7 |
2.70×10−4 |
4.10×10−2 |
| GO:0044093 | Positive regulation
of molecular function | CCND1, EGFR, MET,
MYC | 66.7 |
7.60×10−4 |
4.20×10−2 |
| GO:0009891 | Positive regulation
of biosynthetic process | E2F1, EGFR, MYC,
VEGFA | 66.7 |
1.20×10−3 |
4.30×10−2 |
| GO:0022402 | Cell cycle
process | E2F1, CCND1, EGFR,
MYC | 66.7 |
6.80×10−4 |
4.40×10−2 |
| GO:0051325 | Interphase | E2F1, CCND1,
EGFR | 50 |
6.00×10−4 |
4.50×10−2 |
| GO:0007346 | Regulation of
mitotic cell cycle | CCND1, EGFR,
MYC | 50 |
1.20×10−3 |
4.60×10−2 |
| GO:0031328 | Positive regulation
of cellular biosynthetic process | E2F1, EGFR, MYC,
VEGFA | 66.7 |
1.20×10−3 |
4.90×10−2 |
| Table IV.KEGG pathway enrichment analysis for
the seven HCC-associated miRNA target genes. |
Table IV.
KEGG pathway enrichment analysis for
the seven HCC-associated miRNA target genes.
| KEGG ID | Term | Genes |
Percentagea | P-value |
Benjaminib |
|---|
| hsa05219 | Bladder cancer | E2F1, CCND1, EGFR,
MYC, VEGFA | 83.3 |
2.00×10−8 |
7.00×10−7 |
| hsa05200 | Pathways in
cancer | E2F1, CCND1, EGFR,
MET, MYC, VEGFA | 100 |
1.10×10−6 |
1.90×10−5 |
| hsa05218 | Melanoma | E2F1, CCND1, EGFR,
MET | 66.7 |
2.60×10−5 |
3.00×10−4 |
| hsa05212 | Pancreatic
cancer | E2F1, CCND1, EGFR,
VEGFA | 66.7 |
2.70×10−5 |
2.30×10−4 |
