Introduction
Lung cancer has the highest levels of morbidity and
mortality of all malignant tumor types, in China and worldwide, and
adenocarcinoma is most common type of lung cancer (1). The treatment of lung adenocarcinoma
frequently fails, as the majority of patients present with
metastatic disease at the point of diagnosis (2). Despite continuous progress in
chemotherapeutic drug development and the application of molecular
targeted therapy in recent years, the 5-year survival rate of lung
cancer has not significantly increased (3,4).
Therefore, it is necessary to investigate novel anticancer agents
that are highly effective and exhibit low toxicity for clinical
intervention in lung adenocarcinoma.
Dihydroartemisinin (DHA), a more water-soluble
active metabolite of artemisinin isolated from the Chinese herb
Artemisia annua L (also called qinghaosu or sweet wormwood),
is part of a new generation of drugs against fever and chloroquine-
and mefloquine-resistant strains of Plasmodium falciparum
(5). DHA has been revealed to be
capable of inhibiting cell proliferation and inducing apoptosis in
cancer cells, including ovarian, leukemia, osteosarcoma and lung
cancer cells, when used alone or in combination with conventional
chemotherapeutic agents (6–9). In various cell lines, DHA activates
signaling pathways, including the Wnt/β-catenin, nuclear factor-κB
(NF-κB), mammalian target of rapamycin (mTOR) and caspase signaling
pathways (8,10); however, the molecular mechanism
underlying the DHA-induced inhibition of cell proliferation and
induction of apoptosis requires further investigation.
The mitogen-activated protein kinase (MAPK) cascade
is an important signal transducer from extracellular to nuclear and
participates in diverse cellular functions, including cell
proliferation, differentiation and death (11). At least 4 key proteins of signaling
pathways are associated with the MAPK family in eukaryotic cells:
Extracellular signal-regulated protein kinase (ERK), c-Jun
N-terminal kinase (JNK), big MAP kinase and p38MAPK (12). p38MAPK was identified as a kinase
induced by stress signals (13).
Previous studies indicated that p38MAPK may be a positive or
negative regulator of apoptosis (14,15).
Whether p38MAPK signaling is involved in DHA-induced cell
proliferation inhibition and apoptosis in lung adenocarcinoma
requires further investigation.
In the present study, Lewis lung carcinoma (LLC)
cells were incubated with DHA with or without carboplatin (CBP).
The viability of the treated cells was observed by MTT and
clonogenic assays, cell cycle and apoptotic rate were analyzed by
flow cytometry. The expression levels of phosphorylated (p)-p38MAPK
or p-JNK in the cell lines were analyzed with western blotting. The
present study demonstrated that DHA sensitized LLC cells to CBP
therapy via p38MAPK activation, which suggested that a combined
treatment with DHA and CBP maybe a potential novel treatment
regimen for lung adenocarcinoma therapy.
Materials and methods
Agents
DHA was purchased from Wulingshan Pharm (Chongqing,
China) and dissolved in dimethyl sulfoxide (DMSO; Amresco, LLC,
Solon, OH, USA) to make a 500 µg/ml stock solution, with a final
concentration of 0.5% (v/v) DMSO in the following experiments, as
previously described (16). CBP was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and
50 µmol/l was used as the experimental concentration. Horseradish
peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies were
obtained from Pierce (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). The polyclonal rabbit anti-rat antibodies p38MAPK (cat no.
9212) and p-p38MAPK (cat no. 9211) were supplied by Cell Signaling
Technology, Inc., Danvers, MA, USA. The mouse monoclonal anti-human
antibodies JNK (cat no. sc-7345), p-JNK (cat no. sc-293137), mouse
monoclonal anti-human antibody β-actin (cat no. sc-130300),
Protein-G-agarose beads and Alexa Fluor 488-conjugated anti-HA
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA).
Cell culture
The Lewis lung carcinoma (LLC) cell line was
obtained from the American Type Culture Collection (Manassas, VA,
USA) and maintained in RPMI-1640 (Sigma-Aldrich; Merck KGaA)
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.) and antibiotics (100 U/ml penicillin and 100
µg/ml streptomycin). The cell lines were maintained at 37°C in a
humidified air atmosphere with 5% CO2.
Inhibition assays
To investigate whether MAPK served a role in
DHA-induced apoptosis, LLC cells were treated with DHA in the
presence of specific inhibitor of a p38MAPK (SB202190, 10 µmol/l)
or JNK inhibitor (SP600125, 10 µmol/l) (Merck KGaA) for 2 h,
respectively, prior to further experiments.
MTT assay
To detect the viability of cells following
treatment, an MTT assay was performed. Briefly, cells were seeded
at a density of 1×104 cells/well in 96-well plates.
