Introduction
The α-thalassemia/mental retardation syndrome
X-linked (ATRX) protein encoded by the ATRX gene (NM_000489:
c.6130C>T, p.Leu2044Phe) belongs to the SWI/SNF DNA helicase
chromatin remodeling protein family, and contains a plant
homeodomain (PHD)-like domain and an ATPase/helicase domain. It was
first reported to undergo alterations during cell cycle-dependent
phosphorylation, which is closely associated with its nuclear
matrix at interphase, condensed chromatin, particularly at the M
phase onset, and chromosomal segregation in mitosis (1).
Mutations in the ATRX gene are associated with a
wide and clinically heterogeneous spectrum of X-linked mental
retardation (XLMR) syndrome, frequently accompanied by
α-thalassemia (ATRX) syndrome, a difficultly diagnosed genetic
disorder comprising dysmorphic features, microcephaly, severe
intellectual disability, cryptorchidism and mild anemia (1). To date, 66 mutations have been reported,
primarily located in the PHD-like domain, which is regarded as the
major mutation hot-spot (2). These
mutations have been demonstrated to induce distinct variation in
the pattern of DNA methylation, which may provide a connection
between chromatin remodeling and gene expression in dynamic
processes. It has been reported that multiple alternatively spliced
transcript variants encode diverse isoforms. Several lines of
evidence have indicated the effect of ATRX in the regulation of
chromatin remodeling, and protein and DNA interactions (3,4). A total
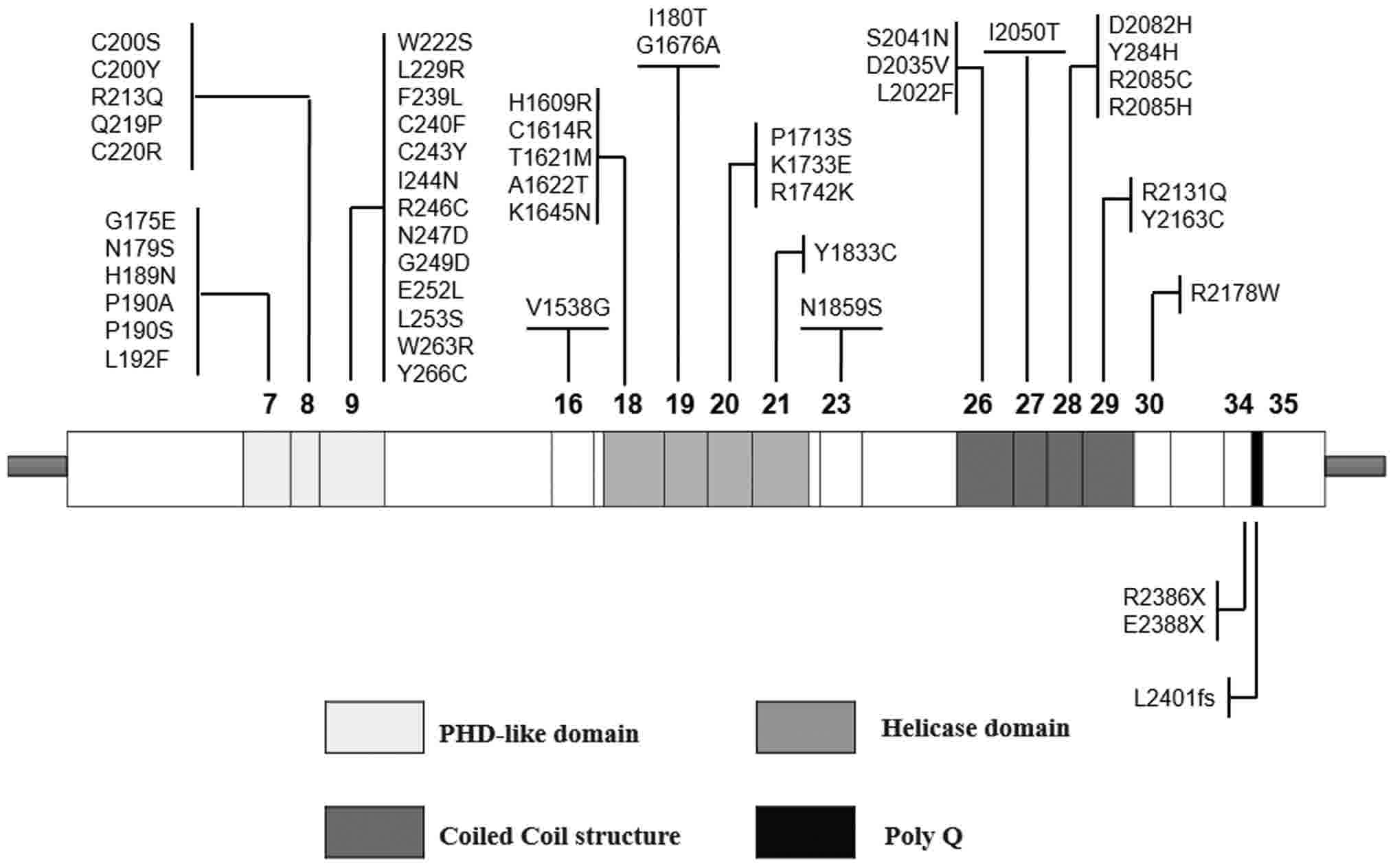
of two primary functional domains have been predicted in the light
of protein structural motifs (Fig.
