Introduction
Systemic sclerosis (SSc), also known as scleroderma,
is a multisystem connective tissue disease, and its pathogenesis is
associated with several factors, including inflammation, autoimmune
antibodies, extensive fibrosis and microvascular changes (1). This disease may lead to life-threatening
complications, including pulmonary hypertension and interstitial
lung disease (ILD) (2). The roles of
the oxidative stress pathways in SSc pathogenesis have been
explored in previously. Bourji et al (3) reported reactive oxygen species (ROS) may
be involved in early skin fibrogenesis. Constitutive intracellular
production of ROS is essential for fibroblast proliferation and
expression of type I collagen (Col-I) in SSc cells (4–6).
Furthermore, the development of fibrosis involves a detrimental
cycle between transforming growth factor-β1 (TGF-β1) and ROS
(7). Emerging evidence indicates that
ROS stimulates fibrosis through modulation of the TGF-β1/Smad2/3
signaling pathway (8,9). Similar to resident epithelial cells and
fibroblasts, TGF-β1 drives fibrocytes to differentiate into
myofibroblasts through activation of the Smad2/3, stress-activated
protein kinase/JUN N-terminal kinase, and mitogen-activated protein
kinase signaling pathways, which stimulate α-smooth muscle actin
(α-SMA) expression (10,11). Due to the complex pathogenesis of SSc,
the availability of effective therapies to treat this disease is
currently limited.
Asiatic acid (AA) is a triterpenoid extracted from
Centella asiatica (12) with a
variety of protective properties, including against inflammation
(13), oxidation (14), and fibrosis (15,16).
Furthermore, AA reduces islet fibrosis, suppresses
mitochondria-mediated inflammasome activation, ameliorates hepatic
lipid accumulation and insulin resistance, and reduces infarct
volume following focal cerebral ischemia (3,17–19). Previous studies have demonstrated AA
alleviates cardiovascular remodeling in hypertensive rats and
protects against diabetic cardiomyopathy (4,5). In
addition, AA may inhibit liver and renal tuberlointersitial
fibrosis (12,20), and bleomycin -induced pulmonary
fibrosis (PF) (21).
Overall, it is essential to characterize the
pathogenesis of SSc and associated interstitial pneumonia, as well
as identify a fibrosis-inhibiting drug that is therapeutically
effective with few side effects. In the present study, whether AA
ameliorates PF, and modulates myofibroblast differentiation, immune
dysfunction and oxidative stress was determined. In addition, it
was determined whether any of these effects are mediated through
the classical TGF-β1/Smad2/3 signaling pathway.
Materials and methods
Animals
A total of 40, six-week-old female-specific
pathogen-free BALB/c mice with a mean weight of 25 g were housed at
the Experimental Animal Center at Wenzhou Medical University
(Zhejiang, China) at 20–24°C with 40–60% humidity and with a
regular light-dark cycle. The animals were allowed free access to
water and standard mouse chow. The present study was approved by
the Institutional Animal Care and Use Committee of Wenzhou Medical
University. Efforts were made to minimize animal suffering and the
number of animals used in experiments.
Reagents
Sodium hypochlorite (NaClO), potassium dihydrogen
phosphate (KH2PO4) solution, sodium
carboxymethylcellulose (CMC-Na) and AA were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). AA was dissolved in
dimethyl sulfoxide, and then diluted with a 0.5% CMC-Na solution to
create separate solutions with concentrations of 0.1 and 0.4 mg/ml.
Advanced oxidation protein products (AOPP; 10572-09m), E-selectin
(E-sel; 10316-09m) and anti-DNA topoisomerase I autoantibody
(TOP1-Ab; 10821-09m) enzyme-linked immunosorbent assay (ELISA) kits
were obtained from Shanghai Boyun Biochemical Institute (Shanghai,
China; http://www.chem-china.net/). Rabbit
antibodies against type I collagen (Col-I; ab21286) and α-SMA
(ab5694), and mouse antibodies against TGF-β1 (ab64715) were
purchased from Abcam (Cambridge, UK). Rabbit antibodies against
GAPDH (cat. no. 8884), Smad2/3 (cat. no. 8685), and phosphorylated
(p)-Smad2/3 (cat. no. 8828) were purchased from Cell Signaling
Technology Inc., (Danvers, MA, USA). Goat anti-rabbit (BA1054) and
goat anti-mouse horseradish peroxidase (HRP)-conjugated IgG
(BA1050) were purchased from Wuhan Boster Biological Technology,
Ltd. (Wuhan, China). A DAB kit and mouse/rabbit plus Polymer HRP
Detection system (PV-6000) were provided by OriGene Technologies,
Inc. (Beijing, China). SuperSignal West Femto Maximum Sensitivity
substrate and a BCA Protein Assay kit were obtained from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). RIPA lysis buffer was
obtained from Beyotime Institute of Biotechonology (Shanghai,
China).
Generation of HOCl
HOCl was synthesized by adding 166 µl NaClO solution
(2.6% active chlorine) to 11.1 ml KH2PO4
solution (100 mM; pH 7.2) as previously described (22). The HOCl concentration was measured
using a spectrophotometer at wavelength 292 nm (molar absorption
coefficient, 350 M−1cm−1).
Experimental groups and
treatments
The mice were randomly divided into control, model,
treatment with a low dose of AA of 2 mg/kg/day (LAA), and treatment
with a high dose of AA of 8 mg/kg/day (HAA) groups (n=10
mice/group). AA was dissolved in a 0.5% CMC-Na solution to create
separate solutions with concentrations of 0.1 and 0.4 mg/ml. Mice
in the model group received 300 µl HOCl subcutaneously and 0.5%
CMC-Na solution by gavage every day for 6 weeks, while mice in the
control group received 300 µl sterilized PBS subcutaneously and
0.5% CMC-Na solution orally every day for 6 weeks. Mice in the LAA
and HAA groups received 0.5 ml AA solution (2 and 8 mg/kg/day,
respectively) by gavage, as well as received HOCl injections as
aforementioned for the model group.
Two weeks after treatment was halted, the mice were
sacrificed, and their lungs and serum harvested. Lower right lung
lobe samples were washed with cold PBS, fixed in 4% buffered
paraformaldehyde at 4°C for 24 h, and then embedded in paraffin for
histological and immunohistochemical studies. The remaining lung
tissue was snap-frozen in liquid nitrogen and stored at −80°C until
processed for protein extraction. Expression of AOPP, E-sel, and
TOP1-Ab was measured in the collected mouse serum.
Histopathological and
immunohistochemical staining
The lower right lobes of the collected lungs were
fixed in 4% paraformaldehyde at 4°C overnight, dehydrated in a
graded ethanol series, embedded in paraffin, and then sectioned
into 5-µm thick slices. Histopathological changes were assessed
using hematoxylin and eosin (H&E) staining. Tissue sections
were deparaffinized in xylene and hydrated gradually through graded
alcohol series and processed with PBS buffer solution. Hematoxylin
was applied for 3 min in room temperature, and then rinsed in
H2O for 5 min. Eosin was used for staining for 1 min in
room temperature, and then H2O for 1 min. The sections
were dehydrated with an ascending alcohol series (75, 85, 95 and
100%) for 3 min each, and then xylene was used twice for 3 min
each. Another set of sections was stained to visualize interstitial
collagen using Masson's trichrome method (23). Primary antibodies against α-SMA and
Col-I were diluted 1:300 in PBS. Tissue were treated with 3%
hydrogen peroxide at 37°C for 10 min, and high pressure cooker,
>120°C for 10 min. Primary antibodies against α-SMA and Col-I
were used and kept overnight at 4°C. After washing with PBS, then
incubated with the secondary antibody of PV-6000 at 37°C for 20
min, and then dyed with DAB developer of PV-6000. The sections were
dehydrated by graded alcohol for 5 min each, and then washed in
xylene twice for 3 min. Pulmonary histopathological changes and
areas that stained positive were observed by light microscopy. Five
random, non-overlapping, high-power (original magnification, ×400)
fields were evaluated on each slide. The integrated optical density
of positively stained cells was measured using Image-ProPlus 6.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Cytokine assays
Expression levels of AOPP, E-sel and TOP1-Ab in
serum were measured using the relevant ELISA kits according to the
manufacturer's protocol. A total of 50 µl of each standard and
sample were added into the appropriate well. 10 µl Biotinylated
antibodies to AOPP and E-sel, and antigen to TOP1 were coated to
samples separately. 50 µl chain enzyme avidin-HRP was added and
incubated at 37°C for 60 min, followed by washing four times with
washing buffer (provided in the kit) for 30 sec. Developer (100 µl)
was added and kept in the dark at 37°C for 10 min. The stop
solution (50 µl) was added to each well to terminate the reaction.
The optical density of each sample was read at 450 nm. The results
were calculated using the linear regression equation based on the
standard curve.
Western blot analysis
The lungs were homogenized in RIPA lysis buffer
containing phenylmethylsulfonyl fluoride (PMSF) (RIPA: PMSF 100:1)
and then centrifuged at 15,000 × g for 30 min at 4°C. The resulting
supernatants were collected and determined using the BCA protein
assay. Subsequently, 20 µg of protein/lane was separated using 10%
SDS-PAGE and then transferred onto polyvinylidene difluoride
membranes. The membranes were then blocked at room temperature for
1 h with 5% milk and incubated overnight at 4°C with primary
antibodies against TGF-β1 (1:500), Smad2/3, and p-Smad2/3 (1:500)
and GAPDH (1:500). Subsequently, the membranes were incubated with
the relevant HRP conjugated secondary antibody (goat anti-rabbit
IgG for GAPDH, 1:10,000; goat anti-mouse IgG for the other primary
antibodies, 1:10,000) at room temperature for 1 h and then
developed using Clarity Western ECL Substrate (cat. no. 1705060;
Bio-Rad Laboratories, Hercules, CA, USA). Images were collected and
analyzed using the Image Lab program 5.0 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
All calculations were performed using SPSS version 19.0 (IBM Corp.,
Armonk, NY, USA). Differences between groups were identified by
one-way analysis of variance (ANOVA) with post hoc contrasts by
Student-Newman-Keuls test. P<0.05 was considered statistically
significant.
Results
Effect of AA on pulmonary interstitial
fibrosis in vivo
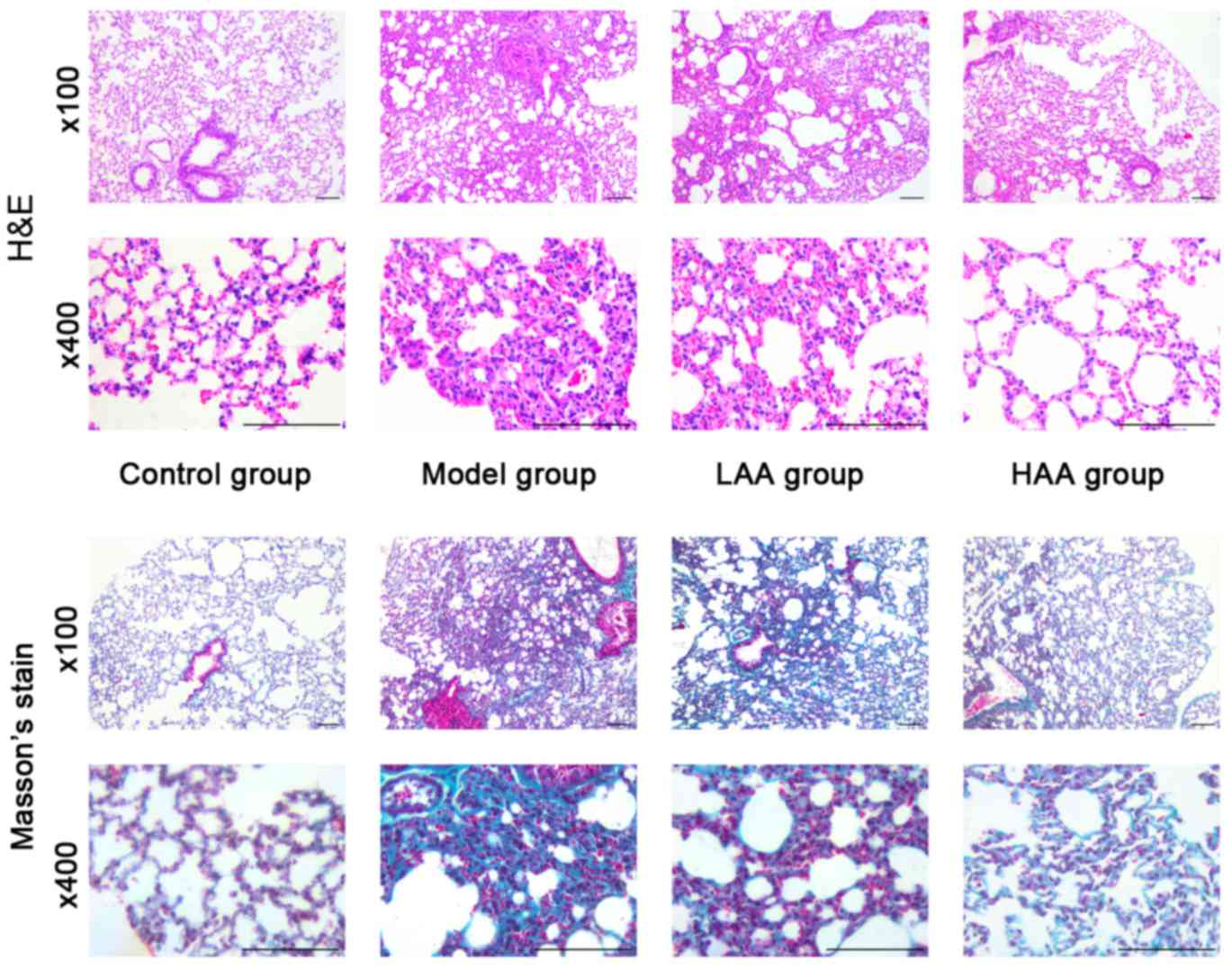
In the model group, the lungs were pale and stiff,
and were harder compared with lungs from the control group
following treatment for 6 weeks with HOCl (Fig. 1). In addition, amongst the groups
treated with AA, the LAA and HAA groups had softer and more
flexible lungs compared with the model group, indicating elasticity
was rescued in these lungs. The effect of AA on remodeling of the
lung tissue in SSc mice was assessed based on histopathological
examination. Lungs in the model group exhibited thickening of their
alveolar walls, broadening of their septum and infiltration of
inflammatory cells as shown in Fig.
1. However, this inflammation was not observed in either the
LAA or HAA groups. Based on Masson's stain, there was also less
collagen fiber deposition in the lungs of AA-treated mice.
Furthermore, the lung structure in the HAA group was more similar
to normal lungs compared with the model group.
AA affects α-SMA and Col-I
expression
The therapeutic antifibrotic effects of AA on lung
pathology was assessed in a HOCl-induced SSc mouse model.
Immunohistochemical staining was used to evaluate α-SMA and Col-I
expression in lungs prior to and following AA treatment (Fig. 2). The expression of α-SMA and Col-I
was significantly reduced in the lungs following AA treatment
compared with the model group. Compared with the control group, the
expression levels of both proteins were significantly higher in the
model group (P<0.05). Compared with the model group, the
expression levels of both proteins were significantly lower in the
HAA group (P<0.05). Notably, α-SMA expression in the HAA group
was significantly lower compared with that of the LAA group
(P<0.05). No significant difference was identified in Col-I
expression between the LAA and HAA groups, but a decreasing trend
was observed.
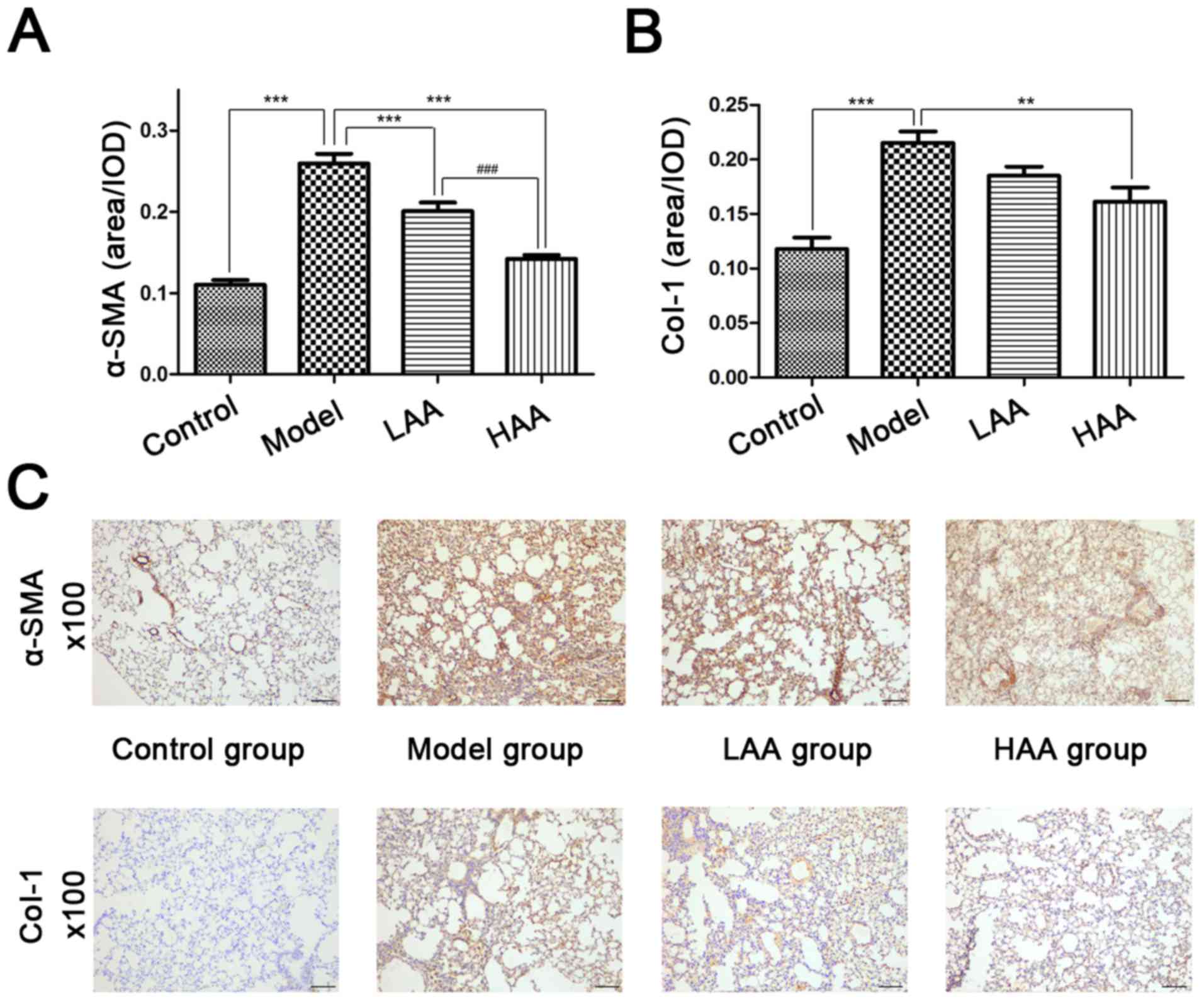 | Figure 2.Effect of AA on α-SMA and Col-I
expression in SSc and normal lungs. The effect of AA on (A) α-SMA
and (B) Col-I expression in each mouse group. (C)
Immunohistochemical staining was used to evaluate α-SMA and Col-I
expression in mouse lungs. Images are presented at magnification,
×100. **P<0.01, and ***P<0.001, compared with the model
group. ###P<0.001, compared with the LAA group.
Image-ProPlus software was used to calculate the relative
expression levels. Scale bar, 100 µm. AA, asiatic acid; LAA, low
dose of AA (2 mg/kg/day); HAA, high dose of AA (8 mg/kg/day);
α-SMA, α-smooth muscle actin; Col-I, type I collagen; OD, optical
density. |
Effect of AA on AOPP, E-sel, and
TOPI-Ab concentrations
To evaluate the effects of AA on oxidative stress,
damage to the epithelium and vessels, and immune response, AOPP,
E-sel and TOPI-Ab expression was evaluated in serum from the mice
(Fig. 3A-C). Compared with the
control group, AOPP, E-sel and TOPI-Ab levels were significantly
higher in the model group (P<0.05). In addition, all three
expression levels were significantly lower in the HAA group
compared with the model group (P<0.05). Expression of E-sel and
TOPI-Ab was significantly lower in the HAA group compared with the
LAA group (P<0.05). No significant difference in E-sel and
TOPI-Ab levels were identified between the LAA and model group.
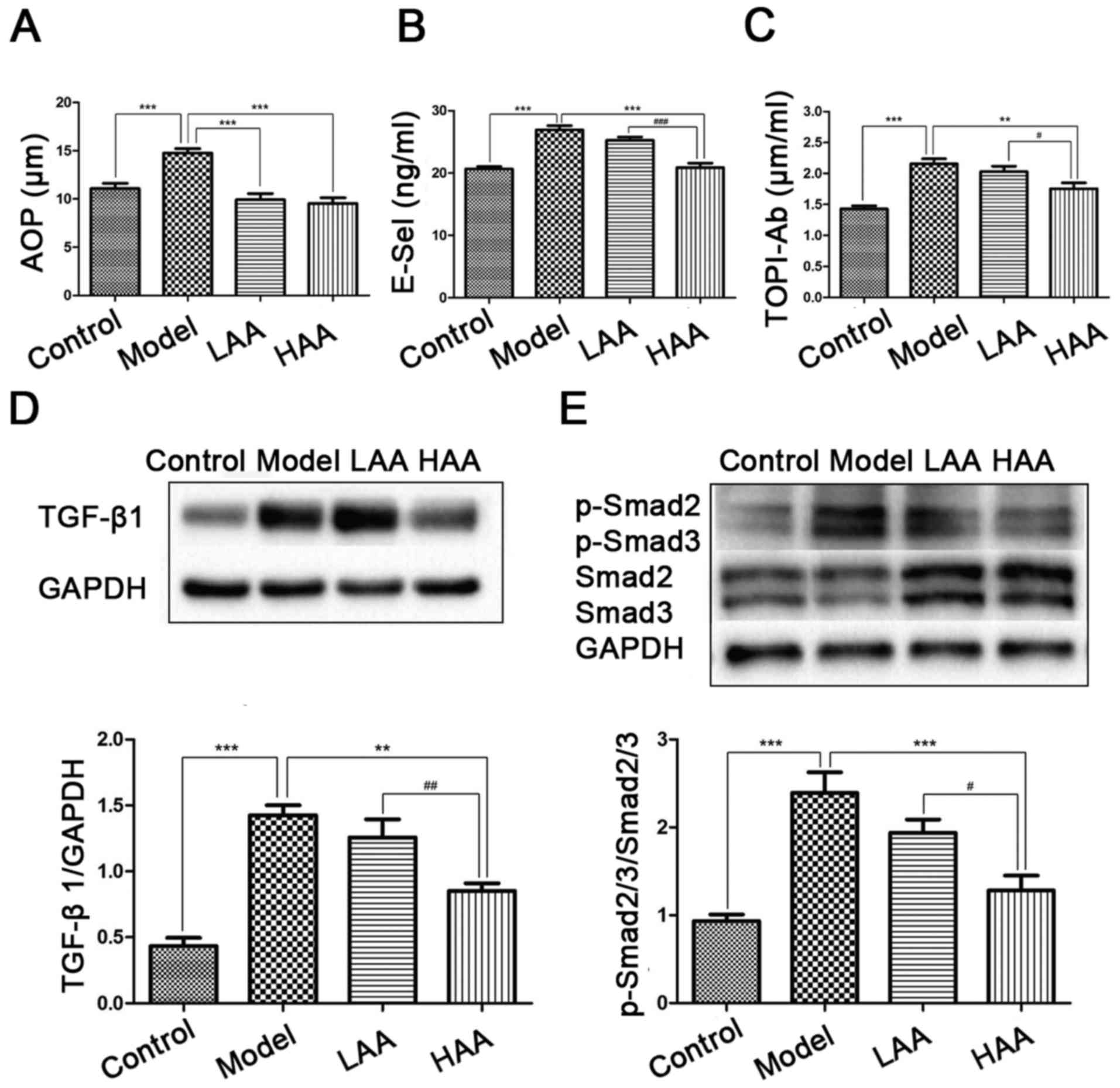 | Figure 3.Effect of AA on AOPP, E-Sel, TOP1-Ab,
TGF-β1 and p-Smad2/3 expression levels. Effects of AA on (A) AOPP,
(B) E-Sel and (C) TOP1-Ab levels in mouse serum. (D) TGF-β1 and (E)
p-Smad2/3/Smad2/3 levels were measured by western blotting.
**P<0.01, and ***P<0.001, compared with the model group.
#P<0.05, ##P<0.01, and
###P<0.001, compared with the LAA group. AA, asiatic
acid; LAA, low dose of AA (2 mg/kg/day); HAA, high dose of AA (8
mg/kg/day); TGF-β1, tumor growth factor-β1; p-, phosphorylated;
AOPP, advanced oxidation protein products; E-sel, E-selectin;
TOP1-Ab, anti-DNA topoisomerase I autoantibody. |
Effect of AA on TGF-β1 and
p-Smad2/3/Smad2/3 expression
To identify the potential mechanism underlying the
effect on AA treatment of mice with SSc and SSc-associated ILD,
TGF-β1/Smad2/3 signaling way was examined by measuring TGF-β1
expression (Fig. 3D) and
p-Smad2/3/Smad2/3 ratios (Fig. 3E). A
higher expression level of TGF-β1 and higher p-Smad2/3/Smad2/3
ratios were identified in the model group compared with the control
(P<0.05). Expression of TGF-β1 was significantly decreased in
the HAA groups compared with the model group (P<0.05), as well
as significantly different between the LAA and HAA groups
(P<0.05).
Discussion
SSc is a multisystem disease with a variable
clinical course (1). Its prognosis is
typically poor, corresponding with the extent of microangiopathy,
and fibrosis of skin and internal organs (1). Although SSc pathogenesis is associated
with fibroblast disorder and leads to multi-organ or systemic
changes, the specific mechanisms underlying this disease have not
been well characterized, and there is still a lack of efficacious
chemical treatments.
It was previously demonstrated that ROS serve an
essential role in SSc-associated ILD pathogenesis (24). Overproduction of ROS, induced by
fibroblasts and endothelial cells, stimulate inflammatory
reactions, oxidization of DNA topoisomerase-I and overexpression of
AOPP (25). The high amounts of AOPP
generated by agents induced by HOCl in the skin spread the fibrosis
from skin to tissues via systemic circulation, and result in
SSc-associated ILD and TOPI-Ab (25,26). The
overexpression of DNA topoisomerase-I induces hydrogen peroxide
production by endothelial cells, and fibroblast proliferation
(25). Furthermore, oxidative stress
leads to T cell infiltration in the lung fibrosis (27). In the present study, ROS administered
in the form of subcutaneously injected HOCl was used to induce SSc
in a mouse model that has been widely used since 2009 (22). Preliminary experiments in SSc mice
were also performed by assessing the effects of different doses of
HOCl as described previously (7,20,28–30), 300
µl was chosen as the optimal dose. This model mimicked the diffuse
cutaneous form of human SSc, which exhibited increased collagen
deposition, inflammatory infiltration in the lungs and autoimmune
activation compared with control mice. Using this mouse model, the
therapeutic efficacy of AA for SSc was assessed.
TGF-β is the most potent profibrogenic cytokine and
its expression is increased in almost all fibrotic diseases
(31). TGF-β1, a member of the TGF
superfamily, is a potent profibrotic factor that induces collagen
synthesis and has been well studied in fibrogenesis (12). It has been demonstrated to serve an
important role in pulmonary fibrosis and airway remodeling
(32,33). Excessive activation of TGF-β1 induces
the phosphorylation of Smad2 and Smad3, subsequently forming a
novel complex on the nuclear membrane (34). Phosphorylation of receptor-regulated
SMADs, in particular Smad2/3 following TGF-β1 activation, binds to
TGF-β type I and II receptors, and activates the TGF-β1/Smad2/3
signaling pathway (8,9). Mesenchymal transition through the
activation of TGF-β1/Smad2/3 signaling pathway may stimulate
myofibroblast proliferation and fibroblast conversion to
myofibroblast (35). The current
study observed significantly high expression of TGF-β1 and high
pSmad2/3/Smad2/3 ratios in the HOCl-induced SSc mouse model
compared with the control group. A study by Xu et al
(11) delineated an underlying
mechanism of lung fibrosis in which epithelial and mesenchymal
cells are activated by TGF-β1/Smad2/3 signaling through Wnt and
β-catenin activation.
AA reduces the occupancy of Smad2/3 elements in
response to TGF-β. Previous studies have reported that AA treatment
inhibits TGF-β1 and Smad2/3 expression in cardiac hypertrophy, and
liver and renal interstitial fibrosis (12,14–16,20).
It was demonstrated AA suppresses TGF-β/Smad signaling in tissue
fibrosis and may be an effective candidate for treatment of acute
injury associated with pulmonary fibrosis (21). To the best of our knowledge, the
present study demonstrated for the first time that high
concentrations of AA may inhibit the phosphorylation of Smad2/3 by
reducing TGF-β1 in SSc-associated PF induced by HOCl. Based on the
data in the current study, in combination with a report by Dong
et al (21), AA-induced
inhibition of SSc-associated PF may be mediated, at least in part,
through the TGF-β/Smad2/3 signaling pathway. Autoimmune antibodies,
including TOP1-Ab, are produced for self-protection (18). E-sel is also released when cell damage
occurs (13,14). This was also observed in the serum of
SSc mice. A previous study demonstrated that serum E-sel level was
positively associated with the presence and extent of pulmonary
fibrosis (36). In addition, during
the early stage of SSc, activated fibrosis produces abundant ROS,
which stimulates Col-I expression and leads to fibrosis (25). The inhibition of the development of
mouse SSc-ILD by AA may explain the decreased expressions of
circulating oxidized proteins in the sera, and especially TOP1-Ab,
E-sel and depression in the concentration of Col-I in the lung of
treated animals. Therefore, we hypothesize the release of highly
toxic ROS by activated fibroblasts and endothelial cells induce
inflammation that triggers the recruitment of inflammatory cells,
the production of cytokines, and increases fibrosis.
In conclusion, the results of the present study
confirmed the presence of pulmonary inflammation and fibrosis in a
murine model of HOCl-induced SSc, and demonstrated that selective
inhibition of ROS reduces PF in this model. By focusing on the
classical TGF-β1/Smad2/3 signaling pathway, it was observed that AA
significantly inhibited the phosphorylation and, thus, the
activation of Smad2/3. This phenotype was even more significant
when high concentrations of AA were used. This pathway may be one
of the most important ways in which AA affects SSc-associated ILD.
A drug similar to AA that regulates the TGF-β1/Smad2/3 signaling
pathway may be a novel therapeutic for treatment of SSc.
Acknowledgements
The authors would like to thank Dr Kate Huang (The
First Affiliated Hospital of Wenzhou Medical University) and Dr
Jianbo Wu (The First Affiliated Hospital of Wenzhou Medical
University) in the Department of Pathology for expert technical
assistance and the Dr Yicheng He (Wenzhou Medical University) and
Dr Zhenni Zhou (Wenzhou Medical University) for help with the mice
experiments.
Funding
The present study was supported by a grant from the
Wenzhou Science and Technology Bureau Project, Zhejiang Province,
China (grant no. Y20140250).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW and YT conceived and designed the experiments; XX
and CD performed the animal experiments; YT, HY performed the
pathological examination; XH analyzed the data; AC contributed
materials and western blot analysis; XX, CD and LW wrote the
paper.
Ethics approval and consent to
participate
The present study was approved by the institutional
Animal Care and Use committee of Wenzhou Medical University
(wydw2016-0067).
Consent for publication
Not applicable.
Competing interests
The authors have declared that they have no
competing interests.
References
|
1
|
Balbir-Gurman A and Braun-Moscovici Y:
Scleroderma-new aspects in pathogenesis and treatment. Best Prac
Res Clin Rheumatol. 26:13–24. 2012. View Article : Google Scholar
|
|
2
|
Tyndall AJ, Bannert B, Vonk M, Airò P,
Cozzi F, Carreira PE, Bancel DF, Allanore Y, Müller-Ladner U,
Distler O, et al: Causes and risk factors for death in systemic
sclerosis: A study from the EULAR Scleroderma Trials and Research
(EUSTAR) database. Ann Rheum Dis. 69:1809–1815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bourji K, Meyer A, Chatelus E, Pincemail
J, Pigatto E, Defraigne JO, Singh F, Charlier C, Geny B, Gottenberg
JE, et al: High reactive oxygen species in fibrotic and nonfibrotic
skin of patients with diffuse cutaneous systemic sclerosis. Free
Radic Biol Med. 87:282–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoshizaki A, Iwata Y, Komura K, Ogawa F,
Hara T, Muroi E, Takenaka M, Shimizu K, Hasegawa M, Fujimoto M, et
al: CD19 regulates skin and lung fibrosis via Toll-like receptor
signaling in a model of bleomycin-induced scleroderma. Am J Pathol.
172:1650–1663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Allanore Y, Borderie D, Lemarechal H,
Ekindjian OG and Kahan A: Acute and sustained effects of
dihydropyridine-type calcium channel antagonists on oxidative
stress in systemic sclerosis. Am J Med. 116:595–600. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sambo P, Baroni SS, Luchetti M, Paroncini
P, Dusi S, Orlandini G and Gabrielli A: Oxidative stress in
scleroderma: Maintenance of scleroderma fibroblast phenotype by the
constitutive up-regulation of reactive oxygen species generation
through the NADPH oxidase complex pathway. Arthritis Rheum.
44:2653–2664. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Varga J and Abraham D: Systemic sclerosis:
A prototypic multisystem fibrotic disorder. J Clin Invest.
117:557–567. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fernandez IE and Eickelberg O: The impact
of TGF-β on lung fibrosis: From targeting to biomarkers. Proc Am
Thorac Soc. 9:111–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu RM and Desai LP: Reciprocal regulation
of TGF-β and reactive oxygen species: A perverse cycle for
fibrosis. Redox Biol. 6:565–577. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pociask DA, Sime PJ and Brody AR:
Asbestos-derived reactive oxygen species activate TGF-beta1. Lab
Invest. 84:1013–1023. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu L, Cui WH, Zhou WC, Li DL, Li LC, Zhao
P, Mo XT, Zhang Z and Gao J: Activation of Wnt/beta-catenin
signalling is required for TGF-β/Smad2/3 signalling during
myofibroblast proliferation. J Cell Mol Med. 21:1545–1554. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bian D, Zhang J, Wu X, Dou Y, Yang Y, Tan
Q, Xia Y, Gong Z and Dai Y: Asiatic acid isolated from Centella
asiatica inhibits TGF-β1-induced collagen expression in human
keloid fibroblasts via PPAR-gamma activation. Int J Biol Sci.
9:1032–1042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meng XM, Zhang Y, Huang XR, Ren GL, Li J
and Lan HY: Treatment of renal fibrosis by rebalancing TGF-β/Smad
signaling with the combination of asiatic acid and naringenin.
Oncotarget. 6:36984–36997. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Si L, Xu J, Yi C, Xu X, Ma C, Yang J, Wang
F, Zhang Y and Wang X: Asiatic acid attenuates the progression of
left ventricular hypertrophy and heart failure induced by pressure
overload by inhibiting myocardial remodeling in mice. J Cardiovasc
Pharmacol. 66:558–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu X, Si L, Xu J, Yi C, Wang F, Gu W,
Zhang Y and Wang X: Asiatic acid inhibits cardiac hypertrophy by
blocking interleukin-1β-activated nuclear factor-κB signaling in
vitro and in vivo. J Thorac Dis. 7:1787–1797. 2015.PubMed/NCBI
|
|
16
|
Xu C, Wang W, Xu M and Zhang J: Asiatic
acid ameliorates tubulointerstitial fibrosis in mice with ureteral
obstruction. Exp Ther Med. 6:731–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramachandran V and Saravanan R: Efficacy
of asiatic acid, a pentacyclic triterpene on attenuating the key
enzymes activities of carbohydrate metabolism in
streptozotocin-induced diabetic rats. Phytomedicine. 20:230–236.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barnes J and Mayes MD: Epidemiology of
systemic sclerosis: Incidence, prevalence, survival, risk factors,
malignancy, and environmental triggers. Curr Opin Rheumatol.
24:165–170. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Highland KB and Silver RM: New
developments in scleroderma interstitial lung disease. Curr Opin
Rheumatol. 17:737–745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong SH, Liu YW, Wei F, Tan HZ and Han ZD:
Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin
(BLM) via suppressing pro-fibrotic and inflammatory signaling
pathways. Biomed Pharmacother. 89:1297–1309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Asano Y, Ihn H, Yamane K, Kubo M and
Tamaki K: Impaired Smad7-Smurf-mediated negative regulation of
TGF-beta signaling in scleroderma fibroblasts. J Clin Invest.
113:253–264. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Ran X, Hu CL, Qin LP, Lu Y and
Peng C: Therapeutic effects of liposome-enveloped ligusticum
chuanxiong essential oil on hypertrophic scars in the rabbit ear
model. PLoS One. 7:e311572012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bei Y, Hua-Huy T, Nicco C, Duong-Quy S,
Le-Dong NN, Tiev KP, Chéreau C, Batteux F and Dinh-Xuan AT:
RhoA/Rho-kinase activation promotes lung fibrosis in an animal
model of systemic sclerosis. Exp Lung Res. 42:44–45. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Servettaz A, Goulvestre C, Kavian N, Nicco
C, Guilpain P, Chéreau C, Vuiblet V, Guillevin L, Mouthon L, Weill
B and Batteux F: Selective oxidation of DNA topoisomerase 1 induces
systemic sclerosis in the mouse. J Immunol. 182:5855–5864. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Servettaz A, Guilpain P, Goulvestre C,
Chéreau C, Hercend C, Nicco C, Guillevin L, Weill B, Mouthon L and
Batteux F: Radical oxygen species production induced by advanced
oxidation protein products predicts clinical evolution and response
to treatment in systemic sclerosis. Ann Rheum Dis. 66:1202–1209.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blackburn RM: Too much of a good thing:
Adenosine overload in adenosine-deaminase-deficient mice. Trends
Pharmacol Sci. 24:66–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HJ, Tashkin DP, Gjertson DW, Brown MS,
Kleerup E, Chong S, Belperio JA, Roth MD, Abtin F, Elashoff R, et
al: Transitions to different patterns of interstitial lung disease
in scleroderma with and without treatment. Ann Rheum Dis.
75:1367–1371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Adtani PN, Narasimhan M, Punnoose AM and
Kambalachenu HR: Antifibrotic effect of Centella asiatica
Linn and asiatic acid on arecoline-induced fibrosis in human buccal
fibroblasts. J Investig Clin Dent. 8:2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J, He T, Lu Q, Shang J, Sun H and
Zhang L: Asiatic acid preserves beta cell mass and mitigates
hyperglycemia in streptozocin-induced diabetic rats. Diabetes Metab
Res Rev. 26:448–454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato M, Hirayama S, Lara-Guerra H, Anraku
M, Waddell TK, Liu M and Keshavjee S: MMP-dependent migration of
extrapulmonary myofibroblast progenitors contributing to
posttransplant airway fibrosis in the lung. Am J Transplant.
9:1027–1036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wolters PJ, Collard HR and Jones KD:
Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol.
9:157–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Su BH, Tseng YL, Shieh GS, Chen YC, Wu P,
Shiau AL and Wu CL: Over-expression of prothymosin-alpha
antagonizes TGFβ signalling to promote the development of
emphysema. J Pathol. 238:412–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang P, Wang Y, Nie X, Braïni C, Bai R and
Chen C: Multiwall carbon nanotubes directly promote
fibroblast-myofibroblast and epithelial-mesenchymal transitions
through the activation of the TGF-β/Smad signaling pathway. Small.
11:446–455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamane K, Ihn H, Kubo M, Yazawa N, Kikuchi
K, Soma Y and Tamaki K: Increased serum levels of soluble vascular
cell adhesion molecule 1 and E-selectin in patients with localized
scleroderma. J Am Acad Dermatol. 42:64–69. 2000. View Article : Google Scholar : PubMed/NCBI
|