Introduction
Uterine sarcoma is a rare but aggressive malignant
gynecological tumor with unknown aetiology and pathogenesis.
Uterine leiomyosarcoma (uLMS) is the most common histological
subtype of uterine sarcoma originating in the smooth muscles of the
myometrium. It accounts for only 1% of all uterine malignancies;
however, it contributes to a considerable proportion of uterine
cancer deaths (1). With poor
biological characteristics, the overall 5-year survival rate for
uLMS is only 25% (2). Surgical
treatment is the mainstay of therapy for uLMS; however, 50–71% of
these patients would develop recurrence (1). Adjuvant radiotherapy and chemotherapy
have minimal effect on improving patient survival (3,4). Hormone
therapy appears to be effective for hormone receptor-positive uLMS;
however, the evidence for this is inadequate (4). Targeted therapy has been developed
rapidly in recent years and it is expected to be a promising
treatment for uLMS. Thus, it is necessary to explore the molecular
aetiology and pathogenesis of uLMS and to search for therapeutic
molecular targets.
The study of uLMS is challenging because of its
rarity. Recently, the genome-wide DNA microarray, which is a
high-throughput platform for the analysis of gene expression, has
been regarded as an efficient tool for detecting molecular changes
in diseases. There are a few microarrays of uLMS in the Gene
Expression Omnibus (GEO) repository; however, no detailed
bioinformatics analysis has been performed. The most relevant study
was conducted by Barlin et al (5). They examined and compared the expression
profiles of uLMS cells, fibroids and normal myometrium for the
identification of molecular subtypes and correlation with clinical
outcomes. In addition, they found that some genes that are related
to cell-cycle regulation, such as CDC7, CDC20, GTSE1, CCNA2,
CCNB1 and CCNB2 were overexpressed in uLMS. However, the
interactions among the differentially expressed genes (DEGs) still
remain poorly understood.
Consequently, we performed bioinformatic analysis to
explore the changes in the expression of mRNA and interactions
among the DEGs during the occurrence of uLMS. To minimize the
frequency of false-positive results of the microarray analysis, we
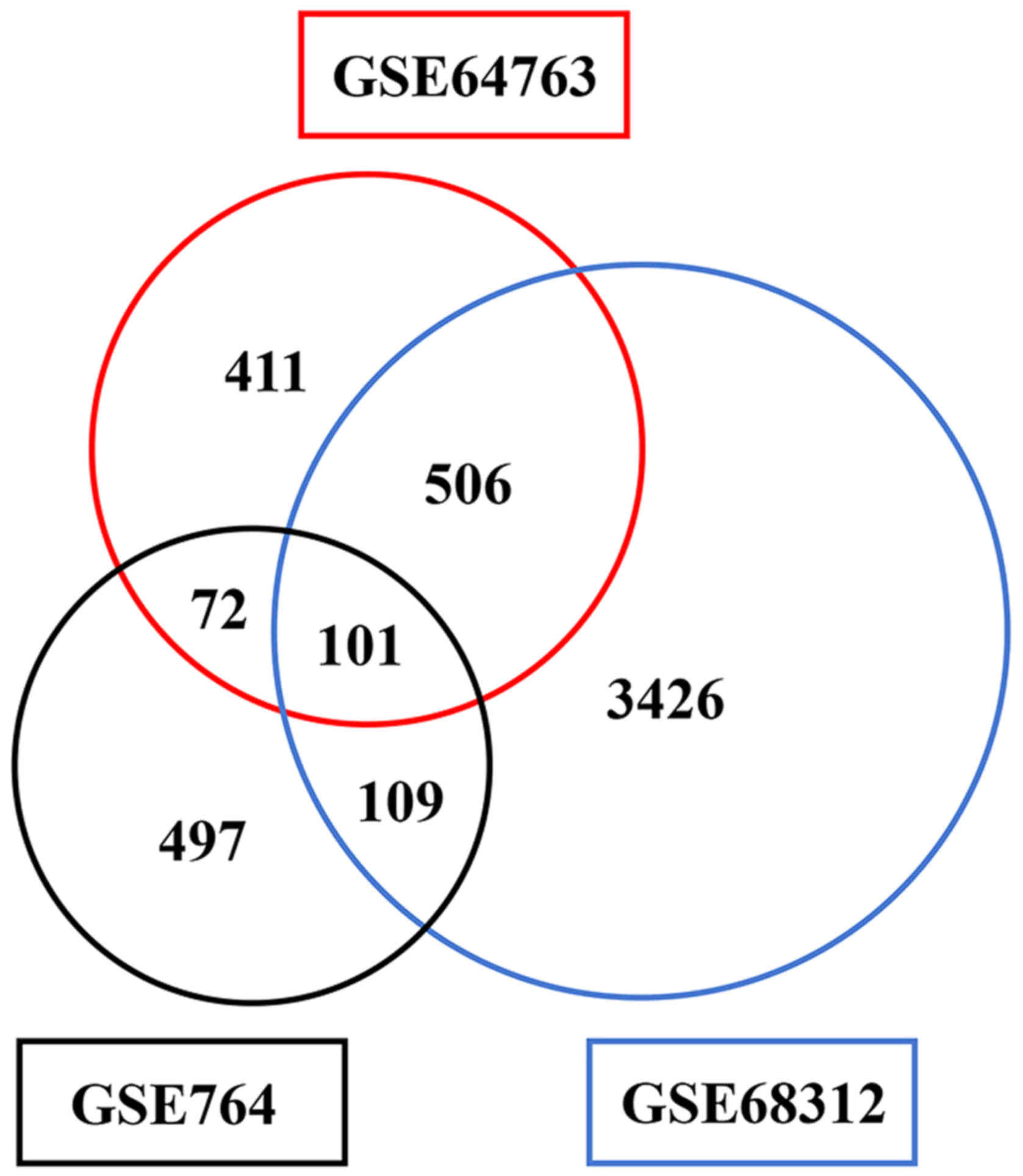
used 3 microarrays including GSE764, GSE64763 and GSE68312 which
were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/). In the present
study, we identified DEGs between leiomyosarcoma samples and normal
myometrial samples using the web tool GEO2R. Then, we performed
Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes
(KEGG) pathway enrichment analyses for identifying enriched
biological functions and pathways. In addition, we constructed
protein-protein interaction (PPI) networks of the DEGs and key
modules in the PPI networks to identify important genes and related
pathways. Our study provides new information on the molecular
aetiology and pathogenesis of uLMS and to provide novel potential
molecular targets for treatment.
Materials and methods
Microarray data
After searching the GEO repository at the National
Centre for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/geo/) for relevant
microarray data on uLMS, the three most eligible gene expression
profiles were found: GSE764, GSE64763 and GSE68312. The GSE764
profile consists of 26 samples, including 4 myometrial samples, 9
uLMS samples and some other leiomyoma and leiomyosarcoma samples.
The GSE64763 profile consists of 25 fibroid samples, 25
leiomyosarcoma samples and 29 normal myometrial samples. The
GSE68312 profile consists of 3 uLMS samples, 3 uterine normal