Subsequently, they were incubated with DHA (final concentrations,
5, 10, 20 and 40 µg/ml) or CBP (final concentration, 25 µg/ml) for
24 h. MTT (20 µl; Amresco, LLC) was added to each well and the
cells were incubated at 37°C for 4 h. Finally, 150 µl DMSO was
added. The optical density (OD) value was then evaluated at 570 nm
using a microplate reader (Thermo Fisher Scientific, Inc.). The
inhibition rate of cell proliferation was calculated as follows:
(OD value of the control group-OD value of the test group) ×100%/OD
value of the control group. The half-maximal inhibitory
concentration (IC50) for DHA and CBP in LLC cells,
defined as the dose of DHA and CBP that inhibits cell growth by
50%, was derived from the dose-response curve.
Clonogenic assay
LLC cells in the exponential phase of growth were
seeded in 6-well plates (1,000 cells per well) and allowed to
adhere. Following treatment with serial concentrations of DHA (5,
10, 20 and 40 µg/ml) and/or CBP (25 µg/ml) for 24 h, the cells were
cultured in fresh medium without drugs until visible colonies
formed. The cell colonies were then fixed with 4% paraformaldehyde
at room temperature for 30 min and stained with 0.5% crystal violet
staining solution at room temperature for 2 h. A total of 5 fields
of view per well were counted under an optical microscope at ×100
magnification.
Cell cycle and apoptosis analysis
Cell cycle and apoptosis were analyzed by flow
cytometry. Briefly, following incubation with DHA (20 µg/ml) or CBP
(25 µg/ml) for 24 h, 1×106 LLC cells were harvested with
trypsin and washed twice with ice-cold PBS. The cells were fixed
with 70% ethanol at 4°C overnight, then treated with propidium
iodide and RNase A at 37°C for 40 min. The samples were then
analyzed by flow cytometry (FACScalibur; BD Biosciences, San Jose,
CA, USA) for the proportion of cells in
G0/G1, S or G2/M cell cycle phase.
The sub-G1 population was scored from hypodiploid DNA
content. Data were analyzed using ModFit LT V3.2 software (Verity
Software House, Topsham, ME, USA). Apoptosis was determined using
the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide
(PI) apoptosis kit (BioVision, Inc., Milpitas, CA, USA), according
to the manufacturer's instructions. The early apoptotic (Annexin
V-FITC-positive) and necrotic/late apoptotic (Annexin
V-FITC-positive, PI-positive) cells were quantified as apoptotic
cells.
Western blotting
For the detection of various proteins (p38MAPK,
p-p38MAPK, JNK or p-JNK), cells were lysed in lysis buffer (Thermo
Fisher Scientific, Inc.) and clarified by centrifugation at 12,000
× g at 4°C for 10 min. The amount of protein was quantified with
the Bradford method. A 50-µg mass of cell proteins were loaded onto
10–15% SDS-PAGE gels for electrophoresis and the separated proteins
were subsequently electro-transferred onto polyvinylidene
membranes. The membranes were blocked with 5% skimmed milk in PBS
with Tween-20 at room temperature for 1h in a container and probed
with various primary antibodies (rabbit polyclonal anti-p38MAPK
antibodies, cat no. 9212; rabbit polyclonal anti-p-p38MAPK, cat no.
9211 at dilutions of 1:250; mouse-polyclonal anti-JNK, cat no.
sc-7345 at a dilution of 1:250; mouse monoclonal anti-p-JNK, cat
no. sc-293137 at a dilution of 1:500) overnight at 4°C, and the
membranes were washed with TBST and incubated with a secondary
rabbit anti-mouse IgG horseradish peroxidase (HRP) antibody (cat
no. sc-358917, 1:2,000; Santa Cruz Biotechnology, Inc.) or mouse
anti-rabbit IgG-HRP antibody (cat no. sc-2357, 1:2,000 dilution;
Santa Cruz Biotechnology, Inc.) diluted in 5% (w/v) dry non-fat
milk in TBST for 1 h at room temperature. Finally, the membranes
were washed and the bands were examined using an enhanced
chemiluminescence western blotting detection system (National
Institutes of Health, Bethesda, MD, USA) and the Bio-Rad ChemiDoc
XRS imager (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data are presented as the mean ± standard
deviation of ≥3 independent experiments. Statistical differences
between means were analyzed by an independent-samples t-test. Rates
were compared using the χ2 test. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using SPSS 17.0 (SPSS Inc.,
Chicago, IL, USA).
Results
Proliferation is inhibited in LLC
cells following treatment with DHA and CBP
An MTT assay was performed to determine the
viability of LLC cells. Following treatment with DHA or CBP, the
proliferation of LLC cells was inhibited with a dose-dependent
manner. The inhibitory rates of DHA or CBP at various
concentrations on LLC cells are described in Table I. The IC50 values for DHA
and CBP in LLC cells in our unpublished materials were 21.94 (18–25
µg/ml) and 26.98 µg/ml (22–30 µg/ml) respectively, thus 20 µg/ml
DHA and 25 µg/ml CBP were selected for use in the subsequent
experiments.
 | Table I.Effect of DHA or CBP on Lewis lung
carcinoma cell proliferation. |
Table I.