1): On the one hand, a PHD-like domain, encoded by exons 7, 8
and 9 (5# part); and on the other hand, a helicase domain located
at the C-terminus, encoded by exons 18, 19, 20 and 21. These two
domains have been reported to serve a role as chromatin remodelers
(5). A third domain, predicted as a
coiled coil structure, has been demonstrated to interact
particularly with EZH2, a protein belonging to the polycomb
multigenic family that is involved in histone methylation and
deacetylation (6). Mutations in the
ATRX gene have been identified in recent whole genome and/or whole
exome genomic studies in sarcomas, particularly osteosarcoma
(7–10).
Telomeres, the short, non-protein coding repeated
hexameric TTAGGG sequences at each end of a chromosome, are
involved in the control of genome integrity and chromosomal
stability, and the development of cancer cells (11). During chromosome replication in
somatic cells, telomeres shorten at each duplication, as the
synthesis of Okazaki fragments requires RNA primers to attach ahead
on the lagging strand so that the gap between the final RNA primer
and the end of the chromosome cannot be completed, and 3′ overhangs
occur (12). Cells may be alive
without division until meeting with a barrier called ‘crisis’, at
which a majority of them will undergo programmed cell death.
However, pre-malignant cells may cross the crisis barrier by
altering the telomere length pathway, leading to cancer initiation
by inducing chromosomal instability (13,14).
Telomerase activation and the alternative lengthening of telomeres
(ALT) pathway have been proposed as the main telomere maintenance
mechanisms underlying the eternal proliferation capacity, and
blocking genetic disorders induced by telomere dysfunction. The
length of telomeres is primarily maintained by telomerase
activation or, in 10–20% of cases, particularly gliomas and
sarcomas, by the ALT pathway (15).
The activation of ALT reduces tumor relapse in mouse models,
although it remains to be elucidated how the molecular mechanisms
control the activation of ALT (16,17).
Cancer cells, which resist cellular replicative
senescence, become immortal cells by activating telomerase or the
ALT pathway (18,19). ALT exists in 5–15% of all types of
human cancer and it is common in specific types of cancer,
including osteosarcoma and glioblastoma (20), suggesting that ALT contributes to
genomic instability. ATRX is part of a multiprotein complex that
regulates telomere maintenance; the loss of the protein ATRX is
associated with ALT in cancer, which is frequently mutated in
osteosarcoma patients.
Sarcomas are a rare and heterogeneous group of
tumors with an earlier mean onset age than other types of
epithelial cancer, accounting for 20% of childhood cancer and 10%
of adolescent and young adult cancer, in addition to various
clinical manifestations, differentiations, histopathological types,
molecular pathogeneses and prognoses. However, the genetic basis
for musculoskeletal sarcoma remains largely unknown (21). Epidemiological studies have identified
a firm genetic component of sarcomas and a number of familial
cancer syndromes have been reported, which is a characteristic of
sarcomas (22). Recently, a published
genetic-association study concerning 1,162 patients with sarcoma
from four different clinical cohort studies demonstrated that 55%
of these patients had underlying monogenic or polygenic variations.
Except TP53, BRCA2, ATM and ataxia telangiectasia and Rad3-related
kinase (ATR), a surprisingly excess of functional mutations were
identified in ERCC2, which is a transcription factor required for
nucleotide excision repair. The greater the accumulated burden of
multiple pathogenic variants, the earlier the age at which the
cancer was diagnosed. To a certain degree, these data demonstrated
that sarcoma has a genetic etiology (23). The musculoskeletal sarcoma National
Comprehensive Cancer Network guidelines (www.nccn.org) have recommended genetic testing and
consulting for patients with a clinical and/or family history of
genetic cancer syndromes associated with a high risk of the
development of musculoskeletal sarcoma (21,24).
Cytogenetic variants form one of the earliest and
most impactful factors in the classification of musculoskeletal
sarcoma. Since the first discovery of the t(11;22)(q24;q12) variant
in Ewing sarcoma, a cytogenetic catalog of archives of chromosomal
alterations specifying distinct mesenchymal tumor entities has been
created (cgap.nci.nih.gov/Chromosomes/Mitelman) (25–27).
According to cytogenetic variant evidence, sarcomas may be broadly
distributed into two groups: i) Sarcomas with simple karyotypes,
frequently involving specific genetic alterations, including
specific oncogenic mutations and reciprocal chromosomal
translocations; and ii) sarcomas with complex unbalanced
karyotypes, usually involving non-specific genetic abnormalities,
including chromosomal number alterations, translocations, and large
amplifications or deletions that may be illustrated [karyotyping,
fluorescence in situ hybridization (FISH)]. The present
review summarizes recent whole genome and/or whole exome genomic
studies, in addition to ATRX immunohistochemistry and ALT FISH, in
sarcomas of various subtypes and in diverse sites, including
osteosarcoma, leiomyosarcoma, liposarcoma, angiosarcoma and
chondrosarcoma. Additionally, the present review involves a few
studies associated with the molecular mechanisms underlying the
loss of ATRX controlling the activation of ALT in sarcomas. Testing
for the loss of ATRX and ALT in sarcomas may facilitate the
identification of novel targets for the treatment of aggressive
sarcomas.
