Introduction
Lung cancer is among the most widely diagnosed
cancers and the leading cause of cancer-associated mortality
worldwide (1). Human lung cancer is
an umbrella term that predominantly includes small cell lung cancer
(SCLC) and non-small cell lung cancer (NSCLC), with the latter
comprising over 80–85% of all lung cancer diagnoses (2). The majority of NSCLC patients are
diagnosed at either the mid- or terminal stages, resulting in low
survival rates (3).
Currently, only small improvements in the treatment
of NSCLC have been made, with early diagnosis considered one of the
most effective means of improving five-year survival rates
(4). The presence of validated
prognostic biomarkers has the potential to provide a valuable tool
for this early diagnosis (4).
Intensive work over previous years has shown that aberrant DNA
methylation in the promoter regions containing CpG-rich areas
(termed CpG islands) is one of the most well-defined epigenetic
changes found in human cancers (5).
Not only could this feature be utilized to distinguish cancer cells
from normal tissue, but it could also be exploited for use in early
cancer diagnosis.
At present, there are various methylation detection
methods that are in use, including methylation-specific polymerase
chain reaction (6), methylight
(7), methylation-sensitive
high-resolution melting (MS-HRM) (8),
pyrosequencing (9), and microarray
technologies (10). Known as a
sensitive and more efficient technology for detecting single
nucleotide variations (11), MS-HRM
is considered to be a novel technology that would provide
clinically-relevant sensitivity and rapidity for gene methylation
screening. MS-HRM identifies PCR-amplified products by monitoring
the melting temperature (Tm) of the double-stranded DNA
helix (12). This process does not
require product movement into another system, which ensures a
closed-tube procedure, and makes it an appropriate method for
methylation detection in patient populations.
In the present study, six target genes, consisting
of protocadherin γ subfamily B, 6 (PCDHGB6), homeobox A9 (HOXA9),
O6-methylguanine-DNA methyltransferase (MGMT), microRNA
(miR)-126, suppressor of cytokine signaling 3 (SOCS3) and Ras
association domain family member 5, also termed NORE1A, were
selected to analyze the promoter methylation status for NSCLC
detection, based on previous studies (4,13–16). PCDHGB6 is located on chromosome 5, and
is a member of the protocadherin γ gene cluster and has an
immunoglobulin-like organization (17). Hypermethylation of PCDHGB6 has been
found to be significantly associated with stage I NSCLC (18). Overexpression of HOXA9 has been shown
to significantly inhibit invasion of cell lines and may therefore
be a potential gene marker for NSCLC diagnosis (19). miR-126 is a metastasis-suppressing
gene and has been shown to be downregulated in a variety of
inherited diseases (20). As a tumor
suppressor, the promoter hypermethylation of miR-126 in lung tumors
has a key role in decreased expression of miR-126, leading to
oncogenesis of lung tissue (21,22). The
translation product of MGMT genes is
O6-methylguanine-DNA-methylotrans, which is both a
critical enzyme in repairing DNA alkylation damages and is
considered a common DNA repair gene (23). In primary lung carcinomas, MGMT
inactivation caused by promoter methylation has been shown to be
more prevalent in advanced stages (24). The SOCS3 gene is a tumor suppressor
gene, with aberrant methylation of SOCS3 occurring frequently in
several types of human cancers (25,26).
Similarly, NORE1A also performs as a tumor suppressor and has been
shown to be generally inactivated in cancer tumors (27). Finally, hypermethylation of CpG
islands in the NORE1A promoter has been found in primary tumors,
including NSCLC and SCLC (28).
In the present study, the promoter methylation
status in the target sequences of six selected genes was analyzed
using MS-HRM as the technology platform. Through the establishment
of standard curves of control DNA samples, the gene methylation
status of 54 lung cancer tissue samples and 54 corresponding
non-tumorous tissue (NT) samples was investigated. As a result, the
present study found frequent methylation on PCDHGB6, HOXA9 and
miR-126, infrequent methylation on MGMT, and no methylation on
either SOCS3 or NORE1A. The combination of PCDHGB6, HOXA9, miR-126
and MGMT reached 85.2% sensitivity and 81.5% specificity, with an
area under the curve (AUC) value of 0.891. In addition, high
consistency was shown between MS-HRM and pyrosequencing.
Furthermore, potential clinical values have been excavated in the
early diagnosis of NSCLC.
Materials and methods
Patient samples
In the present study, 54 pairs of lung cancer and
adjacent NT samples were obtained from 54 patients who underwent
surgical resection from January 2014 to June 2014 at the Department
of Pathology, Shanghai Zhongshan Hospital (Shanghai, China). Of
these patients, 12 were diagnosed with squamous cell carcinoma and
42 were diagnosed with adenocarcinoma. All these samples were
obtained with informed consent and were stored at −80°C until later
total DNA extraction. This research was approved by the
Institutional Review Board of Shanghai Zhongshan Hospital
(Shanghai, China).
DNA extraction and bisulfite
conversion
No more than 30 mg of tissue was obtained using
sterilized operating scissors. Tissue was then ground in tissue
lyser (Jingxin, Shanghai, China) for 80 sec at 65 Hz. Whole-genome
DNA extraction was then performed using ALL Prep DNA/RNA Mini kit
(Qiagen, Hilden, Germany), according to the manufacturer's
instructions. Sodium bisulfite was used to convert the extracted
DNA using an EZ DNA Methylation-Gold kit (Zymo Research Corp.,
Irvine, CA, USA), in which ~500 ng of DNA was used as the proper
addition, according to specification. The final elution volume was
10 µl.
MS-HRM assay
Converted DNA was amplified using a LightCycler 480
(Roche Diagnostics GmbH, Mannheim, Germany). The whole reaction
volume was 10 µl, which consisted of, using the MGMT gene as an
example: 1 ng/µl DNA template, 1X Master Mix (Roche Diagnostics
GmbH), 0.5 µM primers and 4 mM magnesium ions. Polymerase chain
reaction (PCR)-grade water was used to bring the final reaction
volume to 10 µl. The detailed amplification thermocycling protocol
was set to the following: Preheat for 10 min at 95°C before
starting a 50-cycle process involving 10 sec at 95°C, 20 sec at a
temperature between 61 to 55°C for 20 sec (temperature dropped
2.2°C per sec), and 20 sec at 72°C. The following MS-HRM melting
protocol was used: Heating at 95°C for 1 min, followed by 40°C for
1 min (29), 65°C for 1 sec, and
continuous heating to 95°C at a ramp rate of 0.02°C per second. To
ensure veracity and repeatability, each reaction was conducted in
duplicate. 5-Aza-dc-treated Jurkat Genomic DNA and CpG Methylated
Jurkat Genomic DNA (New England Biolabs, Ipswich, MA, USA) that had
been subjected to bisulfite conversion were used as fully
methylated and unmethylated control DNA samples, respectively. The
former was added into the latter in gradient proportions (0, 5, 10,
25, 50, 75, 90 and 100%) and used as artificial DNA standards of
different methylation levels. These DNA standards were used in each
assay to establish gradient standard curves. These curves were then
used to evaluate the methylation levels of the tumor and normal
samples.