myometrial samples and some samples of uterine leiomyoma tissues
and cell lines of human uLMS as well as methylated forms of all
these samples.
Identification of DEGs
After the irrelevant samples were excluded, the uLMS
samples were compared with the normal myometrial samples using
GEO2R. GEO2R, which is an interactive web tool, is designed for the
identification of genes that are differentially expressed across
experimental conditions by comparing two or more groups of samples
in a GEO series. When analysed by the GEO2R, results were presented
as a table of genes ordered by significance, then we can choose to
view profile graphs of the top 250 genes or save the complete
results table. In this way, the DEGs of the three gene expression
profiles were identified. The genes with |log FC| ≥1 were regarded
to be differentially expressed and P<0.05 was considered to
indicate a statistically significant difference. Then, the genes
that were differentially expressed in all the three microarrays
with identical expression patterns were deemed as DEGs in this
study.
GO analysis and KEGG pathway
enrichment analysis of DEGs
The GO repository (http://geneontology.org/) consists of a large set of
annotation terms and is commonly used for annotating genes and
identifying the characteristic biological attributes for microarray
data. The KEGG database (http://www.genome.jp/) contains data on known genes
and their biochemical functions and is used for identifying
functional and metabolic pathways. By performing the GO and KEGG
analysis at the functional level, we can gain a better
understanding of the roles of these DEGs in the initiation and in
the progression of uLMS. The Database for Annotation, Visualization
and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) is an online resource
that provides tools for functional annotation and bioinformatics
microarray analysis. Both GO categories and KEGG pathway enrichment
analysis were performed using DAVID to reveal the functions of
these DEGs. P<0.05 was considered to indicate a statistically
significant difference.
Protein-protein interaction (PPI)
network construction and module analysis
The Search Tool for the Retrieval of Interacting
Genes (STRING) database (https://string-db.org/) is a web resource of PPIs.
Cytoscape (http://www.cytoscape.org/) is an open
source software that allows for the visualization of molecular
interaction networks and the integration of these networks with
gene annotations and expression profiles. All the identified DEGs
were uploaded to STRING (version 10.0) for the analysis of their
interactions. Comprehensive information on the interactions of
these DEGs was downloaded from STRING and the interactions with a
combined score >0.4 were selected for constructing the PPI
networks using the Cytoscape software. Next, significant modules
from the PPI network were extracted using the plugin, Molecular
Complex Detection (MCODE) with cut-off criteria of MCODE scores
>3 and number of nodes >5. Then, the functional and pathway
enrichment analyses of the genes in these modules were performed.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Identification of the DEGs
Analysed with GEO2R, genes which were differentially
expressed between uLMS samples and normal myometrial samples were
screened based on the data of GSE764, GSE64763 and GSE68312
profiles. Totally 779, 1,090 and 4,142 DEGs were identified,
respectively. Among them, 101 genes were identified in all of the
three datasets and 95 genes including 21 upregulated genes and 74
downregulated genes exhibited identical expression patterns
(Fig. 1).