Loss of ATRX in sarcomas
Mutations in the ATRX gene have been discovered in a
series of sarcomas, including osteosarcoma, leiomyosarcoma and
chondrosarcoma. The first report of diagnosed osteosarcoma in two
brothers with ATRX syndrome suggested a potential increased risk of
cancer in patients with this disorder (28). A 22-year-old Caucasian man was
reported in 2017 to exhibit a previously unidentified mutation in
ATRX associated with osteosarcoma (29). All these data suggested that patients
with ATRX syndrome may be at a potential increased risk of
developing osteosarcoma, although the molecular mechanism of ATRX
loss-of-function mutations in osteosarcoma remains unclear at
present (30,31).
Long-term studies by Liau et al have provided
evidence of the frequency of ATRX expression loss in 519 sarcomas
samples (30). Those studies
identified 85 tumors in those samples (85/519, 16%) arising from
ATRX loss, consisting of 83 sarcomas with complex cytogenetics and
two sarcomas with fusion genes; the details of the sarcoma types
are presented in Table I. The loss of
ATRX expression with complex cytogenetics was significantly more
frequent in sarcoma compared with fusion-associated sarcoma
(30). The previous studies of Liau
et al reported additional details. For example, the loss of
ATRX in leiomyosarcoma was correlated with cell modalism, poor
differentiation, necrosis, estrogen receptor expression, lower
patient age and smaller tumor size. There was no significant
association with tumor site, compared among uterine and
non-uterine, non-retroperitoneal or non-intra-abdominal sites
(32).
 | Table I.The proportions of ATRX loss and ALT
status in sarcomas with complex cytogenetics and fusion genes. |
Table I.
The proportions of ATRX loss and ALT
status in sarcomas with complex cytogenetics and fusion genes.
|
|
|
| ALT (+) |
|---|
|
|
|
|
|
|---|
| Sarcomas with
complex cytogenetics | N | ATRX loss (%) | N (%) | ATRX loss (%) |
|---|
| Leiomyosarcoma | 92 | 30 (33) | 51/86 (59) | 28/51 (55) |
| Angiosarcoma | 88 | 16 (18) | 17/70 (24) | 15/17 (88) |
| Dedifferentiated
liposarcoma | 52 | 13 (25) | 14/46 (30) | 13/14 (93) |
| Undifferentiated
pleomorphic sarcoma | 35 | 12 (34) | 22/34 (65) | 12/22 (55) |
|
Myxofibrosarcoma | 27 | 1 (4) | 19/25 (76) | 1/19 (5) |
|
Radiation-associated sarcoma | 20 | 0 (0) | 3/15 (20) | 0/3 (0) |
| Osteosarcoma | 18 | 4 (22) | N/A | N/A |
| Malignant
peripheral nerve sheath tumor | 17 | 1 (6) | 3/14 (21) | 1/3 (33) |
| Embryonal
rhabdomyosarcoma | 9 | 1 (11) | 1/8 (13) | 1/1 (100) |
| Pleomorphic
liposarcoma | 11 | 5 (45) | 8/10 (80) | 5/8 (63) |
| Sarcomas with
fusion genes |
|
|
|
|
|
Epithelioid
hemangioendothelioma | 11 | 1 (9) | 1/7 (14) | 1/1 (100) |
|
Gastrointestinal stromal
tumor | 23 | 1 (4) | 1/16 (6) | 1/1 (100) |
Furthermore, an osteosarcoma discovery cohort
identified the somatic mutation landscapes of 34 primary and
metastatic pediatric osteosarcomas via whole-genome sequencing,
reported Chen et al (33). A
total of five osteosarcomas exhibited ATRX point mutations, and
five exhibited structural variations or focal deletions impacting
the ATRX gene coding regions. In addition, upon analysis with
immunohistochemistry (IHC), 69% (13/19) of the tumors were
ATRX-positive.
A series of studies have used a next generation
sequencing (NGS) panel to examine common cancer-associated genetic
alterations. Lee et al (34)
reported that 25 leiomyosarcomas occurring in multiple sites were
associated with the frequent gene alterations in the following
proportions: TP53 (36%), ATM and ATRX (16%), and EGFR and RB1
(12%). Furthermore, Mäkinen et al (35) reported that 43 genes exhibited
mutations in 19 uterine leiomyosarcoma (ULMS) tumors, including the
following frequently mutant genes: TP53 (6/19, 33%), ATRX (5/19,
26%), and mediator complex subunit 12 (MED12; 4/19, 21%),
demonstrated by whole-exome sequencing. However, all the ATRX
alterations were either frameshift or nonsense mutations, opposite
to TP53 and MED12 which are all the identified alterations. In
addition, ATRX protein expression levels were analyzed by IHC in a
total of 44 ULMS tumors, indicating markedly reduced ATRX
expression in 23 tumors (23/44, 52%). Yang et al (36) analyzed the genetic alterations in 44
cancer-associated genes via NGS in 54 leiomyosarcomas. The most
frequently mutated genes were identified, including TP53 mutations
in 19 of the leiomyosarcomas (19/54, 35%) and ATRX mutations in 9
of the tumors (9/54, 17%). Notably, the ATRX mutations were
associated with low-differentiation or undifferentiated
leiomyosarcomas (P=0.028), and the existence of tumor necrosis
(P=0.015). In addition, leiomyosarcoma patients with ATRX mutations
exhibited a poorer prognosis compared with ATRX-wild-type patients,
as demonstrated by Kaplan-Meier survival analysis.
Hartmann et al (37) reported that isocitrate dehydrogenase
(IDH)-1 or −2 mutations have been detected in gliomas (60–80%) and
cholangiocarcinomas (7–28%). Notably, the loss of ATRX occurred in
the IDH-mutant gliomas. However, without relevance in
cholangiocarcinomas was evaluated by IHC 2 of 36 (5.6%) and usual
chondrosarcomas showed complete negative of ATRX (38).
Loss of ATRX is associated with the ALT
pathway in sarcoma
Previous studies have suggested that the ALT pathway
exists in the majority of cancer types. Previous studies analyzed
the frequency of ALT correlating with the loss of ATRX in soft
tissue sarcomas. Liau et al characterized the significant
association between the ALT pathway and ATRX status in 519 sarcoma
samples (P<0.001) (30). It was
reported that the association was relatively similar in
undifferentiated pleomorphic sarcoma, leiomyosarcoma and
pleomorphic liposarcoma (Table I).
Notably, over 50% tumors are identified to be ALT-positive in which
ATRX deficient were observed about 50%. By contrast, only a few
malignant peripheral nerve sheath tumors and embryonal
rhabdomyosarcomas were ALT-positive or exhibited a loss of ATRX (or
both), which indicated that ALT status and the loss of ATRX were
likely to be associated with katagenesis and dedifferentiation
variation (30). Additionally, these
previous studies included certain essential information. For
example, deletion of ATRX was observed in 33% of the
leiomyosarcomas, and all of them were ALT-positive except for two
ALT-negative tumors out of all the ATRX-deletion tumors. The ALT
phenotype and ATRX deletion are both associated with cell
morphology, tumor necrosis and differentiation. In addition,
ALT-positivity and younger patient age were independent
poor-prognostic risk factors in the multivariate analysis (32). Furthermore, previous studies have
reported that ALT was detected in 20–30% of liposarcomas (20,39).
Notably, Lee et al (34)
provided evidence of a perfect correlation between the loss of
either ATRX or death domain-associated protein and the ALT status
in 46 dedifferentiated liposarcoma samples (DDLS) via
telomere-specific FISH and IHC, and the ALT status significantly
indicated poor clinical outcomes, including poor overall survival
and short progression-free survival (40,41).
Notably, all the well-differentiated (WD) liposarcomas were
ALT-negative (40,42).
Yang et al (36) reported that all ATRX-mutated
leiomyosarcoma samples exhibited the ALT phenotype (P=0.008).
Although the ATRX mutation was not correlated with the loss of ATRX
expression, all the tumors with nonsense and frame shift mutations
were ATRX-negative, according to IHC (36). Likewise, the loss of ATRX expression
was confirmed in all uterine leiomyosarcoma samples. The loss of
ATRX expression has been associated with ALT, as analyzed by
telomere-specific FISH (35).
Similarly, myxofibrosarcoma was frequently positive
for the ALT pathway (43). Recently,
a KRAS-mutated KIT/PDGFRA-wild-type gastrointestinal stromal tumor
was revealed to exhibit ATRX mutations (44). This tumor also exhibited anaplastic
histological features and behaved aggressively, which indicates
that the loss of ATRX expression and/or the acquisition of the ALT
pathway may be associated with dedifferentiation and/or
transdifferentiation of gastrointestinal stromal tumors, indicating
a poor prognosis.
Similarly, Chen et al (33) demonstrated that 85% (12/14) of
pediatric osteosarcomas used the ALT mechanism to maintain their
telomeres detected by the WGS data, telomeres quantitative
polymerase chain reaction and telomere-specific FISH. Furthermore,
58% of 12 ALT-positive osteosarcomas exhibited ATRX mutations.
By contrast, a previous study by Heaphy et al
(20) reported that two out of six
epithelioid sarcomas were ALT-positive, and none of these tumors
were revealed to be ALT and ATRX deficient, implying that marked
genetic alterations may have promoted tumor formation instead of
the ALT mechanism in this series of tumors. Consequently, these
were more similar to the fusion-associated sarcomas and
gastrointestinal stromal tumors in a study undertaken by Liau et
al (30).