Effect of DHA or CBP on Lewis lung
carcinoma cell proliferation.
| Group | Absorbance
value | Inhibitory rate
(%) |
|---|
| Control | 0.54±0.06 | 0 |
| CBP (25 µg/ml) | 0.45±0.04 | 16.67 |
| DHA (20 µg/ml) | 0.48±0.03 | 11.11 |
| DHA (5 µg/ml) + CBP
(25 µg/ml) | 0.43±0.05 | 20.37 |
| DHA (10 µg/ml) +
CBP (25 µg/ml) | 0.34±0.03 | 37.04 |
| DHA (20 µg/ml) +
CBP (25 µg/ml) | 0.29±0.04 | 46.30 |
| DHA (40 µg/ml) +
CBP (25 µg/ml) | 0.19±0.05 | 64.81 |
To determine whether DHA enhanced the cytotoxicity
of CBP, LLC cells were co-treated with 25 µg/ml CBP and 5, 10, 20
or 40 µg/ml DHA for 24 h. Following the treatment, the inhibitory
rate of LLC cells increased to 64.81%, which was higher compared
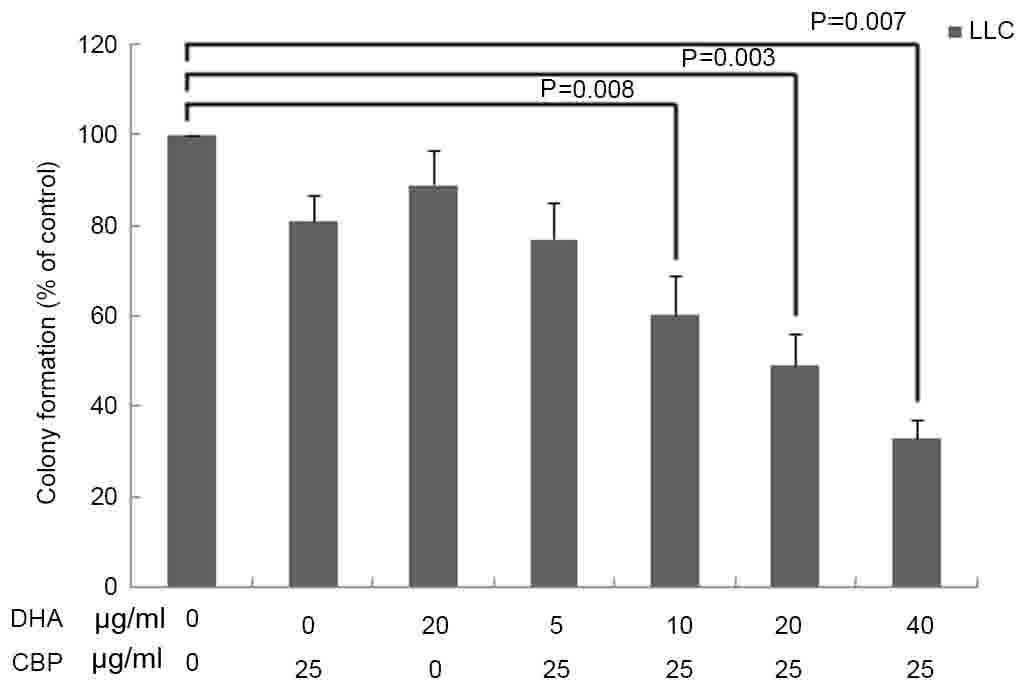
with DHA or CBP used alone (P<0.05; Table I). In addition, the clonogenic assay
revealed that DHA, CBP and DHA+CBP treatments significantly
decreased the colony numbers of LLC cells compared with the control
group (P<0.05; Fig. 1). There was
no significant difference between the group of DHA (20 µg/ml) or
CBP (25 µg/ml) used alone compared with the control group
(P>0.05); however, the colony formation of LLC cells was
significantly inhibited by various concentrations of DHA (5, 10, 20
and 40 µg/ml) combined with CBP (25 µg/ml; P<0.05).