| hsa05210 | Colorectal
cancer | CCND1, EGFR, MET,
MYC | 66.7 |
4.20×10−5 |
3.00×10−4 |
| hsa04510 | Focal adhesion | CCND1, EGFR, MET,
VEGFA | 66.7 |
5.70×10−4 |
3.30×10−3 |
| hsa05213 | Endometrial
cancer | CCND1, EGFR,
MYC | 50 |
1.00×10−3 |
5.00×10−3 |
| hsa05223 | Non-small cell lung
cancer | E2F1, CCND1,
EGFR | 50 |
1.10×10−3 |
4.70×10−3 |
| hsa05214 | Glioma | E2F1, CCND1,
EGFR | 50 |
1.50×10−3 |
5.70×10−3 |
| hsa05220 | Chronic myeloid
leukemia | E2F1, CCND1,
MYC | 50 |
2.10×10−3 |
7.30×10−3 |
| hsa05222 | Small cell lung
cancer | E2F1, CCND1,
MYC | 50 |
2.60×10−3 |
8.30×10−3 |
| hsa05212 | Prostate
cancer | E2F1, CCND1,
EGFR | 50 |
2.90×10−3 |
8.50×10−3 |
| hsa04110 | Cell cycle | E2F1, CCND1,
MYC | 50 |
5.70×10−3 |
1.50×10−2 |
Prediction of the transcription
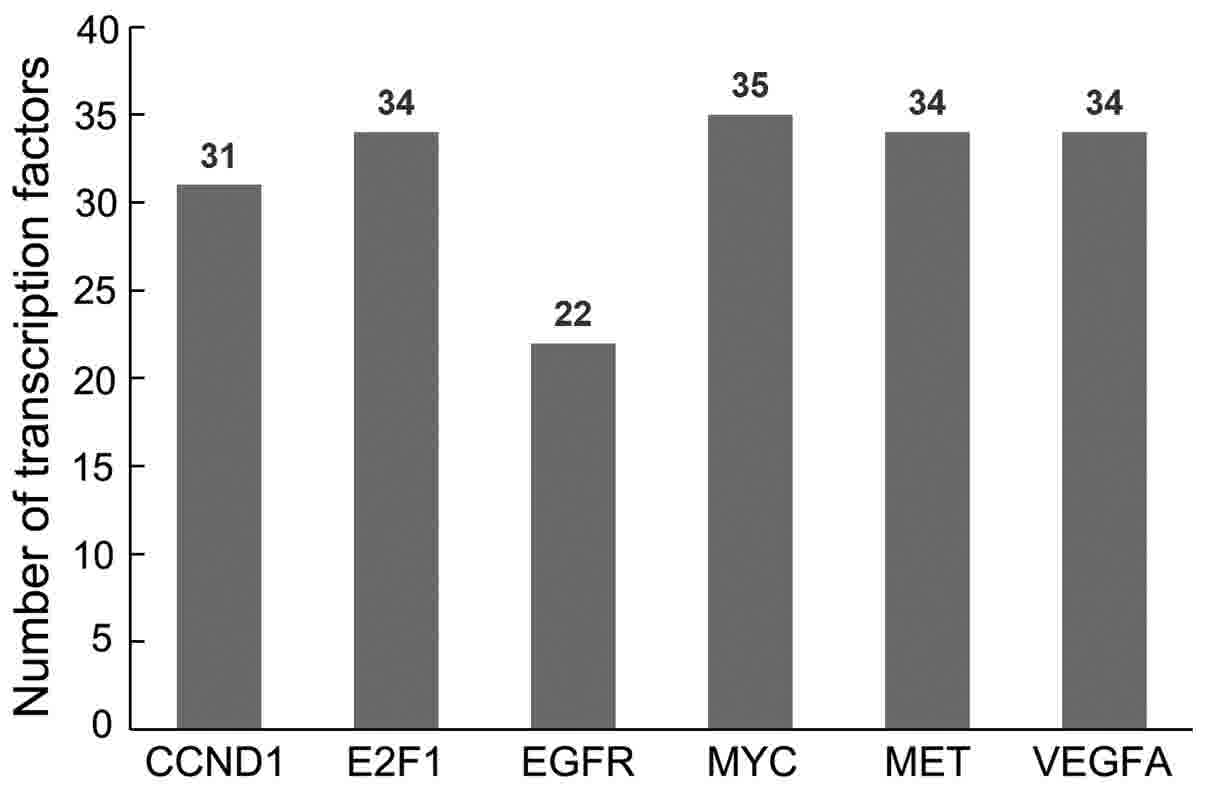
factors for the HCC-associated miRNAs
The transcription factors for the HCC-associated
miRNAs were predicted using ConSite (Fig.
8). The results demonstrated that snail family transcriptional
repressor 1 (Snail) was a common transcription factor for the six
target genes and was involved in the progression of HCC (Table V) (41).
| Table V.Prediction of the overlapping
transcription factors for the six target genes using ConSite. |
Table V.
