Introduction
Immune cells play an important role in the control
of malignancy. Thus, the challenge for immunotherapy is to use
advances in cellular and molecular immunology to develop strategies
that effectively and safely augment antitumor responses. However,
the immune system has effects on tumor growth in several ways. It
is thought that CD8+ T cell (CTL) is particularly
important in eradicating a tumor mass and these tumor-specific CTL
activated by dendritic cells (DC) bearing tumor antigens can kill
tumor cells directly. As far as this pathway is concerned, the aim
is to channel the tumor antigens into the DC-presentation pathway
and to induce the DC to differentiate into a potent
immunostimulatory cell (1,2). The key events that lead to this immune
response are: I) The ability of the (tumor) antigens to gain access
to DC, where they display large amounts of MHC-peptide complexes at
the surface; II) the subsequent maturation of DC to a fully
activated state in which antigen is presented with appropriate
co-stimulation; and III) involvement of activated, antigens-loaded
dendritic cells to traffic out of the tumor to peripheral lymphoid
tissue, where they liaise with and activate antigen-specific T
cells, these T cells can be recruited for tumor cell killing
(3,4).
A promising alternative to non-cell based vaccines
is vaccination using derivatives of whole tumor cells with a whole
array of tumor associated antigens (TAAs) that are both defined and
undefined. This strategy seemed to overcome the weak immunogenicity
of epitope-specific vaccines due to multiclonality of tumors and
their ability to downregulate specific antigens (5–7). Among
cell based vaccines, the method of preparing whole dead tumor cell
by administering a lethal dose of ultraviolet (UV) ray irradiation
has been an attractive approach to cancer therapy. However, the
area of irradiated cancer cell vaccines has been characterized by
numerous set-backs. For example, Canvax in, an irradiated whole
cell vaccine, demonstrated positive results in non-randomized
studies, but in phase III trials actually failed to provide a
survival advantage over BCG (8). The
major underlying reason for this phenomenon can be attributed to
the lack of spontaneous DC maturation after pulsing with irradiated
tumor cells. Given that tumor cells express a large load of self
antigens and have evolutionally adapted to induce immune tolerance,
new generation of the whole cell vaccines to prevent DC suppression
became critically needed.
Genetically modified tumor vaccines are based on the
genetic modification of either autologous or allogeneic tumor
cells, such modification may change the phenotype of the vaccine
making it more immunogenic. Up to date, numerous gene-modified
whole cells are being intensively tested in experimental or
clinical trials, such as autologous or allogeneic cells modified
with gene(s) encoding cytokines (9),
growth factors (10), human leukocyte
antigen (HLA) (11), co-stimulatory
molecules (12). Despite enormous
effort and promising data in animal studies and clinical trials,
however, clinical testing of therapeutic cancer vaccines is still
under debate. The failure of the majority of therapeutic cancer
vaccines is a reflection of the fact that the design of most of
these vaccines preceded a mature understanding of where individual
tumor types tend to fall on tumor immunogenicity (13). Therefore, a full understanding of the
interaction between developing tumors and the immune system is
vital to the logical design of a therapeutic cancer vaccine.
Previously, we have developed an adenoviral-mediated
genetically engineered hepatoma cells vaccine expressing HBx and
demonstrated that the irradiated HBx-modified hepatoma cell
vaccines could elicit significant specific antitumor immunity
against HCC. In addition, a considerable number of autophagosomes
or autolysosomes was observed in irradiated HBx-modified tumor
cells compared with control groups (14), but the mechanism by which autophagy
contributes to enhancing antitumor immune responses have yet to be
fully elucidated. In the present study, we examined how autophagy
induced by this vaccines influence the generation of danger signal
by hepatoma tumor cells and the subsequent activation of the
immunoresponse.
Materials and methods
Ethical statement
Ethical approval for the present study was provided
by the Ethics Committee of State Key Laboratory of Biotherapy,
Sichuan University.
Cell culture
The murine HCC cell line Hepa1-6 (H-2Kb)
and H22 (H-2Kd) were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA). All cell lines were
maintained in Dulbecco's modified Eagle's medium (DMEM) containing
10% fetal bovine serum (FBS) (both from Gibco-BRL, Carlsbad, CA,
USA), 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin. Stable Hepa1-6 cell line expressing HBx (Hepa1-6/HBx)
was constructed as previously described and cultured in complete
medium containing 200 µg/ml G418 (14). The cells mentioned above were
propagated at 37°C in humidified 5% CO2 conditions.
Bone marrow-derived DC and vaccine
preparation
C57BL/6 (H-2Kb) mouse bone marrow-derived DCs were
generated as previously described (15). Briefly, bone marrow cells were
harvested from the femur and tibia of 8- to 12-week-old mice. Cells
were cultured in RPMI-1640 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with recombinant murine GM-CSF (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at the concentration of 20 ng/ml.
The medium was refreshed every 2–3 days. After 7–10 days of
culture, non-adherent and loosely adherenT cells were harvested and
stained with anti-CD11c for purity verification. The purity was
~90%, and the cells were ready to use as DCs. Hepa1-6 cells
expressing HBx (Hepa1-6/HBx) were digested, washed 3 times with
fresh serum-free medium, irradiated with 50 Gy, and then was
centrifuged at 300 × g for 10 min to remove cells and large-cell
debris. Thereafter, the resulting suspension was centrifuged for 15
min at 12,000 × g to separate supernatant containing cytosolic
components and Dribbles (vaccine), the latter was collected and
incubated with purified DCs for 6 h. Then, the cells were collected
and washed with phosphate-buffered saline (PBS), then stained with
anti-CD40-FITC, anti-PD-L1-FITC, anti-CD80-FITC, and anti-CD86-FITC
(BD Biosciences, Franklin Lakes, NJ, USA) for phenotype analysis.
IL-12 and IFN-γ release in harvested supernatants of mature DCs
were subsequently assessed using a standard sandwich enzyme-linked
immunosorbent assay (ELISA) kit (R&D Systems, Shanghai, China)
according to the manufacturer's instructions. All samples were run
in triplicate.
Cell proliferation assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide (MTT)
was performed to test the toxicity of 3-MA, Hepa1-6 or DCs were
plated in 96-well culture clusters at a density of 1×105
cells/well in triplicate. After 24 h incubation, MTT (5.0 mg/l; 15
µl) was added to each well for 3 h, followed by the addition of 150
µl dimethyl sulfoxide (DMSO) into each well. The absorbance (A) of
the formazan product was determined at 570 nm using a plate
microreader and calculated using the following formula:
Cell viability (%) = (Asample -
Ablank)/(Acontrol - Ablank) ×
100
T cells proliferation assay
Spleens of 8- to 12-week-old mice were aseptically
removed, naïve T lymphocytes were isolated from single-cell
suspensions with Nylon Fiber Column T (Wako Chemicals USA, Inc.,
Richmond, VA, USA). To verify the T-cells proliferative ability
after co-incubation with stimulated DCs, naïve T-cells were
isolated from spleens of C57BL/6 mice and labeled by 0.5 µM CFSE
for 10 min at 37°C in 5% CO2. Then, an equal volume of
fetal calf serum (FCS) was added to stop the reaction. The cells
were centrifuged at 400 × g for 5 min and washed twice with
complete medium. CFSE-labeled cells were resuspended in 10%
RPMI-1640 medium, adjusted to 2×105 cells/ml and
stimulated by pulsed DCs at ratios of 2:1. The mixed cells were
co-incubated in 6-well plates at 37°C for 7 days. The labeled
dilution was measured on FACS Calibur and the divisive generations
were assayed via FlowJo software version 7.6.1 (Tree Star, Inc.,
San Carlos, CA, USA). Proliferation index was conveyed as the
percentage of cells that had divided and the average number of cell
divisions.