Apoptosis is induced in LLC cells by
DHA combined with CBP
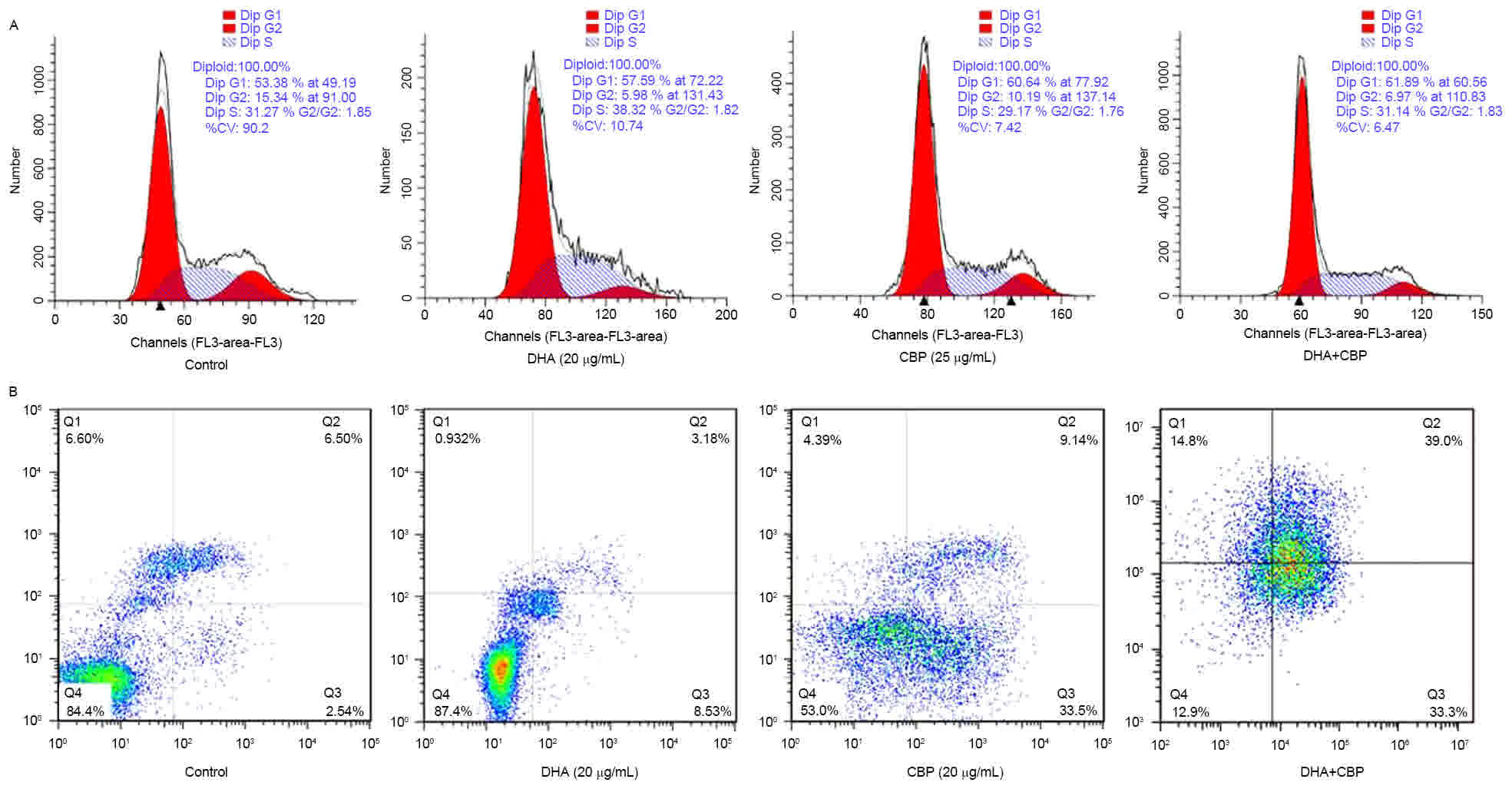
In order to investigate the mechanisms underlying
the reduction in cell proliferation induced by DHA (20 µg/ml)
combined with CBP (25 µg/ml), the cell cycle distribution and
apoptosis rates of LLC cells were assessed by analysis of flow
cytometry. As presented in Table II,
compared with the control group, the number of cells in the
G0/G1 cell cycle phase increased, whereas
cells in the S and G2/M cell cycle phases markedly
decreased (P<0.05; Fig. 2A), and
apoptotic peaks emerged in the DHA, CBP and the combination groups.
The apoptotic rates in the DHA, CBP and the combination groups were
7.45, 32.41 and 36.14%, respectively (Fig. 2B). The apoptotic rate of the
combination group was distinctly different compared with the
control and DHA groups (P<0.05; Table
II).
| Table II.Analysis for the effect of DHA or/and
CBP on cell cycle and apoptosis in Lewis lung carcinoma cells by
flow cytometry. |
Table II.
Analysis for the effect of DHA or/and
CBP on cell cycle and apoptosis in Lewis lung carcinoma cells by
flow cytometry.
| Group |
G0/G1 phase | S phase | G2/M
phase | Apoptotic rate
(%) |
|---|
| Control | 52.90±5.25 | 39.25±4.32 | 5.85±1.28 | 2.95±0.84 |
| DHA (20 µg/ml) | 56.65±6.34 |
38.46±2.10a |
4.89±1.15b |
7.45±3.19a |
| CBP (25 µg/ml) |
61.94±3.37a | 34.08±3.54 | 4.21±1.12 |
32.41±3.73b |
| DHA (20 µg/ml) +
CBP (25 µg/ml) |
65.07±7.40a |
31.79±6.27a |
3.64±0.88a |
36.14±4.98b |
DHA sensitizes LLC cells to
CBP-induced apoptosis via p38MAPK activation
To investigate a potential role for MAPKs in the
therapeutic effect of DHA combined with CBP in LLC cells, the
levels of p-p38MAPK and p-JNK in each treatment group were examined
by western blot analysis. LLC cells exhibited constitutive
p-p38MAPK activity following mono-DHA or mono-CBP treatment.
Furthermore, compared with the control, DHA or CBP groups,
p-p38MAPK was upregulated in the combined treatment group.
Conversely, induction of JNK phosphorylation was not observed in
all four groups. In addition, no p-p38MAPK signal was detected in
the SB202190 group (Fig. 3). Among
the four groups, there was no difference in p-JNK expression level.
These results suggested that in LLC cells, DHA with CBP
co-treatment in vitro can abrogate mono-DHA or
mono-CBP-induced p38MAPK activity, but not JNK phosphorylation,
which may be associated with the enhanced rate of apoptosis
observed.
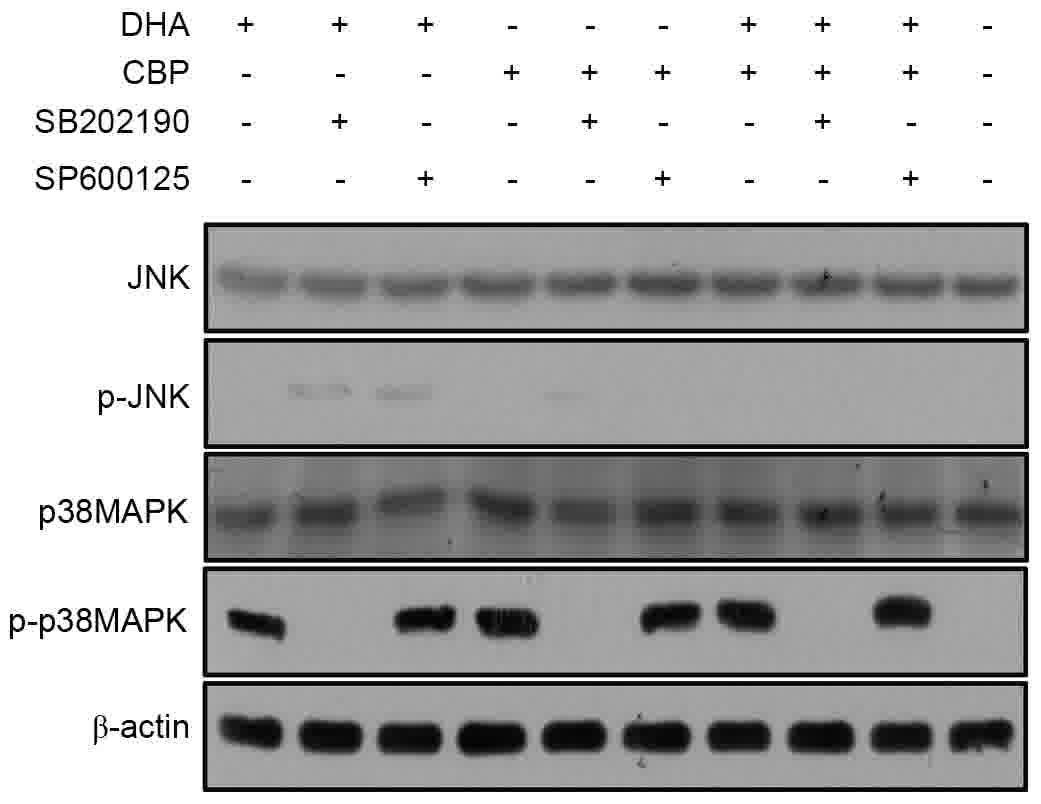 | Figure 3.Potential molecular mechanisms
underlying the DHA sensitization of LLC cells to CBP-induced
apoptosis. The expression levels of p38MAPK-associated proteins
were analyzed by western blotting in LLC cells untreated or treated
with DHA (20 µg/ml), CBP (25 µg/ml), a specific inhibitor of
p38MAPK (SB202190, 10 µmol/l) and JNK inhibitor (SP600125, 10
µmol/l). DHA, dihydroartemisinin; LLC, Lewis lung carcinoma; CBP,
carboplatin; MAPK, mitogen-activated protein kinase; JNK, c-Jun
N-terminal kinase; p, phosphorylated. |
Discussion
The results of the present study revealed that DHA
inhibits cell proliferation and induces apoptosis in LLC cells in a
concentration-dependent manner. Artemisinin, isolated from the
traditional Chinese herb Artemisia annua is an effective
novel antimalarial drug with low toxicity. The derivatives of
artemisinin include DHA, artemether, artemotil and artesunate.
Among them, DHA has the highest water-solubility and antimalarial
effect (17). Besides the
antimalarial effect, DHA can selectively induce the proliferation
inhibition and apoptosis of certain types of tumor cell (6,18).
Although these previous studies have observed the antitumor effect
of artemisinin and its derivatives, the underlying mechanism has
not been fully elucidated. The possible mechanisms underlying
DHA-induced apoptosis may be as follows: i) The reduction of the
Bcl-2/Bcl-associated X, Bcl-2/BH3-interacting domain death agonist
expression ratio; ii) reducing the expression level of survivin;
iii) increasing the intracellular Ca2+ concentration of
tumor cells; iv) activating the death receptor- and
mitochondrion-mediated caspase-dependent signaling pathways
(6,19–22).
Similar to a previous report, the results of the present study
revealed that the proliferation of LLC cells was inhibited by DHA
by the induction of apoptosis (23).
In addition to this result, our previous studies demonstrated that
DHA is not only able to inhibit tumor growth, lymphangiogenesis,
and the nodal and lung metastasis of LLC transplanted tumor by
decreasing VEGF-C expression, but also capable of apoptosis
induction and the inhibition of the migration and formation of
tube-like structures in lymphatic endothelial cells in vitro
(16,23). All these results imply that DHA may be
a promising agent for controlling lung adenocarcinoma growth and
metastasis in vitro and in vivo.
The results of the present study revealed that DHA
increases the effectiveness of CBP by increasing the rate of
apoptosis. Cisplatin, CBP and oxaliplatin are the mainstays of lung
cancer chemotherapy; however, the acquired drug resistance of tumor
cells and the cumulative side effects of these agents present
serious clinical obstacles (24).
Resistance and evasion of apoptosis are critical factors that
contribute to carcinogenesis and drug resistance (25). Therefore, it is necessary to
investigate novel agents and mechanisms of action to enhance the
effects of therapy or sensitize cancer cells to apoptosis. The
present study focused on the anticancer activities of DHA combined
with CBP and revealed that DHA enhanced the inhibitory effects of
CBP on LLC cells. The combined treatment of DHA and CBP induced an
increase in apoptosis, compared with that produced by either
compound alone. These results indicated that DHA may inhibit the
growth of lung adenocarcinoma cells by increasing their apoptosis,
particularly in combination with CBP. In a previous study, it was
demonstrated that DHA markedly inhibited the in vitro and
in vivo growth of ovarian carcinoma alone or in combination
with CBP, and that it enhanced the therapeutic effect of CBP by
increasing apoptosis (6).
Furthermore, Feng et al (10)
revealed that DHA potentiates the anticancer effect of cisplatin
via the inhibition of mTOR in cisplatin-resistant ovarian cancer
cells. Zhao et al (9)
demonstrated the synergistic effect of a treatment combining
gemcitabine with DHA in inducing the apoptosis of A549 cells via
the Bcl2 antagonist/killer 1-mediated intrinsic and the
Fas-caspase-8-mediated extrinsic signaling pathways. Together with
the results of the present study, it has been indicated that DHA
may be an effective anticancer drug, particularly in combination
with conventional chemotherapeutic agents.
Furthermore, the present study demonstrated that
p38MAPK activation may partially mediate the apoptosis of LLC cells
induced by DHA combined with CBP. In 1994, Han et al
(26,27) cloned p38MAPK from a murine liver cDNA
library and detected the expression of p38MAPK mRNA in murine
macrophages, T cells and B cells. p38MAPK is usually activated by
ultraviolet radiation, hyperosmolarity, arsenate compounds, heat
shock, H2O2, cytokines [including
interleukin-1 and tumor necrosis factor-α (TNF-α)] and other
physiological stresses. Following translocation from the nucleus to
the cytoplasm, it initiates the activity of corresponding
transcription factors (28). The role
of p38MAPK in cell survival and apoptosis has been extensively
examined; however, the results of these studies are inconsistent.
For example, p38MAPK activation was previously demonstrated to
induce apoptosis in non-tumor cells, including nerve cells and
fetal brown adipocytes (29,30) and tumor cells (31,32).
However, other studies demonstrated that p38MAPK did not affect
apoptosis (33) or inhibited
apoptosis (34–36). In our previous study, p38MAPK
phosphorylation was detected in TNF-α-treated rat glioma C6 cells
(37). In various types of cell line,
DHA induced apoptosis by activation of signaling pathways,
including the Wnt/β-catenin, NF-κB and caspase signaling pathways.
Lu et al (7) revealed that
DHA-induced apoptosis is dependent on iron and p38MAPK activation,
but not reactive oxygen species, JNK or ERK activation in HL-60
cells. The present study demonstrated a p-p38MAPK signal was
associated with apoptosis induced by DHA combined with CBP in LLC
cells. Following treatment with SB202190 and DHA simultaneously, no
p38MAPK signal was detected by western blotting. The simultaneous
treatment of SP600125 and DHA did not affect apoptosis. These
results suggested that p38MAPK, and not JNK, serves a role in
apoptosis induced by DHA and CBP in LLC cells.