Prediction of the overlapping
transcription factors for the six target genes using ConSite.
| Transcription
factor | Sequence | From
(position) | To (position) | Score |
|---|
| Hunchback | TTTTTTATGC | 6,433 | 6,442 | 12.824 |
| AML-1 | TTTGTGGTT | 2,631 | 2,639 | 12.626 |
| c-REL | GGGGTTTTCC | 8,856 | 8,865 | 12.159 |
| E74A | CCGGAAG | 6,379 | 6,385 | 10.755 |
| Snail | CAGGTG | 2,151 | 2,156 | 10.744 |
| Thing1-E47 | GGTCTGGCTT | 1,529 | 1,538 | 12.164 |
Functional regulatory network of
lncRNA in HCC
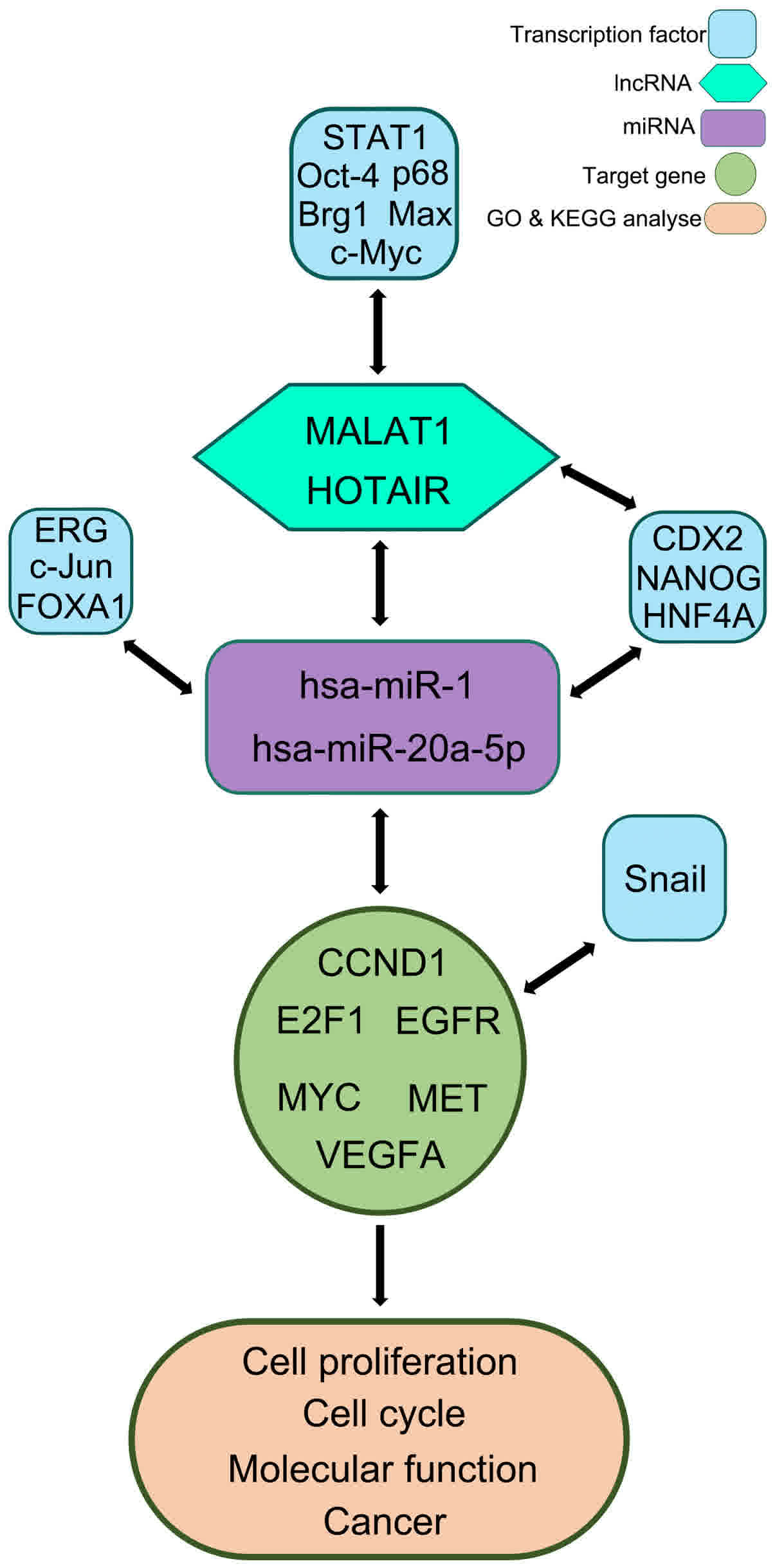
Fig. 9 presents the
proposed regulatory network of lncRNAs in HCC.
Discussion
Following advances in proteomics and genomics, the
function of numerous human ncRNAs has been investigated. On the
basis of their length and biological function, ncRNAs can be
divided into miRNAs, small nucleolar RNA, small nuclear RNAs,
piwi-interacting RNAs, natural antisense transcripts, ribosomal
RNAs, transfer RNAs and lncRNAs (42). A total of 270 lncRNAs have been
documented in the lncRNAdb database (20). Several studies have demonstrated that
lncRNAs may serve notable functions in cell physiological
activities, in the development of various types of tumor and in
epigenetics, transcriptional and post-transcriptional mechanisms
(42–46).