T cells cytotoxicity assay
The T cells cytotoxicity assay was performed as
previously described (14). Primed T
cells were as effector cells. After 5 days of stimulation with
pulsed DCs, effector cells were washed twice and resuspended to a
concentration of 107/ml. The Hepa1-6 cells were used as
target cells, labeled with CFSE (2.5 µM) as described above and
adjusted to a concentration of 105/ml. Effector cells
and target cells were mixed in total volume of 200 µl at E:T ratios
of 40, 20, 10 and 5 in round-bottom polystyrene tubes, centrifuged
at 400 × g for 45–60 sec and incubated at 37°C in 5% CO2
for 4–6 h. The target cells of each sample were 104
cells in 200 µl volume, and only target cells in the tube served as
a negative control. Following incubation, each sample was
supplemented with 20 µl PI (100 µg/ml) on the ice and the
acquisition was performed by flow cytometry within 1 h.
CFSE+/PI+ cells were considered as dead
target cells. Percentage of specific lysis was then expressed as: %
specific lysis = [dead targets in the sample (%) - spontaneously
dead targets (%)/100 - spontaneously dead targets (%)] × 100.
Characterization of T-lymphocyte
subsets
Naïve T lymphocytes were isolated and primed as
mentioned above. The T cells were then harvested and stained by
anti-CD4-APC and anti-IFN-γ-PE or anti-CD8-FITC and anti-IFN-γ-PE
(BD Biosciences). Fluorescence profiles were measured on
FACSCalibur and the data were analyzed by cell quest software (both
from BD Biosciences). To deplete CD8+ and
CD4+ T cells, 500 µg of either anti-CD4 (clone GK1.5,
rat IgG), anti-CD8 (clone 2.43, rat IgG) or isotype controls were
add to T cells 24 h before co-incubation with vaccine pulsed DCs,
after 5 days of stimulation with pulsed DCs, primed T cells were
collected and tested for cytotoxicity assay as mentioned above.
Western blotting
Briefly, 1×106 Hepa1-6 cells were lysed
in lysis buffer. Cells were removed by scraping, and centrifuged at
12,500 rpm for 30 min. The protein concentration of the supernatant
was determined by the Bio-Rad protein assay kit, and whole-cell
lysates after denaturing were separated by 10% SDS-PAGE. Gels were
electroblotted onto a polyvinylidene difluoride membrane. The
membrane blots were blocked at 4°C in 5% non-fat dry milk overnight
and incubated with each antibody at a recommended dilution for 8 h
at 37°C. Followed by rinsing in solution with 10 mM Tris-HCl pH
7.5, 100 mM NaCl, and 0.1% Tween-20 (TBS-T), the gels were
incubated inhorseradish peroxidase-conjugated secondary antibodies
at a dilution of 1:10,000. The immunoreactive bands were detected
by enhanced chemiluminescence (GE Healthcare, Chicago, IL, USA).
Equal loading was confirmed by detection of β-actin.
Adoptive transfer in vivo
Preparation of spleen lymphocytes and sera, and the
procedure for adoptive transfer was performed as previously
reported (10). All animal
experiments were performed in accordance with the guidelines of
Sichuan University and approved by the Animal Care Committee of
Sichuan University (Chengdu, China). Ten 6–8 weeks old C57BL/6
female mice in each group were immunized with 1×106
irradiated AdHBx-infected Hepa1-6 cells, irradiated Adnull-infected
Hepa1-6 cells, irradiated Hepa1-6 cells or NS s.c. in the right
flank 3 on days 1, 14 and 28. Sera were obtained from the mice on
Day 7 after the third immunization and were adoptively transferred
by intravenous injection one day (100 µl/mouse) before another set
of mice were challenged with 1×106 Hepa1-6 cells and
then were treated once per day for 10 days. In addition, isolated
spleen lymphocytes from the immunized mice were adoptively and
intravenously transferred (1×106 cells/100 µl/mouse) and
were then treated twice per week for 2 weeks. Tumor dimensions were
measured with calipers every 4 days for 30 days, and tumor volume
(V) was calculated according to the following formula: V = 0.52 ×
length × width2.
Statistical analyses
Statistical significance of experimental groups was
performed on Statistical Product and Service Solutions Software
(SPSS, V 19.0; IBM Corp., Armonk, NY, USA) and analyzed using a
one-way ANOVA, followed by LSD multiple comparisions or Dunnetts T3
multiple comparisons according to assumptions of equal variances
after homogeneity of variance test. The data were means ± SEM and
P-values <0.05 were considered to indicate statistically
significant differences, presented in figure captions.
Results
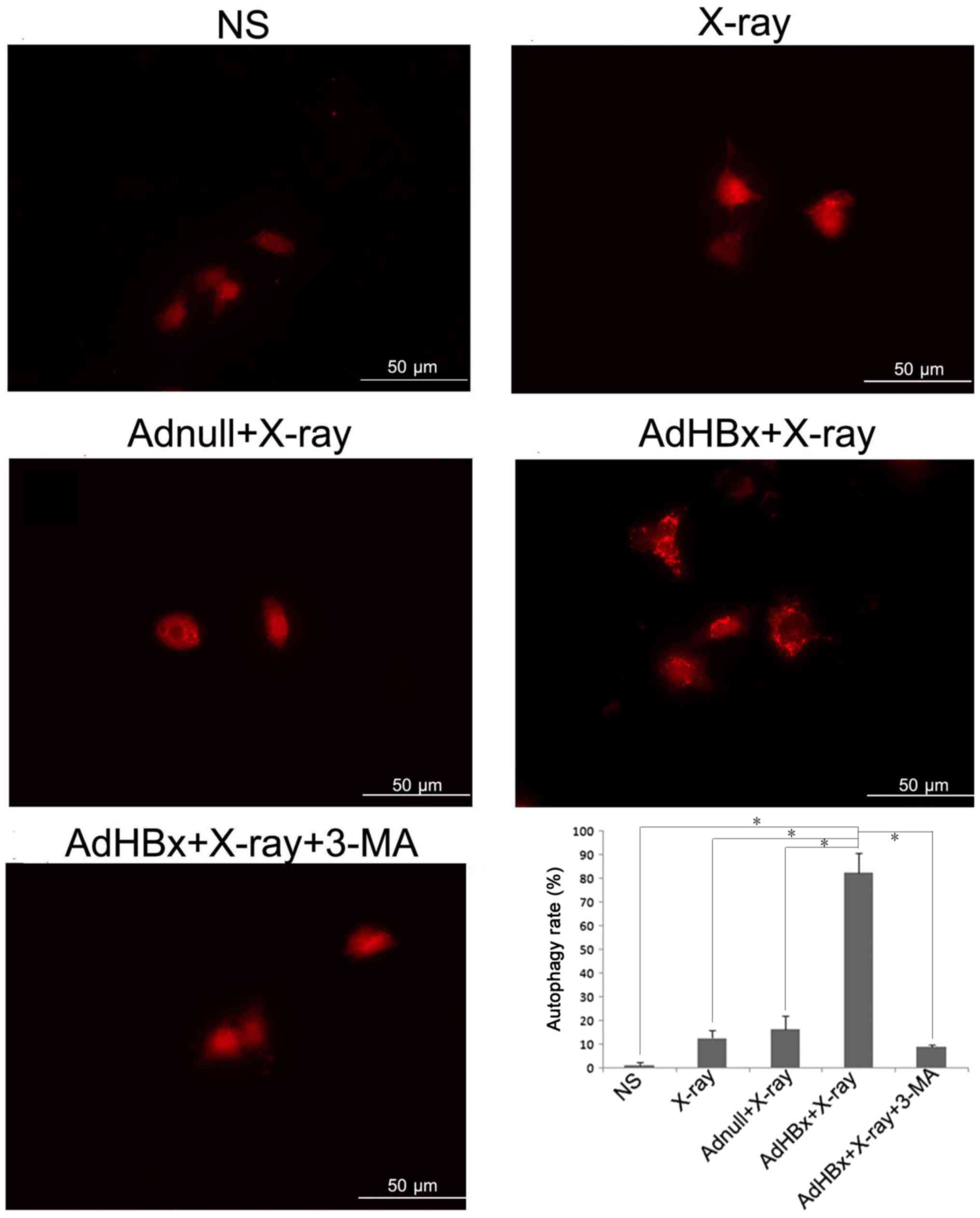
Induction of autophagy in Hepa1-6
cells by HBx plus irradiation
Our previous results have demonstrated that
treatment of Hepa1-6 cells with HBx plus X-ray irradiation has
resulted in numerous auto-phagosomes or auto-lysosomes in Hepa1-6
cells as analyzed by electron microscopy. To further verify the
autophagy in Hepa1-6 cells induced by HBx plus irradiation, Hepa1-6
cells were transfected with RFP-LC3 (a gift from Joanna M. Norman)
(16), which allows a more direct
visualization of autophagy by means of conversion of LC3-I to
LC3-II (change in the levels of RFP-LC3 puncta). As expected,
treatment of Hepa1-6 cells with infection of AdHBx and X-rays
irradiation led to the conversion of autophagy protein Atg8, from
an LC3-I form to an LC3-II form, reflected by significantly high
level of puncta in cells (Fig.
1).
Phenotypic maturation of DC by Hepa1-6
cells treated with HBx plus irradiation
Cell surface marker expression is a widely used
criterion for the assessment of DC maturation. High levels of
co-stimulatory and activation molecules are now considered to be
markers of DC maturation, whereas elevation of PD-L1 favors
inhibition of T-cell responses. Therefore, we focused on 4 cell
surface markers: Co-stimulatory markers (CD80 and CD86), maturation
marker CD40 and inhibitory marker PD-L1. Immature DCs were
generated from BM precursors using 2 ng/ml of GM-CSF. On day 8 of
culture, the cells with CD11c+ were isolated by MACS,
FCM analysis was conducted to test the cell purity of separated
BMDCs. Followed by purification with MACS, the CD11c+
cells were enriched and the results of FCM showed that the purity
of CD11c+ cells approached 90% (data not shown).
Purified CD11c+ immature DCs were further activated with
Hepa1-6 cells treated with HBx plus X-ray irradiation or each
control treatment. Flow cytometric analysis of DCs demonstrated
upregulation of 3 markers (CD80, CD86 and CD40) following
incubation with Hepa1-6 cells treated by HBx plus X-ray
irradiation, compared with control groups. In turn, PD-L1
expression on DCs was only slightly increased after pulsed with
vaccine (Fig. 2A). When vaccine was
co-treated with the phosphoinositide-3-kinase inhibitor, 3-MA, DCs
maturation were significantly reduced. Cytokine (IL-12 and IFN-γ)
production is another functional feature of DCs maturation,
indicating the activation of the Th1 immune response. Thus, we
examined cytokine profiles produced by pulsed DC in each group.
ELISA assay shown that IL-12 and IFN-γ was released in
significantly higher mounts in vaccine pulsed DC group than control
groups (Fig. 2B). Each treated DCs
were >90% viable (determined by propidium iodide (PI) nuclear
staining method) during the incubation (data not shown). Taken
together, these results demonstrate that irradiated HBx gene
modified tumor cells induce the phenotypic maturation of DCs. To
exclude the non-specific toxicity of 3-MA, MTT were performed on
both Hepa1-6 and DCs. It has been shown that 3-MA had no effect on
viability of both Hepa1-6 and DCs (data not shown).
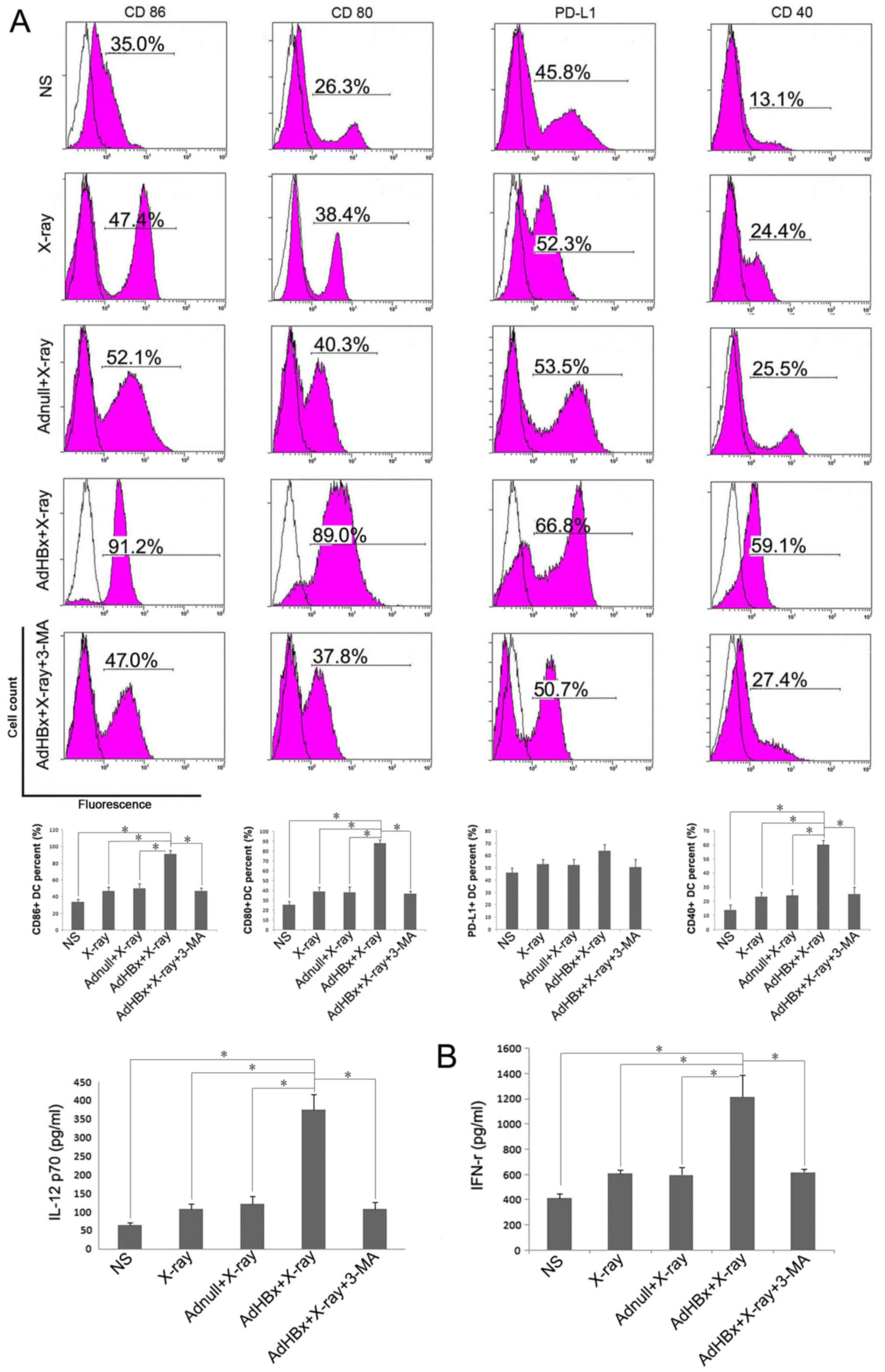 | Figure 2.Hepa1-6 cells treated with HBx plus
irradiation induced maturation of DCs. DCs were generated from
C57BL/6 mouse bone marrow and were cultured in RPMI-1640 with
recombinant murine GM-CSF. After plused with NS, irradiated
Adnull-infected Hepa1-6 cells, irradiated Hepa1-6 cells or
irradiated AdHBx-infected Hepa1-6 cells for 6 h, the cells were
collected and washed with PBS, then stained with anti-CD40-FITC,
anti-PD-L1-FITC, anti-CD80-FITC, and anti-CD86-FITC for phenotype
analysis. Inhibition of autophagy by 3-MA significantly abolished
the maturation of DCs (blank: isotype, pink: sample). (A)
Representative results of 3 independent experiments are shown.
Percentage of the DCs with the expression of each key surface
molecules were shown in bottom of each corresponding rank as mean ±
SEM (*P<0.05). The production of IL-12 and IFN-γ by the DCs
after treatment with NS, irradiated Adnull-infected Hepa1-6 cells,
irradiated Hepa1-6 cells, irradiated AdHBx-infected Hepa1-6 cells
or irradiated AdHBx-infected Hepa1-6 cells + 3-MA. (B) ELISA assay
shown that IL-12 and IFN-γ was released in significantly higher
mounts in vaccine pulsed DC group than control groups
(*P<0.05). |
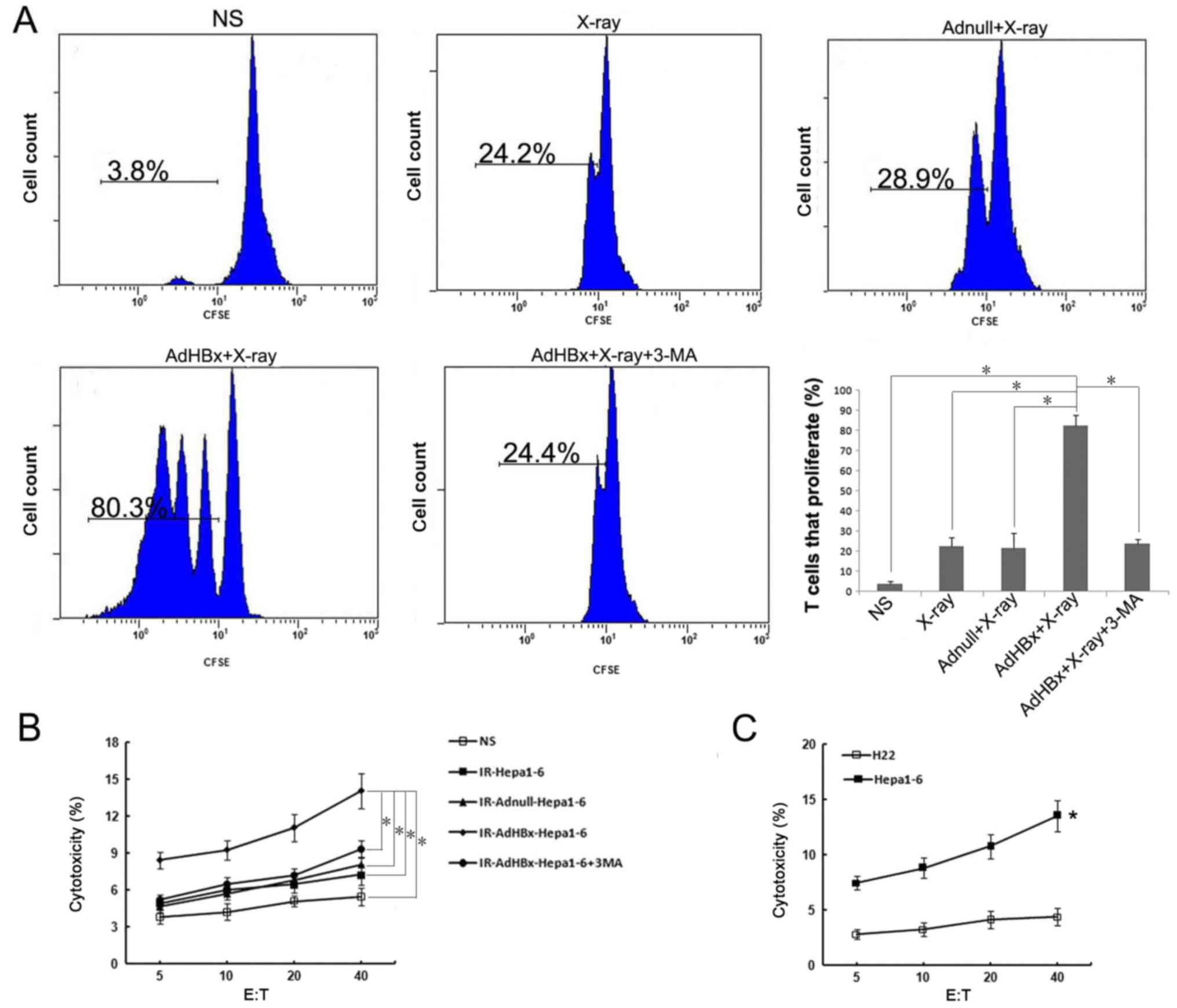
The autophagy in Hepa1-6 cells induced
by HBx plus irradiation affects cross-presentation and function of
primed CTL
Since activation of autophagy in donor cells would
affect cross-presentation of antigens, we tested whether Hepa1-6
cells in autophagic form as the antigen donor after co-incubation
with DCs may lead to the proliferation of naive T cells. Hepa1-6
cells were treated with infection of AdHBx and X-ray irradiation to
induce autophagy, or 3-MA, was added to prevent the initiation of
autophagy. The control groups were treated as mentioned above. Each
treated Hepa1-6 cells were incubated with DCs for 6 h, and then
CFSE-labeled naive T cells were added to the culture. T-cell
proliferation was measured by flow cytometry and analyzed with
FlowJo. Consistent with DC maturation, cross-presentation of
antigen from Hepa1-6 cells was greatly increased after treatment
with AdHBx plus X-ray irradiation as compared with control groups.
In contrast, inhibition of autophagy with 3-MA significantly
decreased cross-presentation of antigen to T-cells, close to the
level of T-cell proliferation with X-ray irradiation treatment
(Fig. 3A). To determine whether
primed lymphocytes could specifically target and kill tumor cells,
the primed lymphocytes in each group were co-cultured with
CFSE-labeled Hepa1-6 for 4 h. As shown in Fig. 3B, lymphocytes primed by pulsed DCs in
vaccine group were able to lyse Hepa1-6 cells (target cells) in an
E:T ratio-dependent manner with statistical significance at E:T
ratios ≥5 as compared with controls (P<0.05). In addition, we
have tested activation of T cells by direct adding of irradiated
tumor cells to naïve T lymphocytes, however, such as all previous
studies (17,18), T cell activation can only occur via
antigen-pulsed DCs (data not shown). To determine the
antigen-specific and MHC-restricted tumor lysis by CTL, the murine
HCC cell line H22 (H-2Kd) was used as target cells, in
contrast to Hepa1-6 cells, there was much lower CTL activity
against H22 target, indicating that the CTLs stimulated by vaccine
pulsed-DCs are antigen-specific and MHC-restricted (P<0.05)
(Fig. 3C). These findings suggested
that DCs co-incubated with irradiated HBx gene modified tumor cells
could lead to efficient cross-presentation and induce a specific
CTL response to recognize and lyse Hepa1-6 cells.
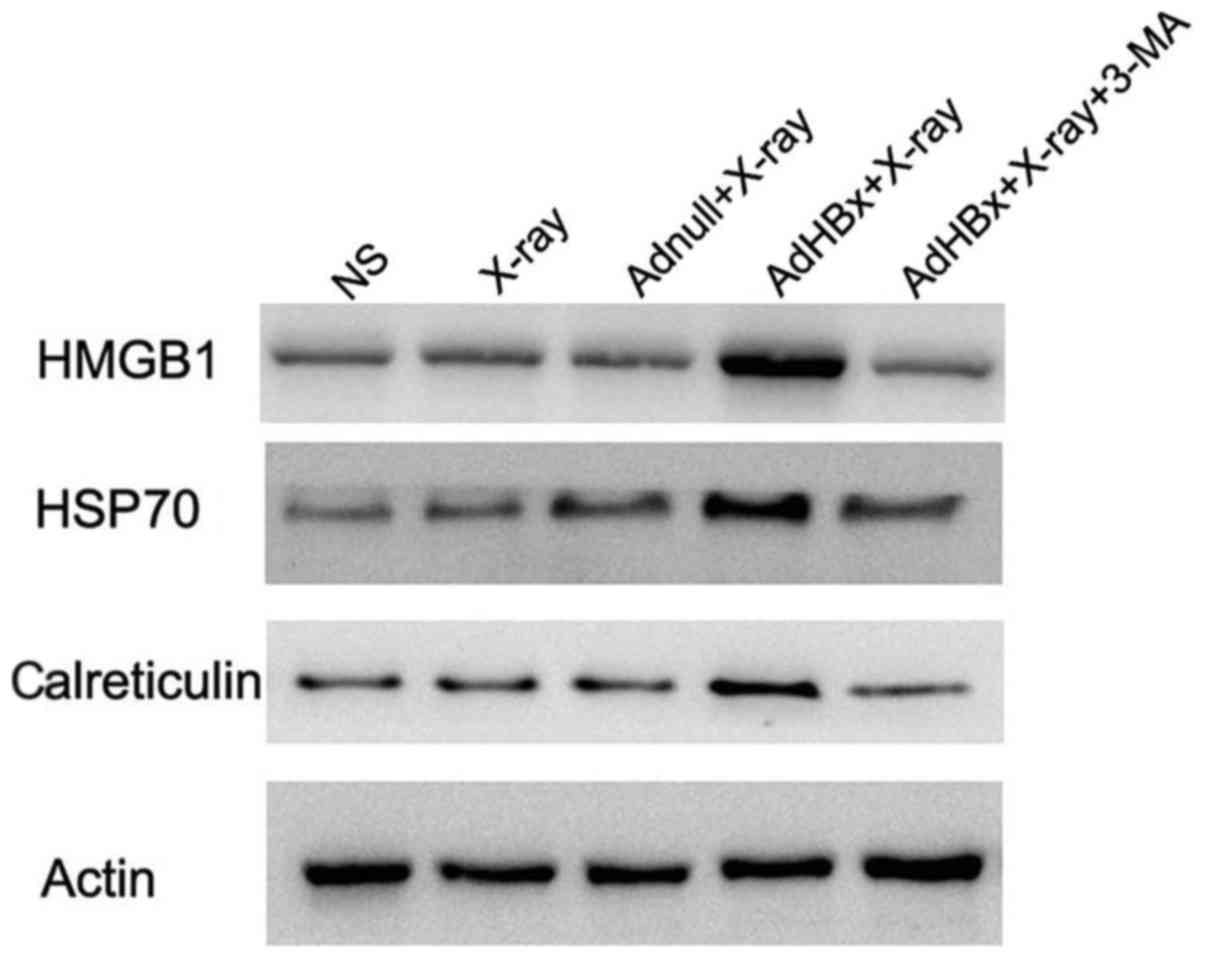
Induction of ‘danger signal’ by
autophagic form of irradiated HBx-modified Hepa1-6 cells
Damage-associated molecular pattern molecules
(DAMPs) are released from injured or stressed dying cells, serving
as endogenous danger signals that contribute to inflammation and
immunity. Release of DAMPs from tumor cells not only alert the
innate immunity but also drive adaptive antitumor immunity during
immunogenic tumor cell death. Autophagy triggered exposure of DAMP,
and in turn is regulated by DAMPs. The interplay between autophagy
and DAMPs shapes the immune response to dying cells, regulating the
efficiency of antitumor treatment. Thus, autophagy and DAMPs are
appropriate natural partners for cancer immunotherapy. For the
reason mentioned above, we tested whether DAMPs were induced in
autophagic form of irradiated HBx-modified hepa1-6 cells. Western
blotting showed that HMGB1, HSP70 and calreticulin were
significantly elevated in the vaccine, whereas they were scarcely
expressed in control groups. Moreover, inhibition of autophagy by
3-MA almost abolished these DAMPs induction (Fig. 4).
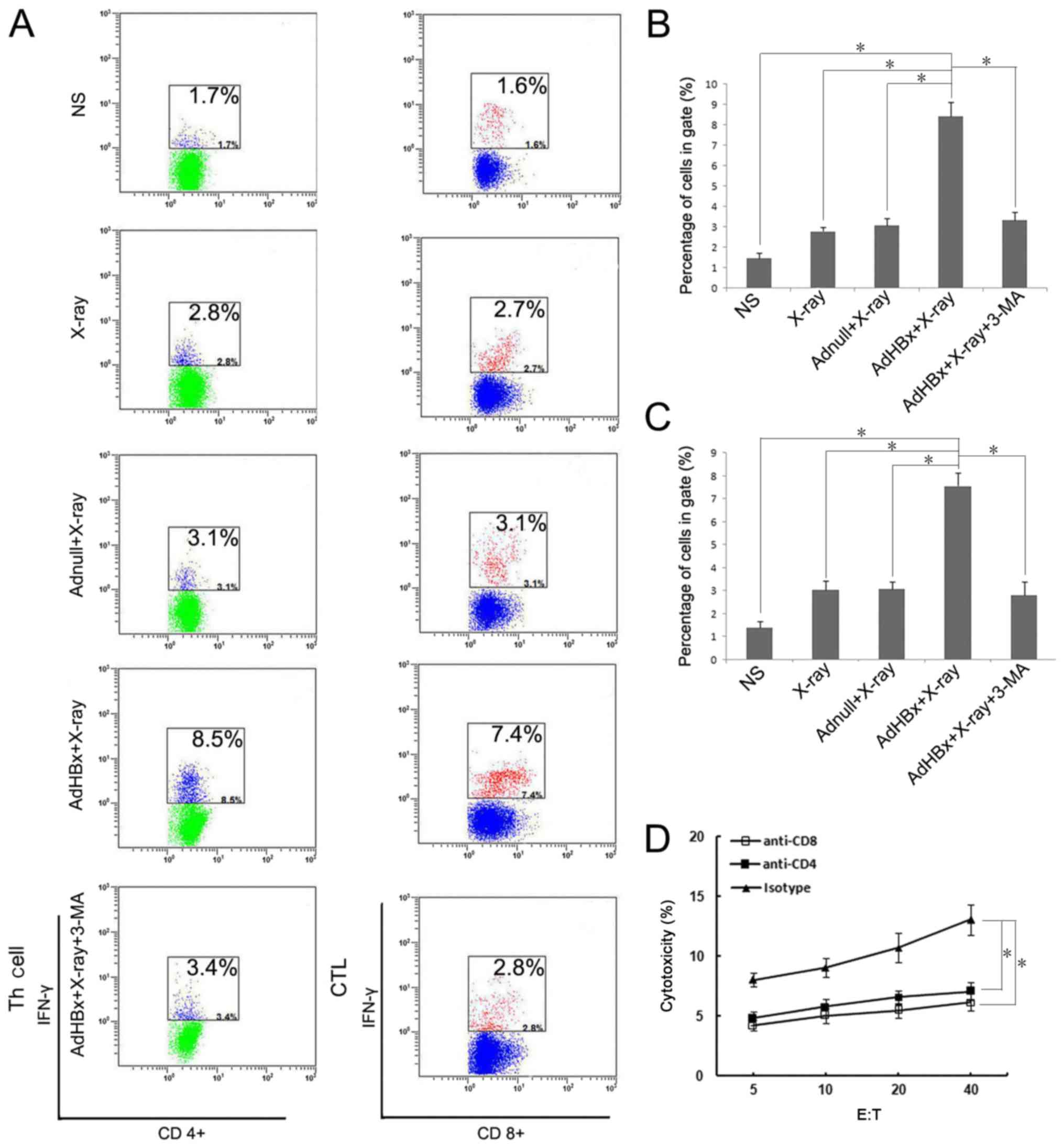
Both CD8+ and
CD4+ lymphocytescontributed to the vaccine-induced
immune response
Our previous study has demonstrated that both
CD8+ and CD4+ T cells participated in the
therapeutic immunological responses in mice vaccinated with
Hepa1-6/AdHBx vaccines. The frequency of IFN-γ secreting
lymphocytes reflected the cellular immune response. For evaluating
both CD8+ and CD4+ T cells response, the
IFN-γ-generating T cells relative to the number of lymphocytes were
detected using flow cytometric analysis (Fig. 5A). In the present study, T cells were
stained by anti-CD4-APC and anti-IFN-γ-PE or anti-CD8-FITC and
anti-IFN-γ-PE after stimulating with pulsed DCs in each group, and
were measured after 48 h. As shown in Fig. 5B, the number of activated
CD8+ cells (CD8+, IFN-γ+) which
stimulated with the vaccine-pulsed DCs were >~4-fold compared
with PBS treatment, and >2-fold compared with groups pulsed with
Hepa1-6 alone or Hepa1-6/AdNull. In a like manner, the activated
CD4+ T cells (CD4+, IFN-γ+) were
5-fold compared with PBS treatment (Fig.
5C). The results further indicated that both CD8+
and CD4+ T lymphocytes participated in antitumor immune
responses induced by the irradiated HBx-modified tumor cell
vaccine. To further assess whether both CD8+ and
CD4+ T cells are necessary for tumor regression,
anti-CD8 or anti-CD4 antibodies were used to deplete corresponding
T-cell subsets one day before co-incubation with pulsed DCs.
Depletion of either CD8+ T cells or CD4+ T
cells during priming almost abrogated the cytotoxicity against
Hepa1-6 (Fig. 5D). Collectively,
these data demonstrated that both CD4+ and
CD8+ T cells were essential for the vaccine-induced
immune response.
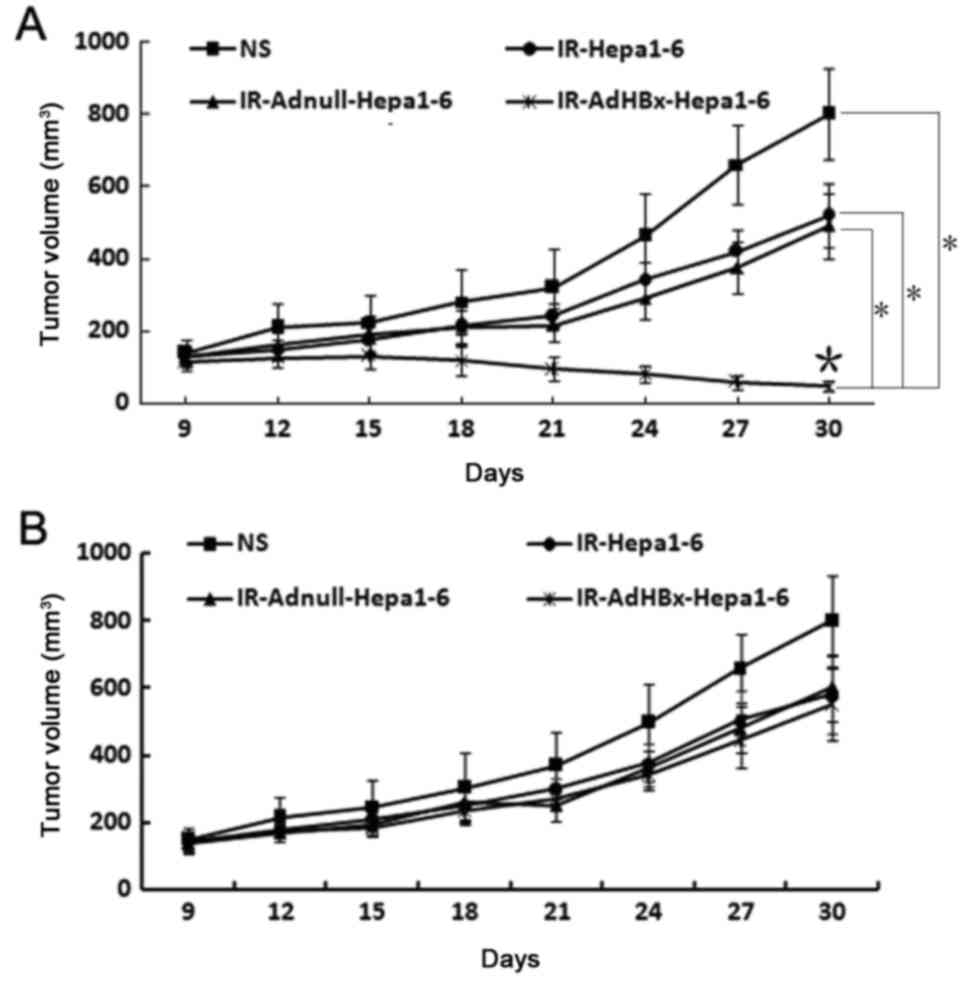
Serum and lymphocyte adoptive transfer
in vivo
Given that the vaccine elicited strong in
vivo antitumor immunresponse, we sought to investigate the
protection from tumor growth in recipient mice after adoptive
transfer of serum and lymphocyte. As expected, treatment with
lymphocytes from the spleens of the mice immunized with the
irradiated AdHBx-infected Hepa1-6 cell vaccine exhibited apparent
protection from tumor growth, compared with those from mice
immunized with controls (Fig. 6A). In
contrast, there was no statistical significance between tumor
volume in each groups after the adoptive transfer of sera from mice
immunized with irradiated HBx-modified tumor cell vaccine or
control groups (Fig. 6B). These
results indicated that the cellular immune responses play an vital
role in antitumor activity induced by the irradiated AdHBx-infected
cell vaccine.
Discussion
HBx is encoded by the HBV genome as a minimum of
open reading frames. It is a multifunctional protein that widely
involved in virus replication, transcription regulation, cell
transformation, apoptosis and cell cycle regulation (19). Numerous studies have confirmed that
HBx also exists multi-epitopes (20,21).
Therefore, when HBx specific CTL responses were elicited, HBx could
act as an effective target for immunotherapy of hepatocellular
carcinoma. In our previous study, we demonstrated that HBx specific
immunity against HCC was elicited by adenovirus vaccine encoding
HBx (22).
In the immune response against malignant tumors, T
cell-mediated immune response plays an extremely important role,
particularly the activation of CD8+ cytotoxic T
lymphocytes (CTLs). Despite presentation of potentially immunogenic
peptides in the context of MHC molecules, tumor cells may fail to
present antigens since they lack the co-stimulatory molecules (such
as B7-1/CD80 and B7-2/CD86) necessary for activation of the T-cell
receptor complex (23). Transgenic
expression of these co-stimulatory molecules on the surface of
tumor cells may therefore enhance their immunogenicity (12,24), but
the dominant mechanism of CTL priming is through the uptake and
presentation of tumor antigens by bone marrow-derived APC including
DC. Thus, cross-priming mechanism (17,18) is
suitable for the notion that many tumors express tumor-specific
antigens capable of being captured and presented to
antigen-specific T cells in context of major histocompatibility
complex (MHC) class I molecules by host professional
antigen-presenting cells. Whole tumor cells death leading to the
efficient cross-presentation is mediated by the exposure of
immunogenic end-stage degradation products and the release of
inflammatory signals (25–27). Autophagy constitutes a mechanism for
the sequestration of organelles and subsequent lysosomal
degradation of autolysosomes which consist of damaged organelles
and invading pathogens (28). In such
context, autophagosomes act as potent tumor-antigen carriers for
efficient cross-presentation. Recent studies suggested that
autophagy shapes cellular immunity far beyond its role as a
cell-intrinsic mechanism to protect against environmental stresses,
including external pathogen attack and internal nutrient depletion
(29,30). It has been shown that in vivo
immunization with cells undergoing autophagy efficiently
facilitated cross-priming of viral and tumor-specific
CD8+ T cells (31,32). In another aspect, previous studies
have found that HBx could sensitize cells to stress or
infection-induced autophagy (33,34). In
light of those discoveries, we have designed a novel tumor
vaccine-irradiated HBx modified hepatocellular carcinoma cell
vaccine, which is prepared from radiation treatment of
adenoviral-mediated genetic engineering of hepatoma cells. Given
that mature and activated DCs are potent antigen-presenting cells
for the priming of naïve T cells, immunization with the irradiated
whole tumor cells could provide a whole array of tumor associated
antigens (TAAs) for as much recognition with TCRs as possible. In
addition, by following this strategy, the majority of naive T cells
proliferate without any prior stimulus, since it is not a recall
response and the stimulus provided is antigen primed BMDC. Our
previous research has shown that this vaccine exerted strong in
vivo antitumor activity by eliciting T cel-mediated immune
response (14).
In the present study, we investigated the mechanism
by which this novel vaccine contributes to enhancing antitumor
immune responses. We found that the advantages of this novel
vaccine lie in: i) Cleverly harness the effect that HBx induced
autophagy in HCC cells, autophagosomes in irradiated HBx-modified
Hepa1-6 cells facilitates efficient cross-presentation of a whole
array of TAAs to T cells. The present study has demonstrated that
IL-12 and IFN-γ was released in significantly higher mounts in
vaccine pulsed DC group than control groups, indicating the
activation of the Th1 immune response. In addition, DCs loaded with
vaccine-derived Ags had significant elevated expression of
co-stimulatory molecules (CD80 and CD86) and maturation marker CD40
compared with control groups. It's been suggested that CD80 mediate
inhibitory effect on T cells through interaction with cytotoxic
T-lymphocyte antigen-4 (CTLA-4/CD152). CD28 and CD152 have crucial
yet opposing functions in T-cell stimulation, in which CD28
promotes but CD152 inhibits T-cell responses. Intriguingly, they
share two ligands, CD80 and CD86, but at present there is no clear
model for understanding whether a ligand may promote or inhibit
responses. In most studies concerning the activation of DCs, CD80
and CD86 are like twins reflecting the mature of DCs (35), in the present study, expression of
both CD80 and CD86 on DCs were elevated significantly upon pulsed
with vaccine, and it will be another good project to test if CD152
blocking plus our vaccine could exert better effect on antitumor
response. Of note, PD-L1 expression was not significantly affected
by vaccine compared with control groups. It's been reported that
stimulatory and inhibitory signal pathways coexist in the process
in which DCs are triggered to stimulate or inhibit T-cells
(36). Our results suggested that
elevation of co-stimulatory molecules provide a sufficiently strong
stimulatory signal to overwhelm the antagonizing signaling pathway
transduced via the PD-1/PD-L1, thus favouring the T cells priming
and avoiding T-cell anergy. In addition, DCs pulsed by irradiated
HBx gene modified Hepa1-6 cells could stimulate CTLs to proliferate
and induce a specific CTL response to recognize and lyse Hepa1-6
cells, which explains the strong specific CTL response in our
previous in-vivo study. ii) Whole tumor cell vaccines is
prepared from autologous tumor cells via radiation inactivation
without defining tumor antigens, the vaccine express a series of
TAAs, including both characterized (HBx inside) and
uncharacterized. These rich sources of antigen containsepitopes of
both CD4+ Th cells and CD8+ CTLs. In this
manner, both MHC class I and II-restricted antigens areparallel
presented, which help to generate stronger adaptive immune response
and long-term memory of CD8+ T-cell via CD4+
T-cell help, thus reducing the chance of tumor escape by antigen
loss variants (37,38). As demonstrated by flow cytometry, both
CD8+ and CD4+ T cells primed by
vaccine-pulsed DCs released high amounts of IFN-γ compared with the
other groups. Furthermore, depletion of either CD8+ or
CD4+ T cells almost abrogated the cytotoxicity against
Hepa1-6, which is consistent with our in-vivo study showing
that both CD8+ and CD4+ T lymphocytes
participated in antitumor immune responses induced by the
irradiated HBx-modified tumor cell vaccine. Induction of a
multi-epitope immune response should be evidenced by recognition of
known tumor antigens or peptides in terms of tetramer staining,
however, there is no reported known epitope sequence of Hepa1-6 in
former literature. We are currently constructing OVA-expressing
Hepa1-6 cells, in our future studies, the immune response can be
detected by tetramer staining using OVA257-264 (SIINFEKL). In the
present study, in order to prove that the killing of tumor cells is
antigen-specific and MHC-restricted, the murine HCC cell line H22
(H-2Kd) was used as target cells, our data showed that
there was much lower CTL activity against H22 target in contrast to
Hepa1-6 cells. In a similar manner, this experiment can also prove
that T cell activation occurs via cross-priming and not direct
tumor cell recognition, through the use of MHC-unmatched T
cells.
The danger model has proposed that
antigen-presenting cells are activated by damage-associated
molecular pattern molecules (DAMPs) that cause tissue stress or
destruction, thereby promoting immunity (39). When autophagic responses in
antigen-donor cells (ADCs) are elicited and are followed by cell
death, the immune responses are stimulated. There are multiple
mechanisms by which autophagy can influence interaction between
immune responses and cell death. One important aspect is that
autophagy regulates DAMPs release and in turn is regulated by
DAMPs. The interplay between DAMPs and autophagy shape ADCs as
immunogenic cell death thereof to be captured by DCs for T cells
priming in an immunostimulatory fashion (40,41). Given
that irradiated HBx modified hepatocellular carcinoma cell vaccine
induced strong cellular immunity, we speculate that DAMPs may be
involved in DCs activation and the following antitumor immune
response due to autophagy induction. As expected, western blotting
shown that the level of HMGB1 (42)
(an chromatin-binding and bending protein which is released from
dying cells and can activate various immune cells), HSP70 (43) (a molecular chaperone that induce
pro-inflammatory cytokine/chemokine release and activation of
adaptive immune system in response to cell stress and injury) and
calreticulin (44) (an ER luminal
resident protein that facilitates the capture of dying tumor cells
by DCs) were significantly elevated in the vaccine. Importantly,
inhibition of autophagy almost abolishes the DAMPs induction,
indicating the elevation of DAMPs is autophagy-dependent.
In conclusion, the present results demonstrated that
irradiated HBx-modified tumor cell vaccine was sufficient for
inducing maturation of DCs and subsequent both CD8+ and
CD4+ T lymphocyte priming. Therefore, our findings have
implications for designing DC-based clinical immunotherapy against
HBV-associated HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81101728), the
Science and Technology Support Program of Sichuan (grant nos.
2014SZ0122 and 2013SZ0044), the National Science and Technology
Major Project for Infectious Diseases Control (grant no.
2017ZX10203206-004).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AH performed the flow cytometric analysis, animal
experiments and drafted the manuscript. JM performed cell culture
and viral preparation experiments. LH contributed to animal
experiments and performed statistical analyses. FY and PC designed
the study.
Ethical approval and consent to
participate
Ethical approval for the study was obtained by the
Ethics Committee of State Key Laboratory of Biotherapy, Sichuan
University.
Consent for publication
All authors have agreed with content for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Steinman RM: The dendritic cell system and
its role in immunogenicity. Annu Rev Immunol. 9:271–296. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hart DN: Dendritic cells: Unique leukocyte
populations which control the primary immune response. Blood.
90:3245–3287. 1997.PubMed/NCBI
|
|
3
|
Rock KL: A new foreign policy: MHC class I
molecules monitor the outside world. Immunol Today. 17:131–137.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fields RC, Shimizu K and Mulè JJ: Murine
dendritic cells pulsed with whole tumor lysates mediate potent
antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci
USA. 95:9482–9487. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Perez CA, Fu A, Onishko H, Hallahan DE and
Geng L: Radiation induces an antitumour immune response to mouse
melanoma. Int J Radiat Biol. 85:1126–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weiss EM, Frey B, Rödel F, Herrmann M,
Schlücker E, Voll RE, Fietkau R and Gaipl US: Ex vivo- and in
vivo-induced dead tumor cells as modulators of antitumor responses.
Ann NY Acad Sci. 1209:109–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Das A and Ali N: Vaccine prospects of
killed but metabolically active Leishmania against visceral
leishmaniasis. Expert Rev Vaccines. 11:783–785. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoshimoto S, Faries MB, Morton DL, Shingai
T, Kuo C, Wang HJ, Elashoff R, Mozzillo N, Kelley MC, Thompson JF,
et al: Assessment of prognostic circulating tumor cells in a phase
III trial of adjuvant immunotherapy after complete resection of
stage IV melanoma. Ann Surg. 255:357–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miguel A, Herrero MJ, Sendra L, Botella R,
Algás R, Sánchez M and Aliño SF: Comparative antitumor effect among
GM-CSF, IL-12 and GM-CSF+IL-12 genetically modified tumor cell
vaccines. Cancer Gene Ther. 20:576–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan HX, Cheng P, Wei HY, Shen GB, Fu LX,
Ni J, Wu Y and Wei YQ: Active immunotherapy for mouse breast cancer
with irradiated whole-cell vaccine expressing VEGFR2. Oncol Rep.
29:1510–1516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meijer SL, Dols A, Jensen SM, Hu HM,
Miller W, Walker E, Romero P, Fox BA and Urba WJ: Induction of
circulating tumor-reactive CD8+ T cells after
vaccination of melanoma patients with the gp100 209-2M peptide. J
Immunother. 30:533–543. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang X, Mei W, Zhang L, Yu H, Zhao X, Fan
X, Qian G and Ge S: Co-expression of P1A35–43/β2m fusion
protein and co-stimulatory molecule CD80 elicits effective
anti-tumor immunity in the P815 mouse mastocytoma tumor model.
Oncol Rep. 22:1213–1220. 2009.PubMed/NCBI
|
|
13
|
Iero M, Filipazzi P, Castelli C, Belli F,
Valdagni R, Parmiani G, Patuzzo R, Santinami M and Rivoltini L:
Modified peptides in anti-cancer vaccines: Are we eventually
improving anti-tumour immunity? Cancer Immunol Immunother.
58:1159–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan Y, Liu N, Lu L, Zang CM, Shao B, Li Y,
Wen Y, Wei Y and Cheng P: Autophagy enhances antitumor immune
responses induced by irradiated hepatocellular carcinoma cells
engineered to express hepatitis B virus X protein. Oncol Rep.
30:993–999. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo C, Shen G, Liu N, Gong F, Wei X, Yao
S, Liu D, Teng X, Ye N, Zhang N, et al: Ammonia drives dendritic
cells into dysfunction. J Immunol. 193:1080–1089. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Norman JM, Cohen GM and Bampton ET: The in
vitro cleavage of the hAtg proteins by cell death proteases.
Autophagy. 6:1042–1056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bevan MJ: Cross-priming for a secondary
cytotoxic response to minor H antigens with H-2 congenic
cells which do not cross-react in the cytotoxic assay. J Exp Med.
143:1283–1288. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang AY, Golumbek P, Ahmadzadeh M, Jaffee
E, Pardoll D and Levitsky H: Role of bone marrow-derived cells in
presenting MHC class I-restricted tumor antigens. Science.
264:961–965. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rehermann B and Nascimbeni M: Immunology
of hepatitis B virus and hepatitis C virus infection. Nat Rev
Immunol. 5:215–229. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Malmassari S, Lone YC, Zhang M, Transy C
and Michel ML: In vivo hierarchy of immunodominant and subdominant
HLA-A*0201-restricted T-cell epitopes of HBx antigen of hepatitis B
virus. Microbes Infect. 7:626–634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding FX, Wang F, Lu YM, Li K, Wang KH, He
XW and Sun SH: Multiepitope peptide-loaded virus-like particles as
a vaccine against hepatitis B virus-related hepatocellular
carcinoma. Hepatology. 49:1492–1502. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Cheng P, Wen Y, Chen P, Yang L, Zhao
X, Lv H, Quan Q, Wu Y, Yang H, et al: T lymphocyte responses
against hepatitis B virus-related hepatocellular carcinoma induced
by adenovirus vaccine encoding HBx. Int J Mol Med. 26:869–876.
2010.PubMed/NCBI
|
|
23
|
Podojil JR and Miller SD: Targeting the B7
family of co-stimulatory molecules: Successes and challenges.
BioDrugs. 27:1–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Townsend SE and Allison JP: Tumor
rejection after direct costimulation of CD8+ T cells by
B7-transfected melanoma cells. Science. 259:368–370. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim R, Emi M and Tanabe K: Cancer
immunoediting from immune surveillance to immune escape.
Immunology. 121:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beatty GL and Gladney WL: Immune escape
mechanisms as a guide for cancer immunotherapy. Clin Cancer Res.
21:687–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rabinovich GA, Gabrilovich D and Sotomayor
EM: Immunosuppressive strategies that are mediated by tumor cells.
Annu Rev Immunol. 25:267–296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Virgin HW and Levine B: Autophagy genes in
immunity. Nat Immunol. 10:461–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Wang LX, Yang G, Hao F, Urba WJ and
Hu HM: Efficient cross-presentation depends on autophagy in tumor
cells. Cancer Res. 68:6889–6895. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uhl M, Kepp O, Jusforgues-Saklani H,
Vicencio JM, Kroemer G and Albert ML: Autophagy within the antigen
donor cell facilitates efficient antigen cross-priming of
virus-specific CD8+ T cells. Cell Death Differ.
16:991–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, Li
F, Wang Y, Tiollais P, Li T and Zhao M: Hepatitis B virus X protein
sensitizes cells to starvation-induced autophagy via up-regulation
of beclin 1 expression. Hepatology. 49:60–71. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sir D, Tian Y, Chen WL, Ann DK, Yen TS and
Ou JH: The early autophagic pathway is activated by hepatitis B
virus and required for viral DNA replication. Proc Natl Acad Sci
USA. 107:4383–4388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Esensten JH, Helou YA, Chopra G, Weiss A
and Bluestone JA: CD28 costimulation: From mechanism to therapy.
Immunity. 44:973–988. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bennett SR, Carbone FR, Karamalis F,
Miller JF and Heath WR: Induction of a CD8+ cytotoxic T
lymphocyte response by cross-priming requires cognate
CD4+ T cell help. J Exp Med. 186:65–70. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cassell D and Forman J: Linked recognition
of helper and cytotoxic antigenic determinants for the generation
of cytotoxic T lymphocytes. Ann NY Acad Sci. 532:51–60. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matzinger P: Tolerance, danger, and the
extended family. Annu Rev Immunol. 12:991–1045. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nickel W and Rabouille C: Mechanisms of
regulated unconventional protein secretion. Nat Rev Mol Cell Biol.
10:148–155. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deretic V, Jiang S and Dupont N: Autophagy
intersections with conventional and unconventional secretion in
tissue development, remodeling and inflammation. Trends Cell Biol.
22:397–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tian J, Avalos AM, Mao SY, Chen B, Senthil
K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al:
Toll-like receptor 9-dependent activation by DNA-containing immune
complexes is mediated by HMGB1 and RAGE. Nat Immunol. 8:487–496.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garg AD, Krysko DV, Vandenabeele P and
Agostinis P: Hypericin-based photodynamic therapy induces surface
exposure of damage-associated molecular patterns like HSP70 and
calreticulin. Cancer Immunol Immunother. 61:215–221. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Obeid M, Tesniere A, Ghiringhelli F, Fimia
GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T,
Casares N, et al: Calreticulin exposure dictates the immunogenicity
of cancer cell death. Nat Med. 13:54–61. 2007. View Article : Google Scholar : PubMed/NCBI
|