In conclusion, the present study demonstrated that
DHA potently induces growth inhibition and apoptosis in lung
adenocarcinoma cells and enhances the therapeutic effect of CBP
in vitro by increasing apoptosis, which may be mediated by
the activation of p38MAPK signaling. Together with previously
reported findings, these results will help to improve the
understanding of the mechanisms underlying DHA as a novel
anticancer agent. Furthermore, the present study provides a basis
for future clinical investigations of DHA in patients with lung
cancer, used alone or in combination with conventional anticancer
drugs. In vivo experiments are also required to verify the
conclusion of the present study.
Acknowledgements
The authors thank the Chinese National Natural
Science Foundation (grant no. 81301631) and a Special Financial
Grant from the China Postdoctoral Science Foundation (grant no.
2014T70977) for the financial support provided.
References
|
1
|
Tiseo M, Bartolotti M, Gelsomino F and
Ardizzoni A: First-line treatment in advanced non-small-cell lung
cancer: The emerging role of the histologic subtype. Expert Rev
Anticancer Ther. 9:425–435. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hou S, Han X and Ji H: Squamous transition
of lung adenocarcinoma and drug resistance. Trends Cancer.
2:463–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loi M, Roche N and Alifano M: Chemotherapy
regimens for non-small cell lung cancer. Minerva Chir. 64:629–641.
2009.PubMed/NCBI
|
|
4
|
Sangodkar J, Katz S, Melville H and Narla
G: Lung adenocarcinoma: Lessons in translation from bench to
bedside. Mt Sinai J Med. 77:597–605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tran TH, Dolecek C, Pham PM, Nguyen TD,
Nguyen TT, Le HT, Dong TH, Tran TT, Stepniewska K, White NJ and
Farrar J: Dihydroartemisinin-piperaquine against
multidrug-resistant Plasmodium falciparum malaria in Vietnam:
Randomised clinical trial. Lancet. 363:18–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen T, Li M, Zhang R and Wang H:
Dihydroartemisinin induces apoptosis and sensitizes human ovarian
cancer cells to carboplatin therapy. J Cell Mol Med. 13:1358–1370.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu JJ, Meng LH, Cai YJ, Chen Q, Tong LJ,
Lin LP and Ding J: Dihydroartemisinin induces apoptosis in HL-60
leukemia cells dependent of iron and p38 mitogen-activated protein
kinase activation but independent of reactive oxygen species.
Cancer Biol Ther. 7:1017–1023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y, Wang W, Xu J, Li L, Dong Q, Shi Q,
Zuo G, Zhou L, Weng Y, Tang M, et al: Dihydroartemisinin inhibits
tumor growth of human osteosarcoma cells by suppressing
Wnt/β-catenin signaling. Oncol Rep. 30:1723–1730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao C, Gao W and Chen T: Synergistic
induction of apoptosis in A549 cells by dihydroartemisinin and
gemcitabine. Apoptosis. 19:668–681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng X, Li L, Jiang H, Jiang K, Jin Y and
Zheng J: Dihydroartemisinin potentiates the anticancer effect of
cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer
cells: Involvement of apoptosis and autophagy. Biochem Biophys Res
Commun. 444:376–381. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown MD and Sacks DB: Protein scaffolds
in MAP kinase signalling. Cell Signal. 21:462–469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rana A, Rana B, Mishra R, Sondarva G,
Rangasamy V, Das S, Viswakarma N and Kanthasamy A: Mixed lineage
kinase-c-Jun N-yerminal kinase axis: A potential therapeutic target
in cancer. Genes Cancer. 4:334–341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fischer S, Koeberle SC and Laufer SA: p38
mitogen-activated protein kinase inhibitors, a patent review
(2005–2011). Expert Opin Ther Pat. 21:1843–1866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zechner D, Craig R, Hanford DS, McDonough
PM, Sabbadini RA and Glembotski CC: MKK6 activates myocardial cell
NF-kappaB and inhibits apoptosis in a p38 mitogen-activated protein
kinase-dependent manner. J Biol Chem. 273:8232–8239. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Remacle-Bonnet MM, Garrouste FL, Heller S,
André F, Marvaldi JL and Pommier GJ: Insulin-like growth factor-I
protects colon cancer cells from death factor-induced apoptosis by
potentiating tumor necrosis factor alpha-induced mitogen-activated
protein kinase and nuclear factor kappaB signaling pathways. Cancer
Res. 60:2007–2017. 2000.PubMed/NCBI
|
|
16
|
Wang J, Guo Y, Zhang BC, Chen ZT and Gao
JF: Induction of apoptosis and inhibition of cell migration and
tube-like formation by dihydroartemisinin in murine lymphatic
endothelial cells. Pharmacology. 80:207–218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Naing C, Mak JW, Aung K and Wong JY:
Efficacy and safety of dihydroartemisinin-piperaquine for treatment
of uncomplicated Plasmodium falciparum malaria in endemic
countries: Meta-analysis of randomised controlled studies. Trans R
Soc Trop Med Hyg. 107:65–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Du XX, Li YJ, Wu CL, Zhou JH, Han Y, Sui
H, Wei XL, Liu L, Huang P, Yuan HH, et al: Initiation of apoptosis,
cell cycle arrest and autophagy of esophageal cancer cells by
dihydroartemisinin. Biomed Pharmacother. 67:417–424. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu YY, Chen TS, Wang XP, Qu JL and Chen M:
The JNK inhibitor SP600125 enhances dihydroartemisinin-induced
apoptosis by accelerating Bax translocation into mitochondria in
human lung adenocarcinoma cells. FEBS Lett. 584:4019–4026. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mao H, Gu H, Qu X, Sun J, Song B, Gao W,
Liu J and Shao Q: Involvement of the mitochondrial pathway and
Bim/Bcl-2 balance in dihydroartemisinin-induced apoptosis in human
breast cancer in vitro. Int J Mol Med. 31:213–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mu D, Chen W, Yu B, Zhang C, Zhang Y and
Qi H: Calcium and survivin are involved in the induction of
apoptosis by dihydroartemisinin in human lung cancer SPC-A-1 cells.