Pyrosequencing validation
In the present study, pyrosequencing was used for 3
of the 6 genes (PCDHGB6, HOXA9 and miR-126), to validate the
accuracy of the present results. Pyrosequencing was performed at
Shanghai Medical College, Fudan University (Shanghai, China).
Table I shows the basic primer
information for both MS-HRM and pyrosequencing. Primers were
designed by MethPrimer (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi),
which is used for designing methylation PCR primers. Subsequent to
bisulfite conversion, cytosine was converted to thymine (except for
CpG islands) and the newfound sequence was used as the template for
primer design.
 | Table I.Primers for MS-HRM and
pyrosequencing. |
Table I.
Primers for MS-HRM and
pyrosequencing.
| A, MS-HRM |
|---|
| Primer | Sequence,
5′-3′ | Amplicon length,
bp | Ensembl
version | Genomic region |
|---|
| PCDHGB6 |
| 171 |
ENST00000520790 |
CHR5:140787402-140787572 |
|
Forward |
AATTTGAGGGGGATGTATATTT |
|
|
|
|
Reverse |
AAAATCCCAAACCAAAAACT |
|
|
|
| HOXA9 |
| 118 |
ENST00000343483 |
CHR7:27200705-27200822 |
|
Forward |
GAGTTGTGGTTGTTTTTTTTTG |
|
|
|
|
Reverse |
ACCTTTCAAAACTCCTTCCTC |
|
|
|
| MGMT |
| 110 |
ENST00000482653 |
chr10:131155459-131155568 |
|
Forward |
GCGTTTCGGATATGTTGGGATAGT |
|
|
|
|
Reverse |
AACGACCCAAACACTCACCAAA |
|
|
|
| miR-126 |
| 93 bp |
ENST00000362291 |
CHR9:139564092-139564184 |
|
Forward |
TGGGTTGGTTTTTGTTAGG |
|
|
|
|
Reverse |
TAACCCTCACCTACTCCACAA |
|
|
|
| SOCS3 |
| 105 bp |
ENST00000330871 |
chr17:76354057-76354161 |
|
Forward |
GAAGGTTTTTTTGTGGATTTTA |
|
|
|
|
Reverse |
ACTAAACCCCCTCRAATCC |
|
|
|
| NORE1A |
| 174 |
ENST00000367117 |
chr1:206680666-206680839 |
|
Forward |
GGAATTTTGTAGTTGTTTTAGGTG |
|
|
|
|
Reverse |
CCTTTAAAAAAACCRCAAC |
|
|
|
|
| B,
Pyrosequencing |
|
| Primer | Sequence,
5′-3′ | Amplicon length,
bp | Ensembl
version | Genomic region |
|
| PCDHGB6 |
| 116 |
ENST00000520790 |
CHR5:140787402-140787572 |
|
Forward |
AATTTGAGGGGGATGTATATTT |
|
|
|
|
Reverse |
biotin-AAAATCCCAAACCAAAAACT |
|
|
|
|
Sequencing primer |
GAATTTAAAATGAAAAAT |
|
|
|
| HOXA9 |
| 101 |
ENST00000343483 |
CHR7:27200705-27200822 |
|
Forward |
GAGTTGTGGTTGTTTTTTTTTG |
|
|
|
|
Reverse |
biotin-ACCTTTCAAAACTCCTTCCTC |
|
|
|
|
Sequencing primer |
TTTTGGGTTTTGTATTTTTT |
|
|
|
| miR-126 |
| 93 |
ENST00000362291 |
CHR9:139564092-139564184 |
|
Forward |
TGGGTTGGTTTTTGTTAGG |
|
|
|
|
Reverse |
biotin-TAACCCTCACCTACTCCACAA |
|
|
|
|
Sequencing primer |