GO term and KEGG pathway enrichment
analysis
The results of GO categories analysis including
biological processes (BP), cellular components (CC) and molecular
functions (MF) are displayed in Table
I. P<0.05 was considered as the cut-off value. When the
number of significant terms was >3, only the 3 predominant terms
were present. Firstly, the upregulated DEGs were annotated with the
BP category, including ‘DNA metabolic process’,
‘nucleobase-containing compound biosynthetic process’ and ‘cellular
macromolecule biosynthetic process’; the downregulated DEGs were
annotated with the GO terms, ‘cellular response to chemical
stimulus’, ‘movement of cell or subcellular component’ and
‘response to inorganic substance’. Secondly, the upregulated DEGs
were annotated with the GO terms of the CC category, namely,
‘cyclin-dependent protein kinase holoenzyme complex’ and the
downregulated DEGs were annotated with ‘sarcolemma’, ‘plasma
membrane region’ and ‘integral component of plasma membrane’.
Thirdly, the upregulated DEGs were annotated with the GO terms of
the MF category, such as ‘transcription factor activity’ (‘RNA
polymerase II transcription factor recruiting’ and ‘transcription
factor recruiting’) and ‘cyclin-dependent protein serine/threonine
kinase regulator activity’; and the downregulated DEGs were
annotated with ‘potassium channel activity’, ‘potassium ion
transmembrane transporter activity’ and ‘calcium-activated
potassium channel activity’. As shown in Table I, the significantly enriched KEGG
pathways of the DEGs with P<0.05 were ‘transcriptional
misregulation in cancer’, ‘proteoglycans in cancer’ and ‘pathways
in cancer’, all of which were pathways of downregulated DEGs. No
significant pathway of upregulated DEGs was identified.
 | Table I.GO and KEGG enrichment analyses of
the DEGs in uLMS. |
Table I.
GO and KEGG enrichment analyses of
the DEGs in uLMS.
| Category | Term | Count | P-value |
|---|
| Upregulated |
|
|
|
|
GOTERM_BP_FAT | GO:0006259~DNA
metabolic process | 5 | 6.93E-03 |
|
GOTERM_BP_FAT |