Bioinformatics is a powerful tool for genomic
research that facilitates the identification of candidate genes and
nucleotide sequences, and predicts the biological functions and
regulatory pathways of the target genes. At present, research into
lncRNAs is limited. In the present study, HCC-associated lncRNAs
(HOTAIR, MALAT1 and HULC) were identified using lncRNA disease
database. According to data from the UCSC Genome Browser and lncRNA
disease database, HOTAIR was located on human chromosome 12 (12649
bp) and exhibited a low expression in the liver, but increased
expression in HCC (47). HOTAIR may
inhibit the expression of interferon-γ and cell cycle-associated
genes, thus promoting cellular invasion in HCC. Knockout of HOTAIR
significantly decreased cellular proliferation and promoted
apoptosis in HCC (48,49). The 3-year recurrence-free survival
rate for patients with HCC that exhibited low expression of HOTAIR
was significant increased compared with that in patients with high
expression of lncRNA following liver transplantation. Therefore,
HOTAIR may be used as an independent indicator for tumor recurrence
following liver transplantation (47). The results of the present study
demonstrated that MALAT1 was located on human chromosome 11q13
(8708 bp), with a length of 870 bp. MALAT1 was identified in
various tissues and exhibited increased expression levels in HCC
(50). One previous study (51) demonstrated that MALAT1 may promote
cellular proliferation in HCC, and patients with HCC with increased
expression of MALAT1 exhibited an increased risk of tumor
recurrence. Therefore, MALAT1 may be used for the prediction of HCC
recurrence. MALAT1 may regulate the migration of HCC cells
(52). The results of the present
study demonstrated that HULC was located on human chromosome
6p24.3. The expression of HULC was increased in the liver but low
in HCC tissues (53).
lncRNAs may interact with miRNAs (54). The results of previous studies have
indicated that the interaction of lncRNAs and miRNAs serve a
notable function in the progression of cancer (55–57). In
the present study, starBase2.0 was used to predict the miRNAs that
were associated with lncRNAs (HOTAIR, MALAT1 and HULC). However, no
miRNA was identified to interact with HULC, so further analyses
were performed on HOTAIR and MALAT1 only. A total of 11 overlaps
were identified between HOTAIR and MALAT1, 9 of which were
HCC-associated miRNAs according to the HMDD database. hsa-miR-1 and
hsa-miR20a-5p were identified to be downregulated in HCC and
selected for further analysis.
ChIPBase was used to predict the transcription
factors for lncRNAs (MALAT1 and HOTAIR) and miRNAs (hsa-miR-1 and
hsa-miR-20a-5p). The results demonstrated that 9 transcription
factors for lncRNAs (STAT1, HNF4A, Oct-4, NANOG, CDX2, p68, Brg1,
Max and c-Myc) were associated with HCC. Additionally, CDX2, ERG,
FOXA1, HNF4A, c-Jun and NANOG were identified to associate with the
target miRNAs and were associated with the progression of HCC.
Among them, CDX2, HNF4A and NANOG were identified as the
transcription factors in common for these lncRNAs and miRNAs.
In the present study, a total of 32 target genes
were identified for hsa-miR-1 and hsa-miR-20a-3p using miRWalk.
Among them, 11 genes were involved in the development of HCC,
according to the OncoDB.HCC database. A total of seven target genes
with increased expression in HCC (cyclin D1, E2F1, EGFR, MYC, MET,
NOS2A, VEGFA) were selected for the further analysis. Additionally,
GO and KEGG enrichment analysis was performed on the seven
HCC-associated miRNA target genes using DAVID. The results
demonstrated that six genes were involved in various cellular
functions, including cell growth, cell cycle progression and
mitosis. KEGG analysis revealed that six genes were involved in the
cell cycle, focal adhesion and disease progression
Snail was predicted to be the transcription factor
in common among the six target genes and was involved in the
progression of HCC according to data from ConSite. A previous study
demonstrated that upregulation of Snail may promote the
proliferation and invasion of HCC cells (58). Therefore, in the present study a
complete regulatory network of lncRNAs in HCC was proposed, whereby
each one of the participants was either directly or indirectly
associated and whose deregulation may result in HCC progression.
Nevertheless, further research is required to confirm these
regulatory associations.
In the present study the function and regulatory
network of HCC-associated lncRNAs (HOTAIR and MALAT1) was predicted
using bioinformatics analysis to understand the molecular
mechanisms underlying the progression of HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the First
Affiliated Hospital of Jinan University (grant no. 2014106), the
Technical New Star of Zhujiang, Pan Yu, Guangzhou (grant nos.
2014-special-15-3.09 and 2013-special-15-6.09), the National
Natural Science Foundation of China (grant no. 81502557), the
Science and Technology Planning Project of Guangdong Province
(grant no. 2017ZC0372) and the Medical and Health Science and
Technology Project of Panyu District, Guangzhou (grant no.
2017-Z04-18).
Availability of data and materials
The data that support results of the present study
are available from lncRNA disease database, starBase, HMDD,
ChIPBase, PubMed, miRWalk, OncoDB.HCC, DAVID and ConSite (webpages
cited in-text).
Authors' contributions
MRC drafted the article. ZPH, JML and JBZ performed
the experiments. MRC and ZPH analysed the data and interpreted the
results. YBL produced the figures and tables. MRC and ZPH revised
the article. JHH and YGL conceived and designed the study. All
authors read and approved the final article.
Ethics approval and consent to publish
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
JTorre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang X, Xie X, Xiao YF, Xie R, Hu CJ, Tang
B, Li BS and Yang SM: The emergence of long non-coding RNAs in the
tumorigenesis of hepatocellular carcinoma. Cancer Lett.
360:119–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anwar SL and Lehmann U: DNA methylation,
microRNAs, and their crosstalk as potential biomarkers in
hepatocellularcarcinoma. World J Gastroenterol. 20:7894–7913. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roderburg C and Luedde T: The role of the
gut microbiome in the development and progression of liver
cirrhosis and hepatocellular carcinoma. Gut Microbes. 5:441–445.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chew CL, Conos SA, Unal B and Tergaonkar
V: Noncoding RNAs: Master regulators of inflammatory signaling.
Trends Mol Med. 24:66–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weidle UH, Birzele F, Kollmorgen G and
Rüger R: Long Non-coding RNAs and their Role in Metastasis. Cancer
Genomics Proteomics. 14:143–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bassett AR, Akhtar A, Barlow DP, Bird AP,
Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras
TR, Haerty W, et al: Considerations when investigating lncRNA
function in vivo. Elife. 3:e030582014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marchese FP, Raimondi I and Huarte M: The
multidimensional mechanisms of long noncoding RNA function. Genome
Biol. 18:2062017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng P, Yin Z, Wu Y, Xu Y, Luo Y and
Zhang TC: LncRNA HOTAIR promotes cell migration and invasion by
regulating MKL1 via inhibition miR206 expression in HeLa cells.
Cell Commun Signal. 16:52018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gutschner T, Richtig G, Haemmerle M and
Pichler M: From biomarkers to therapeutic targets-the promises and
perils of long non-coding RNAs in cancer. Cancer Metastasis Rev.
37:83–105. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
YiRen H, YingCong Y, Sunwu Y, Keqin L,
Xiaochun T, Senrui C, Ende C, XiZhou L and Yanfan C: Long noncoding
RNA MALAT1 regulates autophagy associated chemoresistance via
miR-23b-3p sequestration in gastric cancer. Mol Cancer. 16:1742017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiu L, Tang Q, Li G and Chen K: Long
non-coding RNAs as biomarkers and therapeutic targets: Recent
insights into hepatocellular carcinoma. Life Sci. 191:273–282.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu YR, Tang RX, Huang WT, Ren FH, He RQ,
Yang LH, Luo DZ, Dang YW and Chen G: Long noncoding RNAs in
hepatocellular carcinoma: Novel insights into their mechanism.
World J Hepatol. 7:2781–2791. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He JH, Han ZP, Liu JM, Zhou JB, Zou MX, Lv
YB, Li YG and Cao MR: Overexpression of long non-coding RNA MEG3
inhibits proliferation of hepatocellular carcinoma Huh7 cells via
negative modulation of miRNA-664. J Cell Biochem. 118:3713–3721.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malakar P, Shilo A, Mogilevsky A, Stein I,
Pikarsky E, Nevo Y, Benyamini H, Elgavish S, Zong X, Prasanth KV
and Karni R: Long Noncoding RNA MALAT1 promotes hepatocellular
carcinoma development by SRSF1 upregulation and mTOR activation.
Cancer Res. 77:1155–1167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quek XC, Thomson DW, Maag JL, Bartonicek
N, Signal B, Clark MB, Gloss BS and Dinger ME: lncRNAdb v2.0:
Expanding the reference database for functional long noncoding
RNAs. Nucleic Acids Res. 43:(Database Issue). D168–D173. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42:(Database Issue). D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T
and Cui Q: HMDD v2.0: A database for experimentally supported human
microRNA and disease associations. Nucleic Acids Res. 42:(Database
Issue). D1070–D1074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang JH, Li JH, Jiang S, Zhou H and Qu LH:
ChIPBase: A database for decoding the transcriptional regulation of
long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic
Acids Res. 41:(Database Issue). D177–D187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dweep H, Gretz N and Sticht C: miRWalk
database for miRNA-target interactions. Methods Mol Biol.