Molecular mechanisms of the loss of ATRX
controlling the activation of ALT in sarcomas
As mentioned earlier, the loss of ATRX expression is
associated with ALT, but the molecular mechanisms controlling the
activation of ALT remain unclear. Previous studies into the
molecular mechanisms involved in sarcomas have been rare. Napier
et al (45) primarily reported
the direct and functional evidence that ATRX loss accelerates ALT.
In the aforementioned study, the knockdown of ATRX in pre-crisis
simian virus 40-transformed fibroblasts led to activated ALT or a
significant decrease in the time prior to ALT activation.
Furthermore, transiently upregulated ATRX expression was able to
suppress ALT activity in ALT-positive cells.
Additionally, Flynn et al (46) reported that the loss of ATRX leads to
the formation of a recombinogenic nucleoprotein complex consisting
of ATR, replication protein A (RPA), and telomeric non-coding RNA.
The inhibition of ATR, an essential recombination regulator
recruited by RPA, disrupts ALT, and causes chromosome disruption
and apoptosis in ALT-positive cells. Notably, cell death induced by
ATR inhibitors is highly selective for ALT-positive cancer cells,
proposing that ATR inhibitors may be used to treat ALT-positive
cancer. At present, there have been two highly selective and
effective ATR inhibitors, AZD6738 and VX-970, which have reached
phase I clinical trials. These drugs are being used either as a
monotherapy or combined with a series of genotoxic chemotherapies
for the treatment of ALT-positive solid tumors and refractory
cancer (47).
Furthermore, a series of studies have revealed the
possible mechanism underlying the manner in which ATRX loss leads
to sarcoma formation. McFarlane and Preston (48) proved that the presence of ATRX
inhibited the expression of human cytomegalovirus immediate early
genes instead of herpes simplex virus type 1 in the osteosarcoma
U2OS cell line, and the specific virus phosphoprotein PP71 was able
to withstand the inhibition due to the disassembly of ATRX with
nuclear domain 10 in human fetal foreskin fibroblast 2 cells.
Kovatcheva et al (49)
revealed that the turnover of mouse double minute 2 homolog (MDM2)
and the expression of ATRX were crucial in WD and DDLS cell lines.
Particularly, the effect of the ATRX expression is significant in
cyclin-dependent kinase 4 (CDK4) inhibitors responder inducing
silence or consenescence. ATRX deficiency prevents the loss of
MDM2, which may be blocked by ATRX C-terminal modifications. This
discovery may directly guide the effective enhancement of CDK4
inhibitors in clinical cancer therapy.
Discussion and conclusion
The present review presented screening data for ALT
status and ATRX alteration from a variety of sarcomas (Table II). It was surmised that recent whole
genome and/or whole exome genomic studies, in addition to ATRX
immunohistochemistry and ALT FISH, have been performed for various
sarcomas in diverse sites, including osteosarcoma, leiomyosarcoma,
liposarcoma, angiosarcoma and chondrosarcoma. Considering the
results of these studies, it has been demonstrated that ALT serves
an essential role in telomere maintenance in sarcomas, and is
continually affected by ATRX expression loss. However, the
correlation between ALT and ATRX expression loss differs among the
sarcoma types and the differences are distinct. For certain tumors,
particularly the myxofibrosarcomas, it appears that the mechanisms
may be ascribed to ALT instead of the loss of ATRX expression in
the majority of cases.
| Table II.Overview of studies evaluating
correlation between ATRX and ALT in sarcomas. |
Table II.
Overview of studies evaluating
correlation between ATRX and ALT in sarcomas.
| A,
Osteosarcoma |
|---|
|
|---|
| Authors (year) | Number of
cases | Study
evaluation | (Refs.) |
|---|
| Chen et al
(2014) | 34 cases of
paediatric osteosarcoma | Genome sequencing:
5/34 tumours with point mutations of ATRX and 5/34 tumours
with focal deletions or SVs | (33) |
|
|
| IHC: 6/19 negative
for ATRX |
|
|
|
| FISH: Activation of
ALT found in 12/14, including 7 samples with ATRX
mutation |
|
| Liau et al
(2015) | 18 samples of
osteosarcoma | IHC: Absence of
ATRX-expression in 4/18 samples | (30) |
| Ji et al
(2017) | A case of two
brothers diagnosed osteosarcoma with ATR-X syndrome | Trio exome
sequencing: A nonsense NM_000489.4:c.7156C>T (p.Arg2386*)
mutation in the ATRX gene | (28) |
| Smolle et al
(2017) | A case of a
Caucasian man diagnosed osteosarcoma with ATR-X syndrome | Illumina MiSeq
sequencing: a hemizygous sequence variant within the ATRX gene
(NM_000489: c.6130C>T, p.Leu2044Phe) | (29) |
|
| B, Soft tissue
sarcoma |
|
| Author | Number of
cases | Study
evaluation | (Refs.) |
|
| Liau et al
(2015) | 92 samples of
leiomyosarcoma (46 uterus, 29 Retroperitoneum, 16 other sites) | IHC: Loss of
ATRX-expression in 30/92 | (32) |
|
|
| FISH: ALT
activation present in 51/86 |
|
| Yang et al
(2015) | 54 samples of
leiomyosarcoma (24 uterus, 11 Retroperitoneum, 19 other sites) | Genome sequencing:
ATRX mutations in 9/54 tumors | (36) |
|
|
| IHC: Loss of
ATRX-expression in 19/54 |
|
|
|
| FISH: ALT
activation present in 33/54 |
|
| Lee et al
(2017) | 25 samples of
leiomyosarcoma (9 uterine/pelvic, 5 retroperitoneal/abdominal, 7
somatic soft tissue, 3 vascular, 1 other) | Genome sequencing:
5/25 leiomyosarcomas with mutations of ATRX | (34) |
| Mäkinen et
al (2016) | 19 samples of
Uterine leiomyosarcomas (ULMSs) for Exome sequencing; 44 ULMSs | Exome sequencing:
5/19 Uterine leiomyosarcomas with mutations of ATRX | (35) |
|
|
| IHC: 23/44 ULMSs
with clearly reduced ATRX expression for IHC |
|
| Lee et al
(2015) | 111 cases of
liposarcoma (28 well-differentiated, 52 dedifferentiated, 20
myxoid/round cell, 1/19 myoxid/round cell and 0/16
well-differentiated liposarcomas | IHC: ATRX-loss in
13/52 dedifferentiated liposarcomas and 5/11 pleomorphic
liposarcomas FISH: ALT activation in 8/10 pleomorphic, 14/46
dedifferentiated, 11 pleomorphic) | (40) |
| Liau et al
(2015) | 88 samples of
angiosarcoma (34 skin, 13 liver, 7 breast, 54 others) | IHC: Loss of ATRX
in 16/77 angiosarcomas | (42) |
|
|
| FISH: Activation of
ALT in 49/66 angiosarcoma samples |
|
| Cleven et al
(2017) | 36 samples of
conventional chondrosarcoma | IHC: 2/36 negative
for ATRX | (38) |
| Liau et al
(2015) | 96 samples of
sarcoma (35 Undifferentiated pleomorphic sarcomas, 27
Myxofibrosarcomas, 17 Malignant peripheral nerve sheath tumors, 9
Embryonal rhabdomyosarcomas, 20 Radiation-associated sarcomas, 15
Chondrosarcomas) | IHC: Loss of
ATRX-expression 15/96 sarcomas; | (30) |
|
|
| FISH: ALT
activation present in 48/96 |
|
Recently, the cell death induced by ATR inhibitors,
which were developed as feasible novel drugs for ATRX-deficient
tumors, have demonstrated high selectivity for ALT-positive cancer
cells, indicating that such types of inhibitors may be effective
for the treatment of cancer with an ALT-dependent mechanism
(46,48). ATR was reported to be crucial for
homologous recombination and ALT, and inhibition of this kinase
caused apoptosis in ALT-positive cancer cells. This demonstrates
the significance and development of clinical genome analysis to
assist diagnosis and facilitate individualized precision targeted
therapies for sarcomas with complex pathologies. Identification of
an ALT phenotype and the loss of ATRX are not rarely seen in
sarcomas, suggesting that ATR inhibitors may provide a novel option
to treat these invasive neoplasms (46,48).
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant. no. 81372180) and the Hunan
Provincial Science and Technology Association program (grant no.
2017TJ-Q19) and the Hunan Provincial Science and Technology
Association key-point program (grant no. 2017DK2013).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XR, RM and ZT collected the associated articles
about the ATRX gene. XR and CT analysed and summarized the data. XR
wrote the paper and edited the manuscript and agreed to be
accountable for all aspects of the work ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved. CT analyzed and summarized
the data. XR was a major contributor in writing the manuscript. ZL
revised and checked the manuscript and gave the final approval of
the version to be published. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gibbons RJ, Picketts DJ, Villard L and
Higgs DR: Mutations in a putative global transcriptional regulator
cause X-linked mental retardation with alpha-thalassemia (ATR-X
syndrome). Cell. 80:837–845. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villard L and Fontes M:
Alpha-thalassemia/mental retardation syndrome, X-Linked (ATR-X, MIM
#301040, ATR-X/XNP/XH2 gene MIM #300032). Eur J Hum Genet.
10:223–225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qadeer ZA, Harcharik S, Valle-Garcia D,
Chen C, Birge MB, Vardabasso C, Duarte LF and Bernstein E:
Decreased expression of the chromatin remodeler ATRX associates
with melanoma progression. J Invest Dermatol. 134:1768–1772. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai J, Chen J, Zhang W, Yang P, Zhang C,
Li M, Yao K, Wang H, Li Q, Jiang C and Jiang T: Loss of ATRX,
associated with DNA methylation pattern of chromosome end, impacted
biological behaviors of astrocytic tumors. Oncotarget.
6:18105–18115. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gibbons RJ and Higgs DR:
Molecular-clinical spectrum of the ATR-X syndrome. Am J Med Genet.