GO:0034654~nucleobase-containing compound
biosynthetic process | 9 | 7.24E-03 |
|
GOTERM_BP_FAT | GO:0034645~cellular
macromolecule biosynthetic process | 10 | 7.48E-03 |
|
GOTERM_CC_FAT |
GO:0000307~cyclin-dependent protein kinase
holoenzyme complex | 2 | 3.57E-02 |
|
GOTERM_MF_FAT |
GO:0001135~transcription factor activity,
RNA polymerase II transcription factor recruiting | 2 | 8.53E-03 |
|
GOTERM_MF_FAT |
GO:0001134~transcription factor activity,
transcription factor recruiting | 2 | 1.10E-02 |
|
GOTERM_MF_FAT |
GO:0016538~cyclin-dependent protein
serine/threonine kinase regulator activity | 2 | 2.66E-02 |
| Downregulated |
|
|
|
|
GOTERM_BP_FAT | GO:0070887~cellular
response to chemical stimulus | 26 | 7.35E-09 |
|
GOTERM_BP_FAT | GO:0006928~movement
of cell or subcellular component | 21 | 2.66E-07 |
|
GOTERM_BP_FAT | GO:0010035~response
to inorganic substance | 10 | 4.83E-07 |
|
GOTERM_CC_FAT |
GO:0042383~sarcolemma | 5 | 1.75E-04 |
|
GOTERM_CC_FAT | GO:0098590~plasma
membrane region | 10 | 2.40E-04 |
|
GOTERM_CC_FAT | GO:0005887~integral
component of plasma membrane | 13 | 1.23E-03 |
|
GOTERM_MF_FAT |
GO:0005267~potassium channel activity | 5 | 1.32E-03 |
|
GOTERM_MF_FAT |
GO:0015079~potassium ion transmembrane
transporter activity | 5 | 2.11E-03 |
|
GOTERM_MF_FAT |
GO:0015269~calcium-activated potassium
channel activity | 3 | 2.41E-03 |
|
KEGG_PATHWAY | ptr05202:
Transcriptional misregulation in cancer | 7 | 5.69E-04 |
|
KEGG_PATHWAY | ptr05205:
Proteoglycans in cancer | 6 | 7.61E-03 |
| KEGG_PATHWAY | ptr05200: Pathways
in cancer | 7 | 3.23E-02 |
Hub genes and significant modules
screened from the PPI network
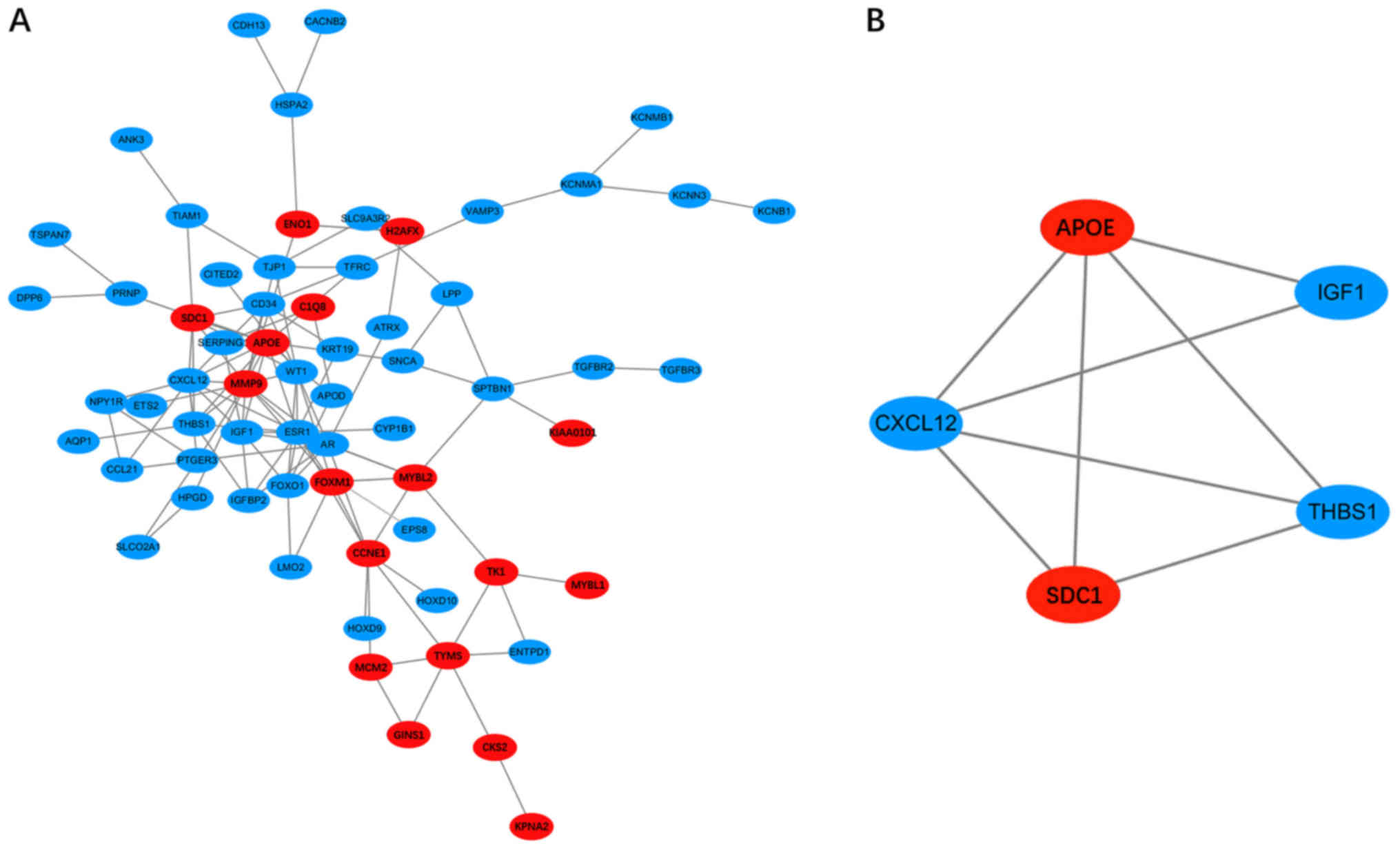
After all the DEGs were uploaded to the online
STRING database, the PPI network with 65 nodes and 119 edges was
constructed using the Cytoscape software (Fig. 2). Sixteen hub DEGs with the node
degree >5 were obtained (Table
II). Among them, MMP9, APOE, CCNE1, SDC1 and
FOXM1 were the major upregulated genes, while ESR1,
CXCL12, AR and WT1 were the major downregulated genes.
Then, one significant module that fulfilled the cut-off criteria,
namely, MCODE scores >3 and number of nodes >5, was screened
(Fig. 2). The SDC1, APOE, IGF1,
THBS1 and CXCL12 genes were identified in the module. GO
analysis of these genes showed that they were annotated with ‘cell
migration’, ‘cell motility’ and ‘localization of cell’ (Table III). In addition, the KEGG
enrichment analysis suggested that these genes were mainly involved
in ‘proteoglycans in cancer’, ‘malaria’, ‘p53 signalling pathway’
and ‘ECM-receptor interaction’ (Table
III). Based on the PPI network and the analysis of the
significant module, the interactions between the DEGs were revealed
clearly.
| Table II.Hub genes of the PPI network with
higher node degrees. |
Table II.
Hub genes of the PPI network with
higher node degrees.
| Hub gene | Gene name | Node degree |
|---|
| Upregulated |
|
|
|
MMP9 | Matrix
metallopeptidase 9 | 15 |
|
APOE | Apolipoprotein
E | 10 |
|
CCNE1 | Cyclin E1 | 8 |
|
SDC1 | Syndecan 1 | 7 |
|
FOXM1 | Forkhead box
M1 | 7 |
|
TYMS | Thymidylate
synthetase | 6 |
|
MYBL2 | MYB proto-oncogene
like 2 | 6 |
| Downregulated |
|
|
|
ESR1 | Estrogen receptor
1 | 14 |
|
CXCL12 | C-X-C motif
chemokine ligand 12 | 10 |
| AR | Androgen
receptor | 10 |
| WT | Wilms tumor 1 | 9 |
|
IGF1 | Insulin like growth
factor 1 | 8 |
|
THBS1 | Thrombospondin
1 | 7 |
|
FOXO1 | Forkhead box
O1 | 6 |
|
PTGER3 | Prostaglandin E
receptor 3 | 6 |
|
CD34 | Cluster of
differentiation | 6 |
|
| 34 molecule |
|
| Table III.GO and KEGG enrichment analyses of
genes in the significant module of the PPI network. |
Table III.
GO and KEGG enrichment analyses of
genes in the significant module of the PPI network.
| Category | Term | Count | P-value | Genes |
|---|
| GOTERM_BP_FAT | GO:0016477~cell
migration | 5 | 1.23E-05 | SDC1, APOE, IGF1,
THBS1, CXCL12 |
| GOTERM_BP_FAT | GO:0048870~cell
motility | 5 | 1.93E-05 | SDC1, APOE, IGF1,
THBS1, CXCL12 |
| GOTERM_BP_FAT |
GO:0051674~localization of cell | 5 | 1.93E-05 | SDC1, APOE, IGF1,
THBS1, CXCL12 |
| GOTERM_CC_FAT | GO:0009897~external
side of plasma membrane | 3 | 1.23E-03 | SDC1, THBS1,
CXCL12 |
| GOTERM_CC_FAT |
GO:0031988~membrane-bounded vesicle | 5 | 2.18E-03 | SDC1, APOE, IGF1,
THBS1, CXCL12 |
| GOTERM_CC_FAT | GO:0098552~side of
membrane | 3 | 3.63E-03 | SDC1, THBS1,
CXCL12 |
| GOTERM_MF_FAT |
GO:0071813~lipoprotein particle
binding | 2 | 4.77E-03 | APOE, THBS1 |
| GOTERM_MF_FAT |
GO:0071814~protein-lipid complex
binding | 2 | 4.77E-03 | APOE, THBS1 |
| GOTERM_MF_FAT | GO:0005102~receptor
binding | 3 | 1.05E-02 | APOE, IGF1,
CXCL12 |
| KEGG_PATHWAY | ssc05205:
Proteoglycans in cancer | 3 | 4.21E-03 | SDC1, IGF1,
THBS1 |
| KEGG_PATHWAY | ssc05144:
Malaria | 2 | 2.93E-02 | SDC1, THBS1 |
| KEGG_PATHWAY | ssc04115: p53
signaling pathway | 2 | 4.04E-02 | IGF1, THBS1 |
| KEGG_PATHWAY | ssc04512:
ECM-receptor interaction | 2 | 4.53E-02 | SDC1, THBS1 |
Discussion
uLMS is a rare tumor with unknown aetiology. All
current therapies for uLMS have some limitations because of its
high rates of recurrence and metastasis. Therefore, the recently
emerging targeted therapy shows importance. Bioinformatics analysis
of data from high-throughput sequencing and microarrays can
accurately reveal the potential molecular mechanisms of the
development of uLMS and predict therapeutic targets by comparing
sarcoma lesions with normal tissues. In the present study, based on
the profiles, GSE764, GSE64763 and GSE68312 from the GEO
repository, 21 upregulated and 74 downregulated DEGs were
identified by comparing uLMS samples with normal myometrial
samples. Further analysis was performed in order to explore the
relations among the DEGs and the interactions of their protein
products.