1182:289–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Su WH, Chao CC, Yeh SH, Chen DS, Chen PJ
and Jou YS: OncoDB. HCC: An integrated oncogenomic database of
hepatocellular carcinoma revealed aberrant cancer target genes and
loci. Nucleic Acids Res. 35:(Database Issue). D727–D731. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
27
|
Sandelin A, Wasserman WW and Lenhard B:
ConSite: Web-based prediction of regulatory elements using
cross-species comparison. Nucleic Acids Res. 32:(Web Server Issue).
W249–W252. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin Z, Hu Y, Lai S, Xue M, Lin J, Qian Y,
Zhuo W, Chen S, Si J and Wang L: Long Noncoding RNA: Its partners
and their roles in cancer. Neoplasma. 62:846–854. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen G, Wang H, Xie S, Ma J and Wang G:
STAT1 negatively regulates hepatocellular carcinoma cell
proliferation. Oncol Rep. 29:2303–2310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao D, Peng S and Dai C: The role of
hepatocyte nuclear factor 4alpha in metastatic tumor formation of
hepatocellular carcinoma and its close relationship with the
mesenchymal-epithelial transition markers. BMC Cancer. 13:4322013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chang TS, Wu YC, Chi CC, Su WC, Chang PJ,
Lee KF, Tung TH, Wang J, Liu JJ, Tung SY, et al: Activation of
IL6/IGFIR confers poor prognosis of HBV-related hepatocellular
carcinoma through induction of OCT4/NANOG expression. Clin Cancer
Res. 21:201–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu R, Wong KF, Lee NP, Lee KF and Luk JM:
HNF1α and CDX2 transcriptional factors bind to cadherin-17 (CDH17)
gene promoter and modulate its expression in hepatocellular
carcinoma. J Cell Biochem. 111:618–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kitagawa N, Ojima H, Shirakihara T,
Shimizu H, Kokubu A, Urushidate T, Totoki Y, Kosuge T, Miyagawa S
and Shibata T: Downregulation of the microRNA biogenesis components
and its association with poor prognosis inhepatocellular carcinoma.
Cancer Sci. 104:543–551. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Endo M, Yasui K, Zen Y, Gen Y, Zen K,
Tsuji K, Dohi O, Mitsuyoshi H, Tanaka S, Taniwaki M, et al:
Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and
BRM in hepatocellular carcinoma. Liver Int. 33:105–117. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mauleon I, Lombard MN, Muñoz-Alonso MJ,
Cañelles M and Leon J: Kinetics of myc-max-mad gene expression
during hepatocyte proliferation in vivo: Differential regulation of
mad family and stress-mediated induction of c-myc. Mol Carcinog.
39:85–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang D, Qi J, Liu R, Dai B, Ma W, Zhan Y
and Zhang Y: c-Myc plays a key role in TADs-induced apoptosis and
cell cycle arrest in human hepatocellular carcinoma cells. Am J
Cancer Res. 5:1076–1088. 2015.PubMed/NCBI
|
|
37
|
Hou X, Peng JX, Hao XY, Cai JP, Liang LJ,
Zhai JM, Zhang KS, Lai JM and Yin XY: DNA methylation profiling
identifies EYA4 gene as a prognostic molecular marker in
hepatocellular carcinoma. Ann Surg Oncol. 21:3891–3899. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tu H, Wei G, Cai Q, Chen X, Sun Z, Cheng
C, Zhang L, Feng Y, Zhou H, Zhou B and Zeng T: MicroRNA-212
inhibits hepatocellular carcinoma cell proliferation and induces
apoptosis by targeting FOXA1. Onco Targets Ther. 8:2227–2235.
2015.PubMed/NCBI
|
|
39
|
Abdou AG, Abd-Elwahed M, Badr M, Helmy M,
Soliman EA and Maher D: The differential immunohistochemical
expression of p53, c-Jun, c-Myc, and p21 between HCV-related
hepatocellular carcinoma with and without cirrhosis. Appl
Immunohistochem Mol Morphol. 24:75–87. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Du T and Zamore PD: Beginning to
understand microRNA function. Cell Res. 17:6612007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou ZJ, Dai Z, Zhou SL, Hu ZQ, Chen Q,
Zhao YM, Shi YH, Gao Q, Wu WZ, Qiu SJ, Zhou J and Fan J: HNRNPAB
induces epithelial-mesenchymal transition and promotes metastasis
of hepatocellularcarcinoma by transcriptionally activating SNAIL.
Cancer Res. 74:2750–2762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Struhl K: Transcriptional noise and the
fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol.
14:103–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Caley DP, Pink RC, Trujillano D and Carter
DR: Long noncoding RNAs, chromatin, and development.
ScientificWorldJournal. 10:90–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vizoso M and Esteller M: The activatory
long non-coding RNA DBE-T reveals the epigenetic etiology of
facioscapulohumeral muscular dystrophy. Cell Res. 22:1413–1415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ishigaki S, Masuda A, Fujioka Y, Iguchi Y,
Katsuno M, Shibata A, Urano F, Sobue G and Ohno K:
Position-dependent FUS-RNA interactions regulate alternative
splicing events and transcriptions. Sci Rep. 2:5292012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY,
Zhang F and Zheng SS: Overexpression of long non-coding RNA HOTAIR
predicts tumor recurrence in hepatocellular carcinoma patients
following liver transplantation. Ann Surg Oncol. 18:1243–1250.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Geng YJ, Xie SL, Li Q, Ma J and Wang GY:
Large intervening non-coding RNA HOTAIR is associated with
hepatocellular carcinoma progression. J Int Med Res. 39:2119–2128.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Niinuma T, Suzuki H, Nojima M, Nosho K,
Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki
Y, et al: Upregulation of miR-196a and HOTAIR drive malignant
character in gastrointestinal stromal tumors. Cancer Res.
72:1126–1136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tano K and Akimitsu N: Long non-coding
RNAs in cancer progression. Front Genet. 3:2192012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kiss T: Small nucleolar RNA-guided
post-transcriptional modification of cellular RNAs. EMBO J.
20:3617–3622. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY,
Zhang F, Wu LM, Chen LM and Zheng SS: Long non-coding RNA MALAT-1
overexpression predicts tumor recurrence of hepatocellular
carcinoma after liver transplantation. Med Oncol. 29:1810–1816.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Du Y, Kong G, You X, Zhang S, Zhang T, Gao
Y, Ye L and Zhang X: Elevation of highly up-regulated in liver
cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell
proliferation via down-regulating p18. J Biol Chem.
287:26302–26311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Takeshita F, Patrawala L, Osaki M,
Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG,
Brown D and Ochiya T: Systemic delivery of synthetic microRNA-16
lnhibits the growth of metastatic prostate tumors via dowregulation
of multiple cell-cycle genes. Mol Ther. 18:181–187. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fu WM, Zhu X, Wang WM, Lu YF, Hu BG, Wang
H, Liang WC, Wang SS, Ko CH, Waye MM, et al: Hotair mediates
hepatocarcinogenesis through suppressing miRNA-218 expression and
activating P14 and P16 signaling. J Hepatol. 63:886–895. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cui M, Xiao Z, Wang Y, Zheng M, Song T,
Cai X, Sun B, Ye L and Zhang X: Long noncoding RNA HULC modulates
abnormal lipid metabolism in hepatoma cells through an
miR-9-mediated RXRA signaling pathway. Cancer Res. 75:846–857.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tang Y, Jin X, Xiang Y, Chen Y, Shen CX,
Zhang YC and Li YG: The lncRNA MALAT1 protects the endothelium
against ox-LDL-induced dysfunction via upregulating the expression
of the miR-22-3p target genes CXCR2 and AKT. FEBS Lett.
589:3189–3196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wan Z, Pan H, Liu S, Zhu J, Qi W, Fu K,
Zhao T and Liang J: Downregulation of SNAIL sensitizes
hepatocellular carcinoma cells to TRAIL-induced apoptosis by
regulating the NF-κB pathway. Oncol Rep. 33:1560–1566. 2015.
View Article : Google Scholar : PubMed/NCBI
|