TGGGTTGGTTTTTGTTAGG |
|
|
|
Statistical analysis
SPSS v17.0 (SPSS, Inc., Chicago, IL, USA) was used
for all statistical analysis. The dependence between the
HRM-assessed methylation status and temperature difference was
analyzed using simple linear regression. To verify the sensitivity
and specificity of high and low methylation status, the
receiver-operating characteristic (ROC) curve was generated and the
AUC was calculated to obtain the highest specificity and
sensitivity. Differences between tumor and surrounding tissue were
evaluated by t-test and P<0.05 was considered to indicate a
statistically significant difference. Additionally, Pearson's
correlation coefficient was evaluated to examine the consistency
between HRM assay and pyrosequencing of PCDHGB6, HOXA9 and
miR-126.
Results
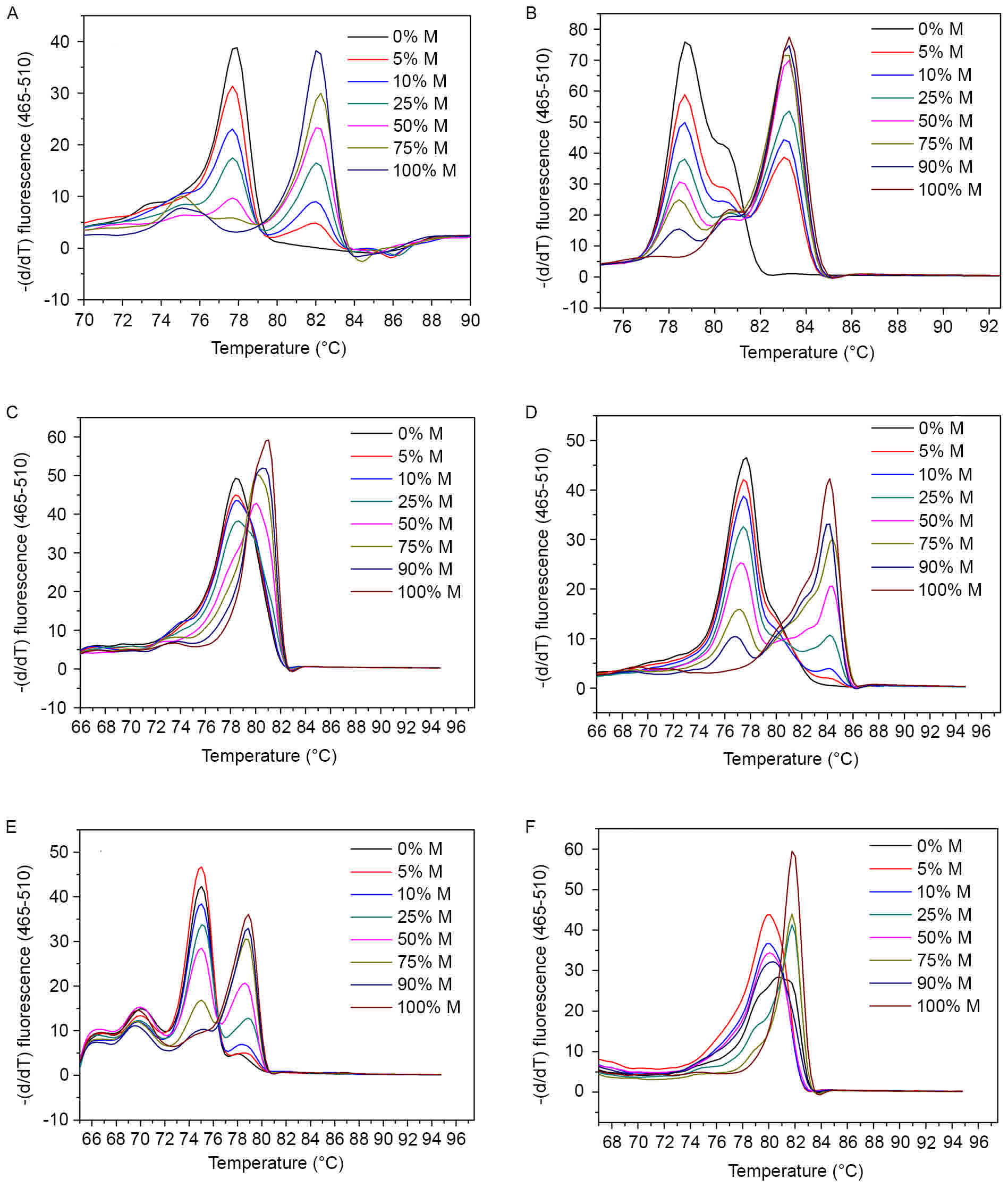
MS-HRM melting curves for each
gene
In the present MS-HRM assay, melting temperature was
obtained as a Tm value in the Tm calling
analysis. This refers to the temperature at which half of the
double-stranded DNA melts into single strands, thus undergoing a
sharp decline in fluorescence intensity (8,9). Fig. 1 shows the melting curves of control
samples of the six genes in the Tm calling analysis. A
gradient of diluted methylated DNA with unmethylated DNA (0–100%)
subsequent to bisulfite conversion were used for each gene as
controls. Melting profiles of all these gene promoters showed
higher Tm in methylated controls and lower Tm
in controls without methylation. Among them, Tm values
of SOCS3 gene exhibited the largest span, from 77 to 84°C (Fig. 1D), whereas small temperature
differences were found in HOXA9 and miR-126 as less than 2°C
(Fig. 1C and F). Based on the
difference in melting temperatures between the amplicons, single
nucleotides can be distinguished (30).
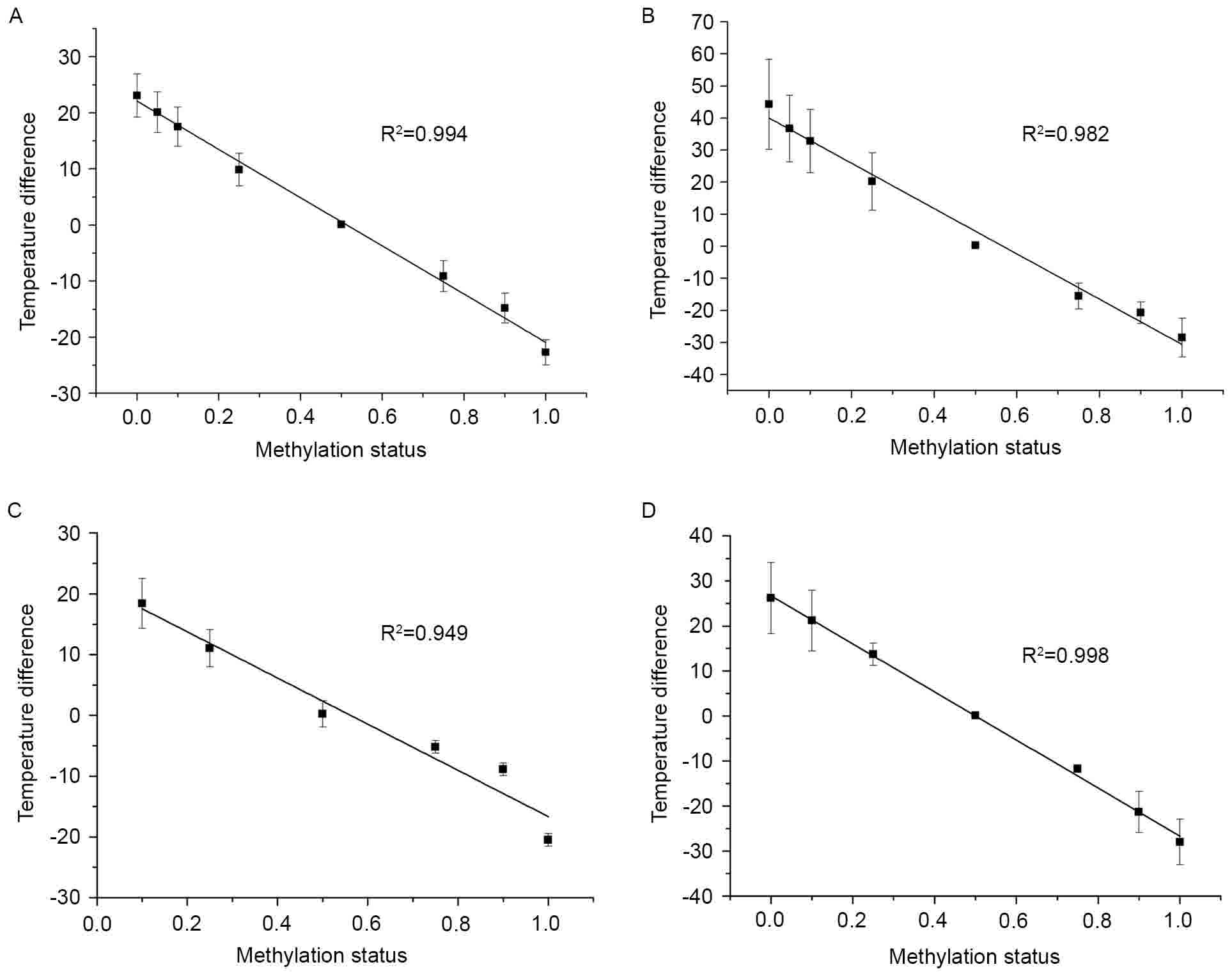
To further explore this association, a correlational
analysis was conducted between temperature difference and
methylation status using a simple linear regression (Fig. 2). Every standard curve yielded a
corresponding temperature difference value. As shown in Fig. 2, there was a negative correlation
between the methylation status of DNA standards in a series of
dilutions and temperatures. HOXA9, PCDHGB6, MGMT and miR-126
exhibited good liner association with significant R2
values of 0.994, 0.982, 0.949 and 0.998, respectively. From these
data, accurate methylation degrees could be calculated.
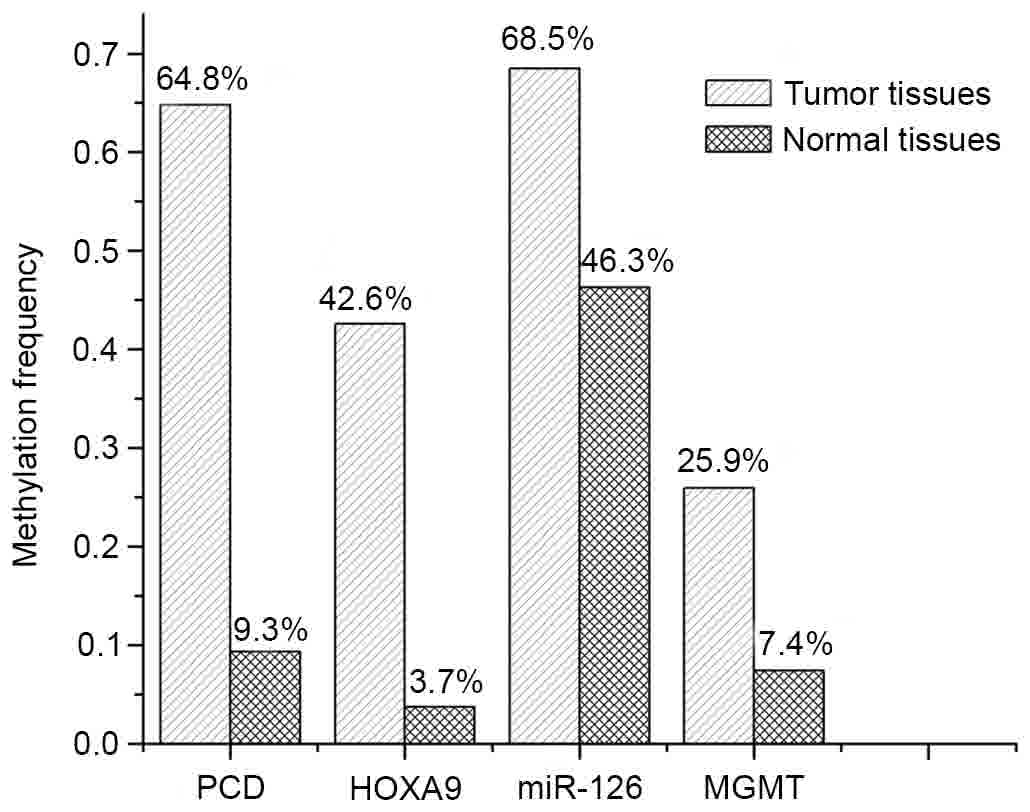
Assessing gene methylation frequency
in tumor and normal tissues
According to the generated standard curves, 54 pairs
of tumor and NT tissues were investigated. Methylation levels of
each gene are shown in Fig. 3. From
the experimental results, PCDHGB6 methylation was found in 35 of
the 54 tumors (64.8%) and HOXA9 methylation was found in 23 of the
54 tumors (42.6%). miR-126 also had a high methylation frequency
(68.5%), but methylation of miR-126 was found to be nonspecific in
normal tissue, with a frequency of 46.3%. Notably, no promoter
methylation of either SOCS3 or NORE1A was identified in the tumor
or normal tissue. As for MGMT, which has been affected in several
malignancies that include colorectal and lung cancer, did not show
any prominent impact (14 positive samples out of the 54 tumor
samples) in the early diagnosis of NSCLC.
Validation of methylation status by
pyrosequencing
The precise methylation values obtained from our
linear regression analysis were then compared with pyrosequencing.
In the present study, methylation values were derived from an
average of all the CpG sites. Ten pairs of malignant and control
tissue from each selected DNA sequence were chosen to detect
methylation degrees of PCDHGB6, HOXA9 and miR-126 by pyrosequencing
with 10, 6 and 5 possible mutations of CpG sites, respectively.
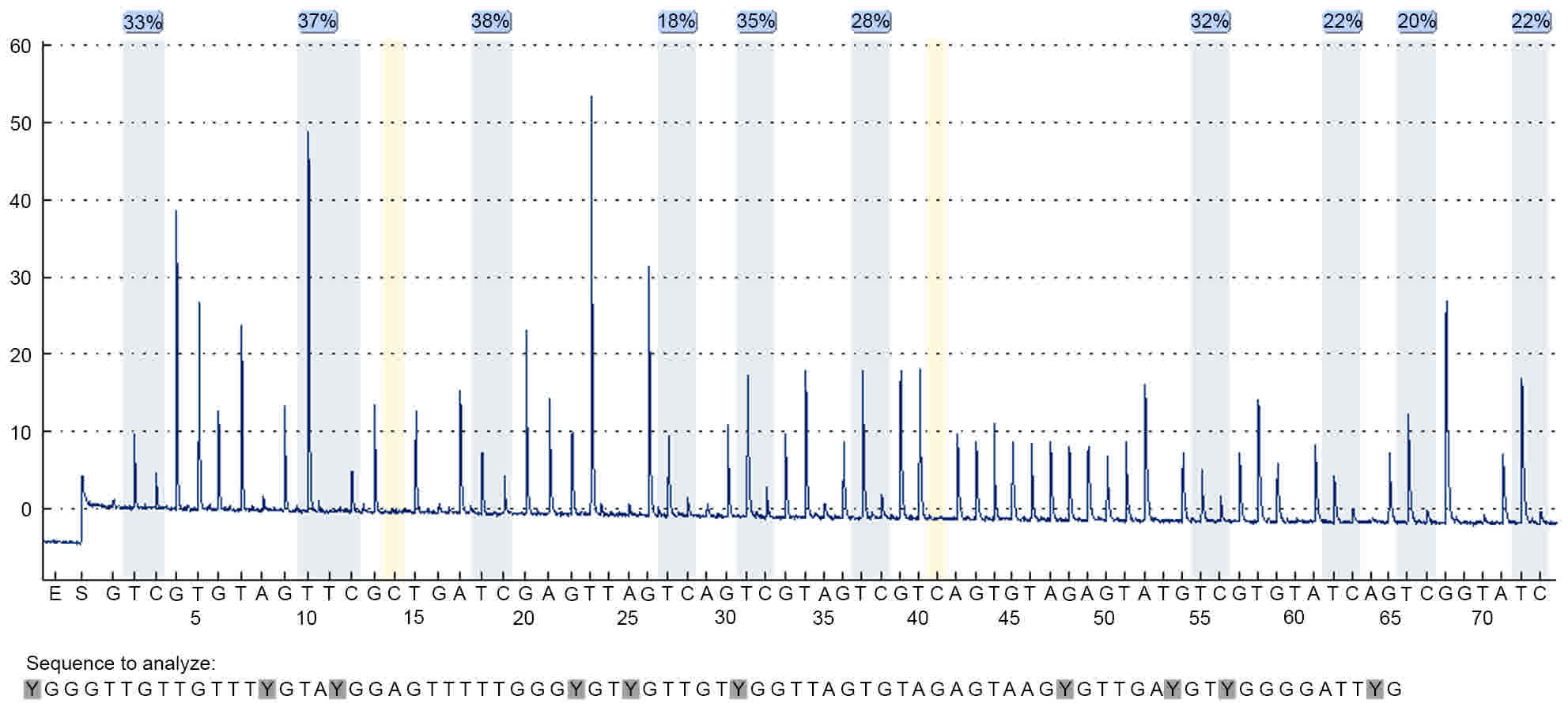
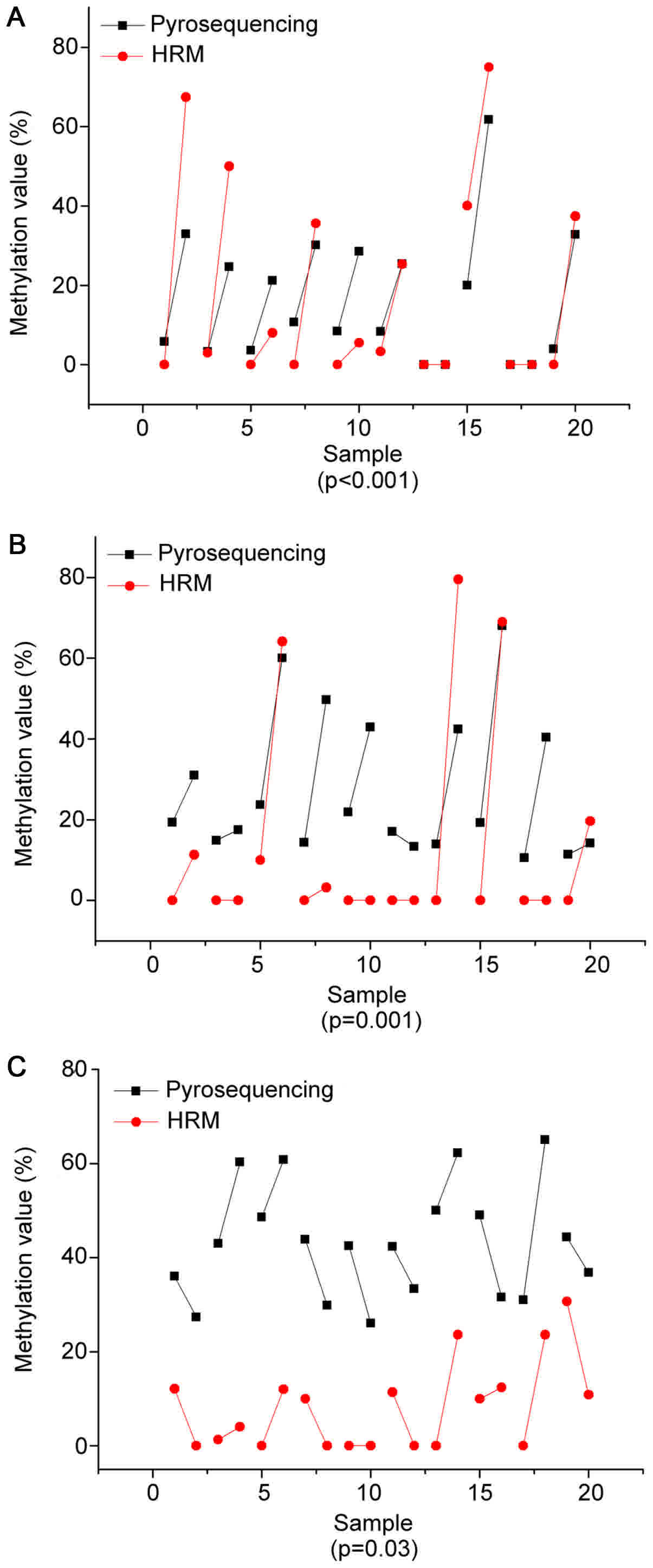
Fig. 4 illustrates the pyrosequencing
results of PCDHGB6 from one tumor tissue sample. The blue sections
indicate successful detection of CpG sites. Fig. 5 shows comparisons of methylation
degrees, as tested by MS-HRM and pyrosequencing. Every two samples
joined by a line represent a pair of tumor and para-carcinoma
tissue excised from the same patient. As shown, PCDHGB6 had the
most closely associated values of the three genes, while HOXA9 came
second. The sensitivity of assessing miR-126 methylation by MS-HRM
was generally lower than by pyrosequencing. To examine statistical
significance, Pearson's correlation coefficient was evaluated for
PCDHGB6, HOXA9 and miR-126, deriving values of P<0.001, P=0.001
and P=0.03, respectively. Collectively, these results demonstrated
a statistically significant coincident tendency between MS-HRM and
pyrosequencing (P<0.05).
Association between clinical features
of NSCLC patients with tumor tissue methylation
The association between clinical manifestations of
NSCLC and the MS-HRM analysis for selected genes are shown in
Table II (31). Based on the present study, the
methylation frequency of PCDHGB6 and HOXA9 increased with disease
progression, while the methylation frequency of PCDHGB6 was higher
than that of HOXA9 in every disease stage. For PCDHGB6, the
frequency was at 53.8% at stage I, with the ratio reaching 83.3% at
later stages. In the case of HOXA9 gene, the ratio increased
between 38.5% at stage I to 66.7% at stages III–IV. When compared
with the PCDHGB6 and HOXA9 genes, MGMT showed no evident
association with tumor-node-metastasis (TNM) stage and possessed
low detection rates in both normal and tumor tissue. For the
miR-126 gene, methylation frequency was significantly increased in
early stages, with 73.1% methylation frequency in stage I.
Additionally, results from all selected genes showed that males
appeared to be more susceptible than females. In the case of
histopathological classification, these four genes appeared to have
increased sensitivity in squamous cell carcinoma, with detection
rates of ≥75%. PCDHGB6 and miR-126 were both reliable biomarkers
for the detection of adenocarcinoma, with positive rates of 54.8
and 61.9%, respectively.
| Table II.The association between clinical
manifestations of patients and study results of selected genes from
methylation-sensitive high-resolution melting analysis. |
Table II.
The association between clinical
manifestations of patients and study results of selected genes from
methylation-sensitive high-resolution melting analysis.
|
|
|
|
|
|
|
|
|
|
|
| Histopathological
classification, n (%) |
|---|
|
| Methylation
frequency, n (%) | Stage, n (%) | Age, n (%) | Gender, n (%) |
|
|---|
|
|
|
|
|
| Squamous |
|---|
| Gene | Tumor tissue | Normal tissue | I | II | III–IV | ≤40 years | >40 years | Male | Female | Adenocarcinoma | cell carcinoma |
|---|
| Total | 54 (100.0) | 54 (100.0) | 26 (100.0) | 22 (100.0) | 6 (100.0) | 10 (100.0) | 44 (100.0) | 32 (100.0 | 22 (100.0) | 42 (100.0) | 12 (100.0) |
| PCDHGB6 | 35 (64.8) | 5 (9.3) | 14 (53.8) | 17 (77.3) | 5 (83.3) | 7 (70.0) | 30 (68.2) | 25 (78.1) | 10 (45.5) | 23 (54.8) | 12 (100.0) |
| HOXA9 | 23 (42.6) | 2 (3.7) | 10 (38.5) | 9 (41.0) | 4 (66.7) | 3 (30.0) | 20 (45.5) | 17 (53.1) | 6 (27.3) | 14 (33.3) | 9 (75.0) |
| MGMT | 14 (25.9) | 4 (7.4) | 7 (26.9) | 3 (13.6) | 1 (16.7) | 6 (60.0) | 7 (15.9) | 9 (28.1) | 4 (18.2) | 10 (23.8) | 2 (16.7) |
| miR-126 | 37 (68.5) | 25 (46.3) | 19 (73.1) | 16 (72.7) | 2 (33.3) | 4 (40.0) | 33 (75.0) | 20 (62.5) | 16 (72.7) | 26 (61.9) | 9 (75.0) |
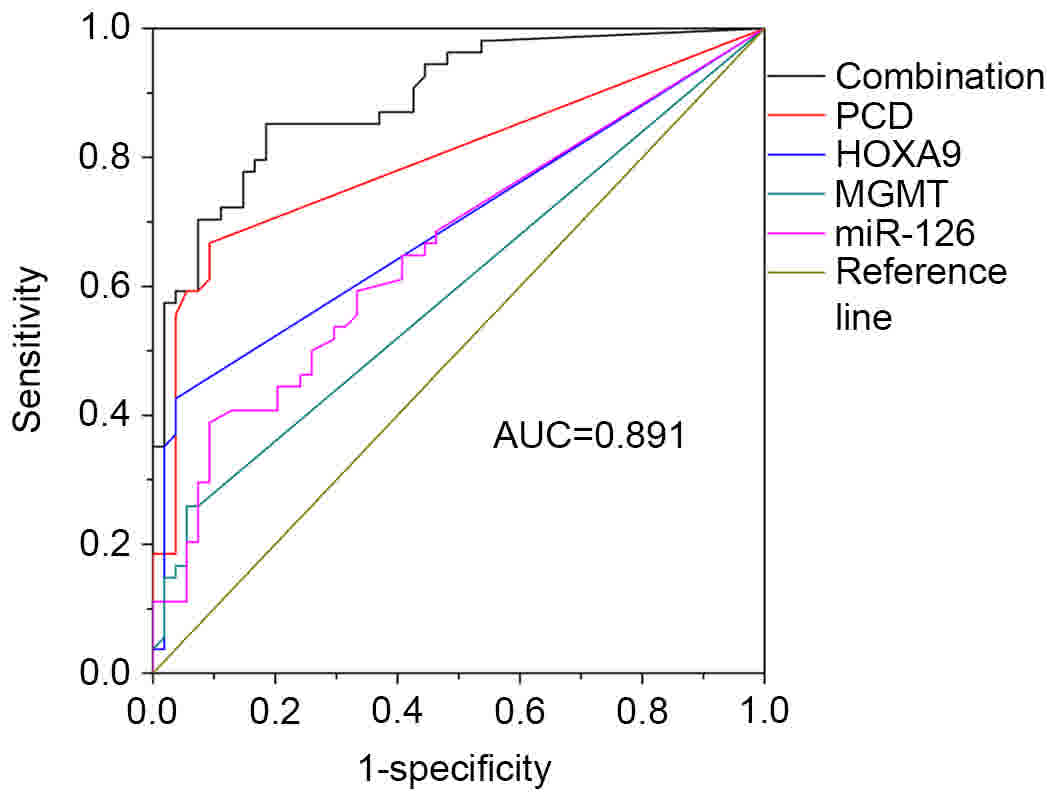
Joint detection of selected genes
A multi-gene analysis was then conducted to evaluate
the sensitivity and specificity of methylation. Results were
analyzed by ROC curve. As shown in Table III, values were calculated using
SPSS software, with various combinations used to acquire the best
result. PCDHGB6 had the highest sensitivity (66.7%) out of the four
genes, with 75.9% of the patients having at least two methylated
genes. Finally, the combination of four genes resulted in the
highest sensitivity and specificity (85.2 and 81.5%, respectively),
with the AUC being 0.891 (Fig.
6).
| Table III.Analysis of different combinations of
four selected genes. |
Table III.
Analysis of different combinations of
four selected genes.
|
| Methylation
frequency, % |
|
|
|
|
|---|
|
|
|
|
|
|
|
|---|
| Genes | Tumor tissues | Normal tissues | Sensitivity, % | Specificity, % | AUC | P-value |
|---|
| PCDHGB6 | 64.8 | 9.3 | 66.7 | 90.7 | 0.796 | P<0.001 |
| HOXA9 | 42.6 | 3.7 | 42.6 | 96.3 | 0.694 | P<0.001 |
| MGMT | 25.9 | 7.4 | 25.9 | 94.4 | 0.594 | P=0.059 |
| miR-126 | 68.5 | 46.3 | 38.9 | 90.7 | 0.658 | P<0.001 |
| PCDHGB6, HOXA9 | 75.9 | 11.1 | 75.9 | 88.9 | 0.835 | P<0.001 |
| PCDHGB6, HOXA9,
miR-126 | 94.4 | 53.7 | 81.5 | 85.2 | 0.872 | P<0.001 |
| PCDHGB6, HOXA9,
MGMT | 81.5 | 18.5 | 81.5 | 81.5 | 0.850 | P<0.001 |
| PCDHGB6, HOXA9,
MGMT, miR-126 | 98.1 | 55.5 | 85.2 | 81.5 | 0.891 | P<0.001 |
Discussion
In the present study, a new combination of target
promoter sequences for the diagnosis of NSCLC was obtained by
MS-HRM analysis. MS-HRM enabled evaluation of the PCR amplicon by
monitoring gradient changes in fluorescence correlated with
sequence-dependent melting properties (32,33). Thus,
an accurate melting status of the PCR amplicon may be identified by
mixing an intercalating dye with the product and monitoring
fluorescence intensity. In the present study, 54 pairs of tumor and
surrounding tissues were selected from NSCLC patients that ranged
between early and advanced TNM stages. Through the HRM diagnostic
system, the promoter methylation status of a series of possible
genes associated with NSCLC, consisting of PCDHGB6, HOXA9, MGMT,
miR-126, SOCS3 and NORE1A, was determined.
Among these tested target genes, Tm
values of the SOCS3 gene exhibited the largest span (77–84°C),
whereas the HOXA9 and miR-126 genes showed small temperature
differences. In addition, and increased Tm difference
between methylated and unmethylated samples indicated a richer CpG
content in selected loci. Based on the establishment of standard
curves that showed highly correlated relations, PCDHGB6 methylation
was found in 35 of the 54 tumors (64.8%) and HOXA9 methylation was
found in 23 of the 54 tumors (42.6%). These values correspond with
previous studies (4,34,35) that
showed a close association between methylation and NSCLC. miR-126
also had a high methylation frequency (68.5%), and has been found
to be significant at stage I–II, indicating that it would be more
sensitive for use in early diagnosis. The only complication was
that methylation of miR-126 was found to exhibit lower specificity
for normal tissues, with a frequency of 46.3%. This may be due to
the presence of methylated CpG distributed in the normal sequence.
Compared with the three hypermethylated genes (PCDHGB6, HOXA9 and
miR-126), it was found that MGMT had a lower detection rate, with
14 of 54 tumor samples (25.9%) found to be MGMT
methylation-positive. However, this is still of clinical value due
to the ubiquity of MGMT methylation in both early and advanced TNM
stages. The promoter methylation observed in SOCS3 and NORE1A
showed no correlation in NSCLC diagnosis. In addition, results from
all selected genes exhibited the tendency that males were more
susceptible than females, which may derive from the increased
incidence of smoking in the male population (36). In the case of histopathological
classification, methylation of these four genes (PCDHGB6, HOXA9,
MGMT and miR-126) were more likely to occur in squamous cell
carcinoma, while PCDHGB6 and miR-126 both appeared to be reliable
biomarkers for adenocarcinoma, and are therefore important for the
typing of NSCLC at diagnosis.
Pyrosequencing is a new DNA sequencing technique and
is a modification of combined bisulfite restriction analysis
(37). Pyrosequencing applies to the
analysis of known, short nucleotide sequences and it is
advantageous in terms of its accuracy, rapidity and repeatability
(38). These qualities make it a gold
standard for evaluating degrees of methylation (39). Pyrosequencing was used to examine the
accuracy of MS-HRM methylation patterns observed in PCDHGB6, HOXA9
and miR-126 by MS-HRM. The present results demonstrated high
consistency between HRM and pyrosequencing data (P<0.05 for all
three genes). It should be noted that the sensitivity of assessing
miR-126 methylation by MS-HRM was generally decreased compared with
pyrosequencing. One possible reason for this is that methylation
sites are distributed on normally selected positions, while by
default the MS-HRM assay regards methylation of normal tissue as
zero. All of these aforementioned results demonstrate the
feasibility of evaluating heterogeneous promoter methylation by
MS-HRM.
Finally, the combination of PCDHGB6, HOXA9, miR-126
and MGMT reached an AUC value of 0.891, with 85.2% sensitivity and
81.5% specificity. This indicated a significant association between
this diagnostic system and NSCLC pathology. Overall, these results
demonstrate that not only does methylation assessment have
statistical significance, but also that conjoint analysis has
improved sensitivity and specificity compared with a single
gene.
In conclusion, a significant joint testing of
relevant target genes was established to evaluate clinical status
of NSCLC by MS-HRM analysis. This research indicates that early
diagnosis of NSCLC is feasible through the monitoring of promoter
methylation using an effective combination of related genes. It
provides a potential valuable and economical method for clinical
applications.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation of China (grant no. 61571429), MOST of
China (grant no. 2017YFA0205300), the STS Project of the Chinese
Academy of Sciences (grant no. KFJ-STS-SCYD-120) and the Science
and Technology Commission of Shanghai Municipality (grant no.
16410711800).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MH and LF conceived and designed the study. LF, ZH,
CZ and BY performed the experiments. LS analyzed the patient data
regarding the TNM stage and histopathological classification. ZQ
and ZJ participated in the data collection and preparation of the
manuscript. LF, WZ and ZL analyzed and interpreted experimental
data. LF wrote the paper. MH, ZQ and ZJ revised the manuscript
critically for important intellectual content. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All samples were obtained with informed consent of
the participants. This research was approved by the Institutional
Review Board of Shanghai Zhongshan Hospital (Shanghai, China).
Consent for publication
All identifying information has been removed from
the manuscript.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SCLC
|
small cell lung cancer
|
|
NSCLC
|
non-small cell lung cancer
|
|
HRM
|
high-resolution melting
|
|
MS-HRM
|
methylation-sensitive high-resolution
melting
|
|
Tm
|
melting temperature
|
|
NT
|
non-tumorous tissue
|
|
ROC
|
receiver operating characteristic
|
|
AUC
|
area under the curve
|
References
|
1
|
Hubaux R, Thu KL, Vucic EA, Pikor LA, Kung
SH, Martinez VD, Mosslemi M, Becker-Santos DD, Gazdar AF, Lam S and
Lam WL: Microtubule affinity-regulating kinase 2 is associated with
DNA damage response and cisplatin resistance in non-small cell lung
cancer. Int J Cancer. 137:2072–2082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peters S, Adjei AA, Gridelli C, Reck M,
Kerr K and Felip E: ESMO Guidelines Working Group: Metastatic
non-small-cell lung cancer (NSCLC): ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 23
Suppl 7:vii56–64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dillman RO, Herndon J, Seagren SL, Eaton
WL Jr and Green MR: Improved survival in stage III non-small-cell
lung cancer: Seven-year follow-up of cancer and leukemia group B
(CALGB) 8433 trial. J Natl Cancer Inst. 88:1210–1215. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sandoval J, Mendez-Gonzalez J, Nadal E,
Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A,
et al: A prognostic DNA methylation signature for stage I
non-small-cell lung cancer. J Clin Oncol. 31:4140–4147. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lister R, Pelizzola M, Dowen RH, Hawkins
RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al:
Human DNA methylomes at base resolution show widespread epigenomic
differences. Nature. 462:315–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: Anovel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C, Shibata D, Danenberg PV and Laird PW: Methy Light: A
high-throughput assay to measure DNA methylation. Nucleic Acids
Res. 28:e322000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du Y, Zhou Y and Wu Q: MS-HRM to detect
serum DNA methylation of intrauterine growth retardation children.
Engineering. 4:106–109. 2012. View Article : Google Scholar
|
|
9
|
Doerks T, Copley RR, Schultz J, Ponting CP
and Bork P: Systematic identification of novel protein domain
families associated with nuclear functions. Genome Res. 12:47–56.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gitan RS, Shi H, Chen CM, Yan PS and Huang
TH: Methylation-specific oligonucleotide microarray: A new
potential for high-throughput methylation analysis. Genome Res.
12:158–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wojdacz TK: Methylation-sensitive
high-resolution melting in the context of legislative requirements
for validation of analytical procedures for diagnostic
applications. Expert Rev Mol Diagn. 12:92012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wojdacz TK and Dobrovic A:
Methylation-sensitive high resolution melting (MS-HRM): A new
approach for sensitive and high-throughput assessment of
methylation. Nucleic Acids Res. 35:e412007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wolf P, Hu YC, Doffek K, Sidransky D and
Ahrendt SA: O6-Methylguanine-DNA methyltransferase promoter
hypermethylation shifts the p53 mutational spectrum in non-small
cell lung cancer. Cancer Res. 61:8113–8117. 2001.PubMed/NCBI
|
|
14
|
Watanabe K, Emoto N, Hamano E, Sunohara M,
Kawakami M, Kage H, Kitano K, Nakajima J, Goto A, Fukayama M, et
al: Genome structure-based screening identified epigenetically
silenced microRNA associated with invasiveness in non-small-cell
lung cancer. Int J Cancer. 130:2580–2590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boosani CS and Agrawal DK: Methylation and
microRNA-mediated epigenetic regulation of SOCS3. Mol Biol Rep.
42:853–872. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Irimia M, Fraga MF, Sanchez-Cespedes M and
Esteller M: CpG island promoter hypermethylation of the
Ras-effector gene NORE1A occurs in the context of a wild-type K-ras
in lung cancer. Oncogene. 23:8695–8699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang KH, Lin CJ, Liu CJ, Liu DW, Huang RL,
Ding DC, Weng CF and Chu TY: Global methylation silencing of
clustered proto-cadherin genes in cervical cancer: Serving as
diagnostic markers comparable to HPV. Cancer Med. 4:43–55. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haller F, Zhang JD, Moskalev EA, Braun A,
Otto C, Geddert H, Riazalhosseini Y, Ward A, Balwierz A, Schaefer
IM, et al: Combined DNA methylation and gene expression profiling
in gastrointestinal stromal tumors reveals hypomethylation of SPP1
as an independent prognostic factor. Int J Cancer. 136:1013–1023.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Son JW, Jeong KJ, Jean WS, Park SY, Jheon
S, Cho HM, Park CG, Lee HY and Kang J: Genome-wide combination
profiling of DNA copy number and methylation for deciphering
biomarkers in non-small cell lung cancer patients. Cancer Lett.
311:29–37. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saito Y, Friedman JM, Chihara Y, Egger G,
Chuang JC and Liang G: Epigenetic therapy upregulates the tumor
suppressor microRNA-126 and its host gene EGFL7 in human cancer
cells. Biochem Biophys Res Commun. 379:726–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang P, Yang D, Zhang H, Wei X, Ma T,
Cheng Z, Hong Q, Hu J, Zhuo H, Song Y, et al: Early detection of
lung cancer in serum by a panel of MicroRNA biomarkers. Clin Lung
Cancer. 16:313–319.e1. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Crawford M, Brawner E, Batte K, Yu L,
Hunter MG, Otterson GA, Nuovo G, Marsh CB and Nana-Sinkam SP:
MicroRNA-126 inhibits invasion in non-small cell lung carcinoma
cell lines. Biochem Biophys Res Commun. 373:607–612. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Esteller M, Garcia-Foncillas J, Andion E,
Goodman SN, Hidalgo OF, Vanaclocha VV, Baylin SB and Herman JG:
Inactivation of the DNA-repair gene MGMT and the clinical response
of gliomas to alkylating agents. New Engl J Med. 343:1350–1354.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu JY, Wang J, Lai JC, Cheng YW, Yeh KT,
Wu TC, Chen CY and Lee H: Association of O6-methylguanine-DNA
methyltransferase (MGMT) promoter methylation with p53 mutation
occurrence in non-small cell lung cancer with different histology,
gender and smoking status. Ann Surg Oncol. 15:3272–3277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barclay JL, Anderson ST, Waters MJ and
Curlewis JD: SOCS3 as a tumor suppressor in breast cancer cells and
its regulation by PRL. Int J Cancer. 124:1756–1766. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rigby RJ, Simmons JG, Greenhalgh CJ,
Alexander WS and Lund PK: Suppressor of cytokine signaling 3
(SOCS3) limits damage-induced crypt hyper-proliferation and
inflammation-associated tumorigenesis in the colon. Oncogene.
26:4833–4841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moshnikova A, Frye J, Shay JW, Minna JD
and Khokhlatchev AV: The growth and tumor suppressor NORE1A is a
cytoskeletal protein that suppresses growth by inhibition of the
ERK pathway. J Biol Chem. 281:8143–8152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hesson L, Dallol A, Minna JD, Maher ER and
Latif F: NORE1A, a homologue of RASSF1A tumour suppressor gene is
inactivated in human cancers. Oncogene. 22:947–954. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin TC, Jiang SS, Chou WC, Hou HA, Lin YM,
Chang CL, Hsu CA, Tien HF and Lin LI: Rapid assessment of the
heterogeneous methylation status of CEBPA in patients with acute
myeloid leukemia by using high-resolution melting profile. J Mol
Diagn. 13:514–519. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Newman M, Blyth BJ, Hussey DJ, Jardine D,
Sykes PJ and Ormsby RJ: Sensitive quantitative analysis of murine
LINE1 DNA methylation using high resolution melt analysis.
Epigenetics. 7:92–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang X, Dai W, Kwong DL, Szeto CY, Wong
EH, Ng WT, Lee AW, Ngan RK, Yau CC, Tung SY and Lung ML: Epigenetic
markers for noninvasive early detection of nasopharyngeal carcinoma
by methylation-sensitive high resolution melting. Int J Cancer.
136:E127–E135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wojdacz TK, Møller TH, Thestrup BB,
Kristensen LS and Hansen LL: Limitations and advantages of MS-HRM
and bisulfite sequencing for single locus methylation studies.
Expert Rev Mol Diagn. 10:575–580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu J and Yao X: Use of DNA methylation
for cancer detection: Promises and challenges. Int J Biochem Cell
Biol. 41:147–154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma Y, Bai Y, Mao H, Hong Q, Yang D, Zhang
H, Liu F, Wu Z, Jin Q, Zhou H, et al: A panel of promoter
methylation markers for invasive and noninvasive early detection of
NSCLC using a quantum dots-based FRET approach. Biosens
Bioelectron. 85:641–648. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hwang JA, Lee BB, Kim Y, Hong SH, Kim YH,
Han J, Shim YM, Yoon CY, Lee YS and Kim DH: HOXA9 inhibits
migration of lung cancer cells and its hypermethylation is
associated with recurrence in non-small cell lung cancer. Mol
Carcinog. 54 Suppl 1:E72–E80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang T, Chen X, Hong Q, Deng Z, Ma H, Xin
Y, Fang Y, Ye H, Wang R, Zhang C, et al: Meta-analyses of gene
methylation and smoking behavior in non-small cell lung cancer
patients. Sci Rep. 5:88972015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Colella S, Shen L, Baggerly KA, Issa JP
and Krahe R: Sensitive and quantitative universal Pyrosequencing
methylation analysis of CpG sites. Biotechniques. 35:146–150.
2003.PubMed/NCBI
|
|
38
|
Candiloro IL, Mikeska T and Dobrovic A:
Assessing combined methylation-sensitive high resolution melting
and pyrosequencing for the analysis of heterogeneous DNA
methylation. Epigenetics. 6:500–507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Quillien V, Lavenu A, Karayan-Tapon L,
Carpentier C, Labussiere M, Lesimple T, Chinot O, Wager M, Honnorat
J, Saikali S, et al: Comparative assessment of 5 methods
(methylation-specific polymerase chain reaction, MethyLight,
pyrosequencing, methylation-sensitive high-resolution melting and
immunohistochemistry) to analyze
O6-methylguanine-DNA-methyltranferase in a series of 100
glioblastoma patients. Cancer. 118:4201–4211. 2012. View Article : Google Scholar : PubMed/NCBI
|