Functional annotation and enrichment analysis were
performed using the online resource DAVID to obtain information on
the biological functions of these DEGs. On the one hand, the
results of the GO analysis suggested that the upregulated DEGs were
annotated with ‘DNA metabolic process’, ‘nucleobase-containing
compound biosynthetic process’ and ‘cellular macromolecule
biosynthetic process’, while the downregulated DEGs were annotated
with ‘cellular response to chemical stimulus’, ‘movement of cell or
subcellular component’ and ‘response to inorganic substance’. The
increased levels of DNA replication and translation are
comprehensible for the uncontrolled proliferation of cancer. On the
other hand, the KEGG analysis showed that the downregulated DEGs
were enriched in ‘transcriptional misregulation in cancer’,
‘proteoglycans in cancer’ and ‘pathways in cancer’. As reported in
the literature, transcriptional deregulation (6) as well as alteration of pathways such as
the p53 signalling pathway, the Wnt signalling pathway and the
PI3K/AKT/mTOR pathway were verified in many types of cancer. For
example, Wnt signalling pathway was proved important in cancer
progression, including tumor initiation, growth, metastasis as well
as cell senescence and death in breast cancer and colonal cancer;
importantly, targeting WNT signalling pathways is potential new
therapy in cancer patients (7). The
expression of PTEN, which is a negative regulator of PI3K/AKT/mTOR
signalling pathway, was significantly reduced in more than one half
of uLMS patients; what's more, the PI3K/AKT/mTOR signaling
inhibitor sapanisertib and serabelisi are currently tested in
clinical trials for sarcomas (8). In
addition, alteration of the expression of proteoglycans during the
development of cancer was also confirmed (9). Therefore, the results of the GO and KEGG
analyses in our study are consistent with those of the previous
studies and could provide a novel understanding of the pathogenesis
of uLMS.
With the information on the interactions from
STRING, the PPI network was constructed to reveal the relations
among the DEGs. MMP9, also called matrix metallopeptidase 9,
was the principal upregulated gene with the highest node degree. It
bears direct interactions with some other key genes such as
ESR1, SDC1, APOE, THBS1, CXCL12, IGF1, FOXM1, AR and
WT1. As the major member of the matrix metallopeptidase
family, MMP9 plays a vital role in tumor progression because of its
ability to degrade the extracellular matrix. It is reported that
activation of ERK drives the upregulation of MMP9 expression and
subsequent MMP9 mediated shedding of SDC1 (syndecan 1) (10). According to the study by Brule et
al (11), the shedding of SDC1
mediated by the MMP9 was accelerated by SDF-1/CXCL12 in HeLa cells
and human primary macrophages. In addition, an MMP9-miR-494-SDC1
regulatory loop was revealed to be associated with
irradiation-induced angiogenesis in medulloblastoma cells. In this
regulatory mechanism, suppression of miR-494 by MMP9 leads to the
enhanced SDC1 shedding and angiogenesis (12). In addition, the overexpression of
FOXM1 can upregulate the MMP9 expression by combining to its
promoter, leading to the promotion of proliferation, migration and
invasion of epithelial ovarian cancer cells (13). In the contrary, MMP9 expression
could be inhibited by THBS1 leading to the suppression of cell
invasion in colon and ovarian cancers (14). Furthermore, MMP9 was also reported to
activate latent cytokines and growth factors (15). In breast carcinoma, it was
demonstrated that fibroblasts could promote angiogenesis and then,
enhance tumor growth by the upregulation of MMP9 via the
MAPK-AP1 signalling axis, which is co-stimulated by TGF-β, TNF-α
and IL-1β (15). In colorectal cancer
(CRC), MMP9 was overexpressed in each clinical stage and
could be used as a diagnostic marker (16). More notably, the significant negative
correlation between the inhibition of Matrigel invasion and
MMP9 levels in SK-UT-1 uLMS cells found by Roomi et
al (17) was in support of our
study. Contrary to MMP9, ESR1, which encodes oestrogen
receptor 1 (ER1), was the principal downregulated gene with the
most connections with other genes. In consonance with our results,
the loss of ER1 activity was reported in uLMS relative to uLMY
(18,19). Moreover, the expression of ER1 was
statistically related to survival in patients with uLMS and IHC
testing of ER1 in these patients was recommended (20). However, as reported by Garcia et
al (21), the expression of ER1
was not significantly correlated with the survival in patients with
uLMS. According to the literature, ER1 was expressed in 40–80% of
patients with uLMS and longer progression-free-survival (PFS) was
observed in patients with advanced uLMS with strongly expressed ER
and progesterone receptor (PR) when treated with aromatase
inhibitors (22). Therefore,
MMP9 and ESR1 may play crucial roles in the
progression of uLMS and may be of great value as prognosis
markers.
Further, module analysis of the PPI network and the
enrichment analyses were performed. Results showed that the main
module was primarily involved in ‘cell migration’ and ‘cell
motility’, while the enrichment pathways were ‘proteoglycans in
cancer’, ‘malaria’, ‘p53 signalling pathway’ and ‘ECM-receptor
interaction’. The upregulated SDC1 and APOE genes and
the downregulated THBS1, CXCL12 and IGF1 genes are
all hub genes of the significant module. Among these genes, only
the expression of THBS1 (thrombospondin 1) has been studied
in uLMS and the results of most studies were consistent with our
results. For instance, THBS1 was identified as a gene
encoded by a BAC clone, whose expression was frequently lost in
uLMS (23); THBS1 expression
was moderate in uLMY but minimal in uLMS (24); THBS1 was less frequently
expressed in uLMS than in uLMY with a significantly negative
correlation between its expression and lymph-vascular space
invasion in uLMS (25). THBS1, which
is a secreted protein, functions as an endogenous anti-angiogenic
agent with the ability to interfere with endothelial cell migration
and survival. It was reported that the loss of THBS1 expression was
related to worsening of PFS due to the abrogation of the
suppressive effects on angiogenesis and metastasis (14). The suppression of THBS1 by the
activation of the β-adrenergic signalling pathway could induce
angiogenesis in prostate cancer (26). What's more, research indicated that
SDC1 could be necessary in coupling between THBS1 and fascin spike
formation, resulting in the promotion of cell spreading and
cytoskeletal organization (27).
SDC1 encodes the type1 transmembrane heparan sulphate
proteoglycan (HSPG) that contributes substantially to the cell-cell
and cell-matrix interactions, cell growth and migration,
neovascularisation and adhesion-dependent signalling pathways. As
reported by Alexander et al (28), SDC1 is essential for
tumorigenesis, which is induced by the Wnt-1 signalling pathway in
mouse mammary gland. By interacting with laminin 332, SDC1
could promote tumor invasion via the PI3 K and RAC1 signalling
pathways (29). Clinically,
SDC1 has proved to be valuable as a prognostic marker in
metastatic CRC patients (30). In
addition, SDC1 was found to internalize Apolipoprotein E-very
low-density lipoproteins (apoE-VLDL) in human fibroblasts through a
low density lipoprotein receptor-related protein (LRP)-independent
pathway (31). ApoE, encoded by gene
APOE, is mainly produced by the liver and the macrophages of
peripheral tissues and astrocytes of the brain. As a major
component of low-density lipoproteins (LDL) and VLDL, ApoE mediates
the metabolism and transport of lipoproteins (32). It is noteworthy that ApoE is related
to tumorigenesis. For example, serum ApoE levels were strikingly
elevated in non-small cell lung cancer (NSCLC) patients (33) as well as in breast cancer patients
(34) relative to normal healthy
controls; meanwhile, the elevated levels of ApoE were correlated
with tumor metastasis and poor prognosis. Furthermore, in gastric
cancer, the DEGs related to ApoE such as the upregulated
transcription factors, signal transducer and activator of
transcription 2 (STAT2) and STAT3 were mainly involved in the
JAK-STAT cascade and the steroid hormone response (35). However, different from current views,
CXCL12 and IGF1, which are usually related to tumor
invasion and metastasis, are downregulated in uLMS which is a
finding of our study. CXCL12, which is also known as stromal
cell-derived factor 1 (SDF1) or pre-B cell stimulating factor
(PBSF), is a chemokine that mediates inflammatory response,
regulates stem cell migration and participates in tumor metastasis.
CXCL12 regulates multiple tumor-related factors via the
CXCL12-CXCR4 axis and finally results in tumor progression in
various cancers. However, Roy et al (36) found that the expression of
CXCL12 was low in pancreatic cancer tissue relative to
healthy tissue and they concluded that the expression of
CXCL12 suppressed tumor growth and metastasis in pancreatic
cancer based on the results of their study. Besides, CXCL12 was
identified as the target of oestrogen in ER-positive ovarian cancer
and breast cancer and ER-positive cell lines could express
CXCL12 with the existence of oestradiol (37). As mentioned previously, the expression
of ER was low in uLMS. Collectively, it is possible that
CXCL12 is downregulated in uLMS; however, further studies
are required for understanding its effect. Insulin-like growth
factor 1 (IGF1) is an important polypeptide growth factor which
could promote proliferation and inhibit apoptosis by activating the
PI3K-Akt and MAPK pathways in cancer. At present, there is no
report on the expression or effect of IGF1 in uLMS. The major
significant genes and pathways were identified based on the results
of the module analysis and the enrichment analysis, which may help
in the better understanding of the mechanism of the development of
uLMS.
In addition to the abovementioned genes, CCNE1,
FOXM1, AR and WT1 are also crucial hub genes.
CCNE1 and FOXM1, both trigger cancer; however, no
study on their role in uLMS has been reported. Cyclin E1 (CCNE1), a
key regulator of the G1/S transition, suppresses the retinoblastoma
(RB) protein by activating cyclin-dependent kinases (CDK), leading
to unrestricted proliferation. It was reported that CCNE1
was overexpressed in breast cancer (38) as well as in ovarian cancer (39) and could be used as a potential target
for ovarian cancer therapy (39,40).
FOXM1, a transcriptional factor with the fork head domain, is
related to proliferation, angiogenesis metastasis, which are vital
events of tumorigenesis and tumor progression (41,42). FOXM1
was identified as a driver of tumor progression and potential
clinical marker in aggressive cancers, such as prostate cancer and
breast cancer and the suppression of FOXM1 was regarded as a
promising method of cancer therapy (41,42). In
contrast to CCNE1 and FOXM1, AR and WT1 were
the downregulated genes identified in our analyses. AR is
the gene that encodes the androgen receptor. In the study by
Koivisto-Korander et al (20),
AR immunoreactivity was absent in all 100 uterine sarcoma samples
including 28 uLMS samples. Similarly, AR was found to be related to
a lower risk of recurrence of uLMS by Leitao et al (43). Wilms tumor-1 (WT1) protein, is a
transcription factor as well as a tumor suppressor. Mutations in
WT1 were reported to relate to childhood tumors of the
kidney (44). Studies suggested that
WT1 was less expressed in uLMS than in uLMY (45) and patients with WT1-negative
high-grade uterine sarcoma had a better prognosis than the patients
with WT1-positive tumors (46). From
the abovementioned findings, we conclude that AR and WT1 could be
used as prognostic markers, while CCNE1 and FOXM1 may
also participate in the development of uLMS and more studies are
required to reveal the underlying mechanisms.
In conclusion, a total of 95 DEGs including 21
upregulated genes and 74 downregulated genes were identified in the
uLMS samples. As per the PPI network and module analysis, the DEGs
are enriched in ‘proteoglycans in cancer’, ‘p53 signalling pathway’
and ‘ECM-receptor interaction’. In addition, MMP9, APOE, SDC1,
CCNE1 and FOXM1 may play predominant roles in the
initiation and progression of uLMS. This study is the first to
identify the key DEGs and related pathways as well as the
interactions among these key genes in uLMS using bioinformatic
analysis, which may provide a novel understanding of the underlying
mechanisms and help in discovering new molecular targets for the
treatment of uLMS. However, there are limitations of the present
study, including relatively small sample size and no targeted
experimental validation. Therefore, further studies are needed in
the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of China (grant nos. 81572568 and 81272863).
Availability of data and materials
The datasets analyzed during the current study are
available in the GEO repository (http://www.ncbi.nlm.nih.gov/geo/).
Authors' contributions
YZa, YW and FX conceived and designed the study.
YZa, LG and YZh performed the study. YZa wrote the paper. YW and FX
revised and edited the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ricci S, Stone RL and Fader AN: Uterine
leiomyosarcoma: Epidemiology, contemporary treatment strategies and
the impact of uterine morcellation. Gynecol Oncol. 145:208–216.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Park JY, Lee JW, Lee HJ, Lee JJ, Moon SH,
Kang SY, Cheon GJ and Chung HH: Prognostic significance of
preoperative 18F-FDG PET/CT in uterine leiomyosarcoma. J
Gynecol Oncol. 28:e282017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ducie JA and Leitao MM Jr: The role of
adjuvant therapy in uterine leiomyosarcoma. Expert Rev Anticancer
Ther. 16:45–55. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cui RR, Wright JD and Hou JY: Uterine
leiomyosarcoma: A review of recent advances in molecular biology,
clinical management and outcome. BJOG. 124:1028–1037. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barlin JN, Zhou QC, Leitao MM, Bisogna M,
Olvera N, Shih KK, Jacobsen A, Schultz N, Tap WD, Hensley ML, et
al: Molecular subtypes of uterine leiomyosarcoma and correlation
with clinical outcome. Neoplasia. 17:183–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee TI and Young RA: Transcriptional
regulation and its misregulation in disease. Cell. 152:1237–1251.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cuppens T, Moisse M, Depreeuw J, Annibali
D, Colas E, Gil-Moreno A, Huvila J, Carpén O, Zikán M, Matias-Guiu
X, et al: Integrated genome analysis of uterine leiomyosarcoma to
identify novel driver genes and targetable pathways. Int J Cancer.
142:1230–1243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iozzo RV and Sanderson RD: Proteoglycans
in cancer biology, tumor microenvironment and angiogenesis. J Cell
Mol Med. 15:1013–1031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Purushothaman A, Babitz SK and Sanderson
RD: Heparanase enhances the insulin receptor signaling pathway to
activate extracellular signal-regulated kinase in multiple myeloma.
J Biol Chem. 287:41288–41296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brule S, Charnaux N, Sutton A, Ledