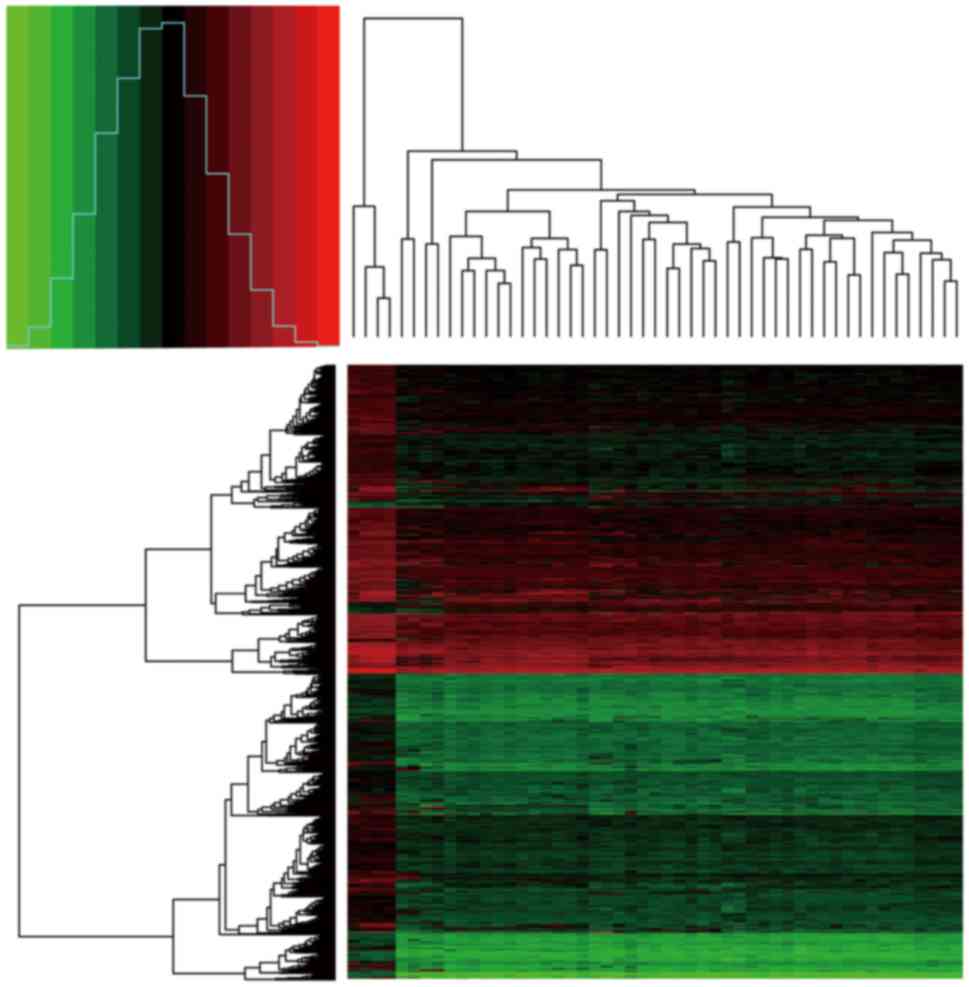
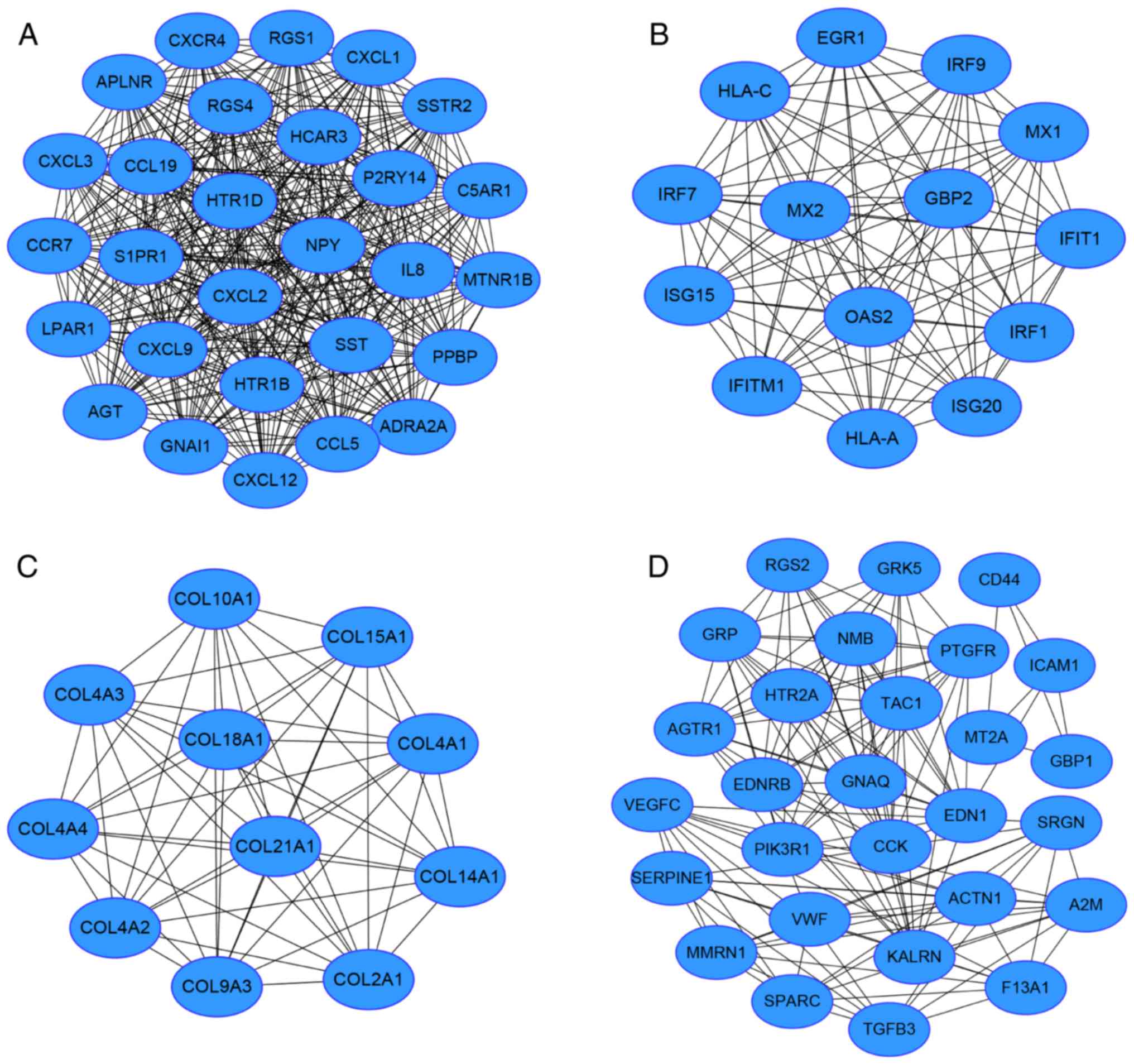
|
1
|
Ostrom QT, Blank PMD, Kruchko C, Petersen
CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio
JJ and Barnholtz-Sloan JS: Alex's lemonade stand foundation infant
and childhood primary brain and central nervous system tumors
diagnosed in the united states in 2007–2011. Neuro Oncol. 16 Suppl
10:x1–x36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang SY, Park CK, Park SH, Kim DG, Chung
YS and Jung HW: Atypical and anaplastic meningiomas: Prognostic
implications of clinicopathological features. J Neurol Neurosurg
Psychiatry. 79:574–580. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Riemenschneider MJ, Perry A and
Reifenberger G: Histological classification and molecular genetics
of meningiomas. Lancet Neurol. 5:1045–1054. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maillo A, Orfao A, Espinosa AB, Sayagués
JM, Merino M, Sousa P, Lara M and Tabernero MD: Early recurrences
in histologically benign/grade I meningiomas are associated with
large tumors and coexistence of monosomy 14 and del(1p36) in the
ancestral tumor cell clone. Neuro Oncol. 9:438–446. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perry A, Scheithauer BW, Stafford SL,
Lohse CM and Wollan PC: ‘Malignancy‘ in meningiomas: A
clinicopathologic study of 116 patients, with grading implications.
Cancer. 85:2046–2056. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mawrin C and Perry A: Pathological
classification and molecular genetics of meningiomas. J Neurooncol.
99:379–391. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lomas J, Bello MJ, Arjona D, Alonso ME,
Martinez-Glez V, Lopez-Marin I, Amiñoso C, de Campos JM, Isla A,
Vaquero J and Rey JA: Genetic and epigenetic alteration of the NF2
gene in sporadic meningiomas. Genes Chromosomes Cancer. 42:314–319.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harada T, Irving RM, Xuereb JH, Barton DE,
Hardy DG, Moffat DA and Maher ER: Molecular genetic investigation
of the neurofibromatosis type 2 tumor suppressor gene in sporadic
meningioma. J Neurosurg. 84:847–851. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ng HK, Lau KM, Tse JY, Lo KW, Wong JH,
Poon WS and Huang DP: Combined molecular genetic studies of
chromosome 22q and the neurofibromatosis type 2 gene in central
nervous system tumors. Neurosurgery. 37:764–773. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choy W, Kim W, Nagasawa D, Stramotas S,
Yew A, Gopen Q, Parsa AT and Yang I: The molecular genetics and
tumor pathogenesis of meningiomas and the future directions of
meningioma treatments. Neurosurg Focus. 30:E62011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lamszus K, Kluwe L, Matschke J, Meissner
H, Laas R and Westphal M: Allelic losses at 1p, 9q, 10q, 14q, and
22q in the progression of aggressive meningiomas and
undifferentiated meningeal sarcomas. Cancer Genet Cytogenet.
110:103–110. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clark VE, Ersonomay EZ, Serin A, Yin J,
Cotney J, Ozduman K, Avşar T, Li J, Murray PB, Henegariu O, et al:
Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7,
KLF4, AKT1, and SMO. Science. 339:1077–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang X, Shi L, Gao F, Russin J, Zeng L,
He S, Chen TC, Giannotta SL, Weisenberger DJ, Zada G, et al:
Genomic and transcriptome analysis revealing an oncogenic
functional module in meningiomas. Neurosurg Focus. 35:E32013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Serna E, Morales JM, Mata M,
Gonzalez-Darder J, Miguel San T, Gil-Benso R, Lopez-Gines C,
Cerda-Nicolas M and Monleon D: Gene expression profiles of
metabolic aggressiveness and tumor recurrence in benign meningioma.
PLoS One. 8:e672912013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Surace EI, Lusis E, Murakami Y,
Scheithauer BW, Perry A and Gutmann DH: Loss of tumor suppressor in
lung cancer-1 (TSLC1) expression in meningioma correlates with
increased malignancy grade and reduced patient survival. J
Neuropathol Exp Neurol. 63:1015–1027. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmidt M, Mock A, Jungk C, Sahm F, Ull
AT, Warta R, Lamszus K, Gousias K, Ketter R, Roesch S, et al:
Transcriptomic analysis of aggressive meningiomas identifies PTTG1
and LEPR as prognostic biomarkers independent of WHO grade.
Oncotarget. 7:14551–14568. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Francesca M, Orzan FN, Eoli M, Farinotti
M, Maderna E, Pisati F, Bianchessi D, Valletta L, Lodrini S, Galli
G, et al: DNA microarray analysis identifies CKS2 and LEPR as
potential markers of meningioma recurrence. Oncologist.
16:1440–1450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:(Database Issue).
D991–D995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tabernero MD, Maillo A, Gilbellosta CJ,
Castrillo A, Sousa P, Merino M and Orfao A: Gene expression
profiles of meningiomas are associated with tumor cytogenetics and
patient outcome. Brain Pathol. 19:409–420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smyth GK: Limma: Linear models for
microarray data. In: Bioinformatics and Computational Biology
Solutions Using R and BioconductorStatistics for Biology and
Health. Gentleman R, Carey V, Huber W, Irizarry RA and Dudoit S:
Springer; New York, NY: pp. 397–420. 2005, View Article : Google Scholar
|
|
23
|
Gene Ontology Consortium: The gene
ontology project in 2008. Nucleic Acids Res. 36:(Database Issue).
D440–D444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nature Protocol. 4:44–57. 2009.
View Article : Google Scholar
|
|
26
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41:(Database Issue). D808–D815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rooprai HK, Martin AJ, King A, Appadu UD,
Jones H, Selway RP, Gullan RW and Pilkington GJ: Comparative gene
expression profiling of ADAMs, MMPs, TIMPs, EMMPRIN, EGF-R and
VEGFA in low grade meningioma. Int J Oncol. 49:2309–2318. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kirches E, Grunewald J, Von-Bossanyi P,
Szibor R, Plate I, Krüger S, Warich-Kirches M and Dietzmann K:
Expression of matrix metalloproteinases in a series of 12
meningiomas. Clin Neuropathol. 20:26–30. 2001.PubMed/NCBI
|
|
30
|
Roy R, Zhang B and Moses MA: Making the
cut: Protease-mediated regulation of angiogenesis. Exp Cell Res.
312:608–622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bourboulia D and Stetler-Stevenson WG:
Matrix metalloproteinases (MMPs) and tissue inhibitors of
metalloproteinases (TIMPs): Positive and negative regulators in
tumor cell adhesion. Semin Cancer Biol. 20:161–168. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Keller A, Ludwig N, Backes C, Romeike BF,
Comtesse N, Henn W, Steudel WI, Mawrin C, Lenhof HP and Meese E:
Genome wide expression profiling identifies specific deregulated
pathways in meningioma. Int J Cancer. 124:346–351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vara Fresno JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC,
Whang-Peng J, Liu JM, Yang DM, Yang WK and Shen CY: PIK3CA as an
oncogene in cervical cancer. Oncogene. 19:2739–2744. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shayesteh L, Lu Y, Kuo WL, Baldocchi R,
Godfrey T, Collins C, Pinkel D, Powell B, Mills GB and Gray JW:
PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet.
21:99–102. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen F: JUN (V-Jun sarcoma virus 17
oncogene homolog (avian)). Atlas Genet Cytogenet Oncol Haematol.
7:85–86. 2003.
|
|
37
|
Wei C, Xiao W, Zhang K, Yin X, Lai J,
Liang L and Chen D: Activation of c-Jun predicts a poor response to
sorafenib in hepatocellular carcinoma: Preliminary clinical
evidence. Sci Rep. 6:229762016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Blau L, Knirsh R, Ben-Dror I, Oren S,
Kuphal S, Hau P, Proescholdt M, Bosserhoff AK and Vardimon L:
Aberrant expression of c-Jun in glioblastoma by internal ribosome
entry site (IRES)-mediated translational activation. Proc Natl Acad
Sci USA. 109:E2875–E2884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ameyar M, Wisniewska M and Weitzman JB: A
role for AP-1 in apoptosis: The case for and against. Biochimie.
85:747–752. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huhe M, Liu S, Zhang Y, Zhang Z and Chen
Z: Expression levels of transcription factors c-Fos and c-Jun and
transmembrane protein HAb18G/CD147 in urothelial carcinoma of the
bladder. Mol Med Rep. 15:2991–3000. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mahner S, Baasch C, Schwarz J, Hein S,
Wölber L, Jänicke F and Milde-Langosch K: C-Fos expression is a
molecular predictor of progression and survival in epithelial
ovarian carcinoma. Brit J Cancer. 99:1269–1275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Winnay JN, Solheim MH, Dirice E, Sakaguchi
M, Noh HL, Kang HJ, Takahashi H, Chudasama KK, Kim JK, Molven A, et
al: PI3-kinase mutation linked to insulin and growth factor
resistance in vivo. J Clin Invest. 126:1401–1412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thauvin-Robinet C, Auclair M, Duplomb L,
Caron-Debarle M, Avila M, St-Onge J, Le Merrer M, Le Luyer B, Héron
D, Mathieu-Dramard M, et al: PIK3R1 mutations cause syndromic
insulin resistance with lipoatrophy. Am J Hum Genet. 93:141–149.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McCurdy CE, Schenk S, Holliday MJ, Philp
A, Houck JA, Patsouris D, MacLean PS, Majka SM, Klemm DJ and
Friedman JE: Attenuated Pik3r1 expression prevents insulin
resistance and adipose tissue macrophage accumulation in
diet-induced obese mice. Diabetes. 61:2495–2505. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Seliger C, Meier CR, Becker C, Jick SS,
Proescholdt M, Bogdahn U, Hau P and Leitzmann MF: Metabolic
syndrome in relation to risk of meningioma. Oncotarget.
8:2284–2292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Williamson CR, Khurana S, Nguyen P, Byrne
CJ and Tai TC: Comparative analysis of renin-angiotensin system
(RAS)-related gene expression between hypertensive and normotensive
rats. Med Sci Monit Basic Res. 23:20–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Campisi J, Gray HE, Pardee AB, Dean M and
Sonenshein GE: Cell-cycle control of c-myc but not c-ras expression
is lost following chemical transformation. Cell. 36:241–247. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fei X, Yuan Y, Xie L, Ran P, Xiang X,
Huang Q, Qi G, Guo X, Xiao C and Zheng S: miRNA-320a inhibits tumor
proliferation and invasion by targeting c-Myc in human
hepatocellular carcinoma. Onco Targets Ther. 10:885–894. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Staal JA, Pei Y and Rood BR: A
proteogenomic approach to understanding MYC function in metastatic
medulloblastoma tumors. Int J Mol Sci. 17:pii: E1744. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Finver SN, Nishikura K, Finger LR, Haluska
FG, Finan J, Nowell PC and Croce CM: Sequence analysis of the MYC
oncogene involved in the t(8;14)(q24;q11) chromosome translocation
in a human leukemia T-cell line indicates that putative regulatory
regions are not altered. Proc Natl Acad Sci USA. 85:3052–3056.
1988. View Article : Google Scholar : PubMed/NCBI
|