97:204–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cardoso C, Timsit S, Villard L,
Khrestchatisky M, Fontès M and Colleaux L: Specific interaction
between the XNP/ATR-X gene product and the SET domain of the human
EZH2 protein. Hum Mol Genet. 7:679–684. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perry JA, Kiezun A, Tonzi P, Van Allen EM,
Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS,
et al: Complementary genomic approaches highlight the PI3K/mTOR
pathway as a common vulnerability in osteosarcoma. Proc Natl Acad
Sci USA. 111:E5564–E5573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reimann E, Kõks S, Ho XD, Maasalu K and
Märtson A: Whole exome sequencing of a single osteosarcoma
case-integrative analysis with whole transcriptome RNA-seq data.
Hum Genomics. 8:202014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bousquet M, Noirot C, Accadbled F, de
Gauzy Sales J, Castex MP, Brousset P and Gomez-Brouchet A:
Whole-exome sequencing in osteosarcoma reveals important
heterogeneity of genetic alterations. Ann Oncol. 27:738–744. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Joseph CG, Hwang H, Jiao Y, Wood LD, Kinde
I, Wu J, Mandahl N, Luo J, Hruban RH, Diaz LA Jr, et al: Exomic
analysis of myxoid liposarcomas, synovial sarcomas, and
osteosarcomas. Genes Chromosomes Cancer. 53:15–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Yuan Q and Wang W: The role of
telomeres in musculoskeletal diseases. J Int Med Res. 40:1242–1250.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ozturk MB, Li Y and Tergaonkar V: Current
insights to regulation and role of telomerase in human diseases.
Antioxidants (Basel). 6:pii: E17. 2017.PubMed/NCBI
|
|
13
|
Henderson ER and Blackburn EH: An
overhanging 3′ terminus is a conserved feature of telomeres. Mol
Cell Biol. 9:345–348. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayflick L and Moorhead PS: The serial
cultivation of human diploid cell strains. Exp Cell Res.
25:585–621. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shay JW: Role of telomeres and telomerase
in aging and cancer. Cancer Discov. 6:584–593. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Günes C and Rudolph KL: The role of
telomeres in stem cells and cancer. Cell. 152:390–393. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harley CB and Villeponteau B: Telomeres
and telomerase in aging and cancer. Curr Opin Genet Dev. 5:249–255.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Artandi SE and DePinho RA: Telomeres and
telomerase in cancer. Carcinogenesis. 31:9–18. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cesare AJ and Reddel RR: Alternative
lengthening of telomeres: Models, mechanisms and implications. Nat
Rev Genet. 11:319–330. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heaphy CM, Subhawong AP, Hong SM, Goggins
MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL,
Westra WH, et al: Prevalence of the alternative lengthening of
telomeres telomere maintenance mechanism in human cancer subtypes.
Am J Pathol. 179:1608–1615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sidaway P: Sarcoma: Genetic determinants
of sarcoma risk revealed. Nat Rev Clin Oncol. 13:5902016.
View Article : Google Scholar
|
|
22
|
Thomas DM and Ballinger ML: Diagnosis and
management of hereditary sarcoma. Recent Results Cancer Res.
205:169–189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ballinger ML, Goode DL, Ray-Coquard I,
James PA, Mitchell G, Niedermayr E, Puri A, Schiffman JD, Dite GS,
Cipponi A, et al: Monogenic and polygenic determinants of sarcoma
risk: An international genetic study. Lancet Oncol. 17:1261–1271.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Antonescu CR: The role of genetic testing
in soft tissue sarcoma. Histopathology. 48:13–21. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bridge JA: The role of cytogenetics and
molecular diagnostics in the diagnosis of soft-tissue tumors. Mod
Pathol. 27 Suppl 1:S80–S97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rosenberg AE: WHO classification of soft
tissue and bone, fourth edition: Summary and commentary. Curr Opin
Oncol. 25:571–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zambo I and Veselý K: WHO classification
of tumours of soft tissue and bone 2013: The main changes compared
to the 3rd edition. Cesk Patol. 50:64–70. 2014.PubMed/NCBI
|
|
28
|
Ji J, Quindipan C, Parham D, Shen L, Ruble
D, Bootwalla M, Maglinte DT, Gai X, Saitta SC, Biegel JA and
Mascarenhas L: Inherited germline ATRX mutation in two brothers
with ATR-X syndrome and osteosarcoma. Am J Med Genet A.
173:1390–1395. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smolle MA, Heitzer E, Geigl JB, Al Kaissi
A, Liegl-Atzwanger B, Seidel MG, Holzer LA and Leithner A: A novel
mutation in ATRX associated with intellectual disability, syndromic
features, and osteosarcoma. Pediatr Blood Cancer. 64:e265222017.
View Article : Google Scholar
|
|
30
|
Liau JY, Lee JC, Tsai JH, Yang CY, Liu TL,
Ke ZL, Hsu HH and Jeng YM: Comprehensive screening of alternative
lengthening of telomeres phenotype and loss of ATRX expression in
sarcomas. Mod Pathol. 28:1545–1554. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kovac M, Blattmann C, Ribi S, Smida J,
Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M,
Krieg A, et al: Exome sequencing of osteosarcoma reveals mutation
signatures reminiscent of BRCA deficiency. Nat Commun. 6:89402015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liau JY, Tsai JH, Jeng YM, Lee JC, Hsu HH
and Yang CY: Leiomyosarcoma with alternative lengthening of
telomeres is associated with aggressive histologic features, loss
of ATRX expression, and poor clinical outcome. Am J Surg Pathol.
39:236–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, Bahrami A, Pappo A, Easton J,
Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al:
Recurrent somatic structural variations contribute to tumorigenesis
in pediatric osteosarcoma. Cell Rep. 7:104–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee PJ, Yoo NS, Hagemann IS, Pfeifer JD,
Cottrell CE, Abel HJ and Duncavage EJ: Spectrum of mutations in
leiomyosarcomas identified by clinical targeted next-generation
sequencing. Exp Mol Pathol. 102:156–161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mäkinen N, Aavikko M, Heikkinen T, Taipale
M, Taipale J, Koivisto-Korander R, Bützow R and Vahteristo P: Exome
sequencing of uterine leiomyosarcomas identifies frequent mutations
in TP53, ATRX, and MED12. PLoS Genet. 12:e10058502016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang CY, Liau JY, Huang WJ, Chang YT,
Chang MC, Lee JC, Tsai JH, Su YN, Hung CC and Jeng YM: Targeted
next-generation sequencing of cancer genes identified frequent TP53
and ATRX mutations in leiomyosarcoma. Am J Transl Res. 7:2072–2081.
2015.PubMed/NCBI
|
|
37
|
Hartmann C, Meyer J, Balss J, Capper D,
Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et
al: Type and frequency of IDH1 and IDH2 mutations are related to
astrocytic and oligodendroglial differentiation and age: A study of
1,010 diffuse gliomas. Acta Neuropathol. 118:469–474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cleven AHG, Suijker J, Agrogiannis G,
Briaire-de Bruijn IH, Frizzell N, Hoekstra AS, Wijers-Koster PM,
Cleton-Jansen AM and Bovée JVMG: IDH1 or −2 mutations do not
predict outcome and do not cause loss of 5-hydroxymethylcytosine or
altered histone modifications in central chondrosarcomas. Clin
Sarcoma Res. 7:82017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Johnson JE, Varkonyi RJ, Schwalm J, Cragle
R, Klein-Szanto A, Patchefsky A, Cukierman E, von Mehren M and
Broccoli D: Multiple mechanisms of telomere maintenance exist in
liposarcomas. Clin Cancer Res. 11:5347–5355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JC, Jeng YM, Liau JY, Tsai JH, Hsu HH
and Yang CY: Alternative lengthening of telomeres and loss of ATRX
are frequent events in pleomorphic and dedifferentiated
liposarcomas. Mod Pathol. 28:1064–1073. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Venturini L, Motta R, Gronchi A, Daidone M
and Zaffaroni N: Prognostic relevance of ALT-associated markers in
liposarcoma: A comparative analysis. BMC Cancer. 10:2542010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liau JY, Tsai JH, Yang CY, Lee JC, Liang
CW, Hsu HH and Jeng YM: Alternative lengthening of telomeres
phenotype in malignant vascular tumors is highly associated with
loss of ATRX expression and is frequently observed in hepatic
angiosarcomas. Hum Pathol. 46:1360–1366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Willems SM, Debiec-Rychter M, Szuhai K,
Hogendoorn PC and Sciot R: Local recurrence of myxofibrosarcoma is
associated with increase in tumour grade and cytogenetic
aberrations, suggesting a multistep tumour progression model. Mod
Pathol. 19:407–416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hechtman JF, Zehir A, Mitchell T, Borsu L,
Singer S, Tap W, Oultache A, Ladanyi M and Nafa K: Novel oncogene
and tumor suppressor mutations in KIT and PDGFRA wild type
gastrointestinal stromal tumors revealed by next generation
sequencing. Genes Chromosomes Cancer. 54:177–184. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Napier CE, Huschtscha LI, Harvey A, Bower
K, Noble JR, Hendrickson EA and Reddel RR: ATRX represses
alternative lengthening of telomeres. Oncotarget. 6:16543–16558.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Flynn RL, Cox KE, Jeitany M, Wakimoto H,
Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suvà ML, Benes CH, et al:
Alternative lengthening of telomeres renders cancer cells
hypersensitive to ATR inhibitors. Science. 347:273–277. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Deeg KI, Chung I, Bauer C and Rippe K:
Cancer cells with alternative lengthening of telomeres do not
display a general hypersensitivity to ATR inhibition. Front Oncol.
6:1862016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
McFarlane S and Preston CM: Human
cytomegalovirus immediate early gene expression in the osteosarcoma
line U2OS is repressed by the cell protein ATRX. Virus Res.
157:47–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kovatcheva M, Liu DD, Dickson MA, Klein
ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S,
et al: MDM2 turnover and expression of ATRX determine the choice
between quiescence and senescence in response to CDK4 inhibition.
Oncotarget. 6:8226–8243. 2015. View Article : Google Scholar : PubMed/NCBI
|