Methods Find Exp Clin Pharmacol. 29:33–38. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mu D, Zhang W, Chu D, Liu T, Xie Y, Fu E
and Jin F: The role of calcium, P38 MAPK in
dihydroartemisinin-induced apoptosis of lung cancer PC-14 cells.
Cancer Chemother Pharmacol. 61:639–645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang J, Zhang BC, Guo Y, Chen ZT and Gao
JF: Inhibition of lymphangiogenesis, nodal and lung metastasis by
dihy-droartemisinin in mice bearing Lewis lung carcinoma. J Med
Coll PLA. 22:272–278. 2007. View Article : Google Scholar
|
|
24
|
Markman JL, Rekechenetskiy A, Holler E and
Ljubimova JY: Nanomedicine therapeutic approaches to overcome
cancer drug resistance. Adv Drug Deliv Rev. 65:1866–1879. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu W and Kavanagh JJ: Anticancer therapy
targeting the apoptotic pathway. Lancet Oncol. 4:721–729. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han J, Lee JD, Tobias PS and Ulevitch RJ:
Endotoxin induces rapid protein tyrosine phosphorylation in 70Z/3
cells expressing CD14. J Biol Chem. 268:25009–25014.
1993.PubMed/NCBI
|
|
27
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
García-Cano J, Roche O, Cimas FJ,
Pascual-Serra R, Ortega-Muelas M, Fernández-Aroca DM and
Sánchez-Prieto R: p38MAPK and Chemotherapy: We always need to hear
both sides of the story. Front Cell Dev Biol. 4:692016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Valladares A, Alvarez AM, Ventura JJ,
Roncero C, Benito M and Porras A: p38 mitogen-activated protein
kinase mediates tumor necrosis factor-alpha-induced apoptosis in
rat fetal brown adipocytes. Endocrinology. 141:4383–4395. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ishii A, Furusho M and Bansal R: Sustained
activation of ERK1/2 MAPK in oligodendrocytes and schwann cells
enhances myelin growth and stimulates oligodendrocyte progenitor
expansion. J Neurosci. 33:175–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park MT, Choi JA, Kim MJ, Um HD, Bae S,
Kang CM, Cho CK, Kang S, Chung HY, Lee YS and Lee SJ: Suppression
of extracellular signal-related kinase and activation of p38 MAPK
are two critical events leading to caspase-8- and
mitochondria-mediated cell death in phytosphingosine-treated human
cancer cells. J Biol Chem. 278:50624–50634. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ozaki I, Tani E, Ikemoto H, Kitagawa H and
Fujikawa H: Activation of stress-activated protein kinase/c-Jun
NH2-terminal kinase and p38 kinase in calphostin C-induced
apoptosis requires caspase-3-like proteases but is dispensable for
cell death. J Biol Chem. 274:5310–5317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kanagasabai R, Karthikeyan K, Vedam K,
Qien W, Zhu Q and Ilangovan G: Hsp27 protects adenocarcinoma cells
from UV-induced apoptosis by Akt and p21-dependent pathways of
survival. Mol Cancer Res. 8:1399–1412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park JG, Yuk Y, Rhim H, Yi SY and Yoo YS:
Role of p38 MAPK in the regulation of apoptosis signaling induced
by TNF-alpha in differentiated PC12 cells. J Biochem Mol Biol.
35:267–272. 2002.PubMed/NCBI
|
|
36
|
Petersen C, Svechnikov K, Fröysa B and
Söder O: The p38 MAPK pathway mediates interleukin-1-induced
Sertoli cell proliferation. Cytokine. 32:51–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang BC, Li Q, Ye J, et al: Role of
p38MAPK in mediating TNF-α-induced apoptosis of rat glioma cell
line C6. J Med Coll PLA. 18:308–311. 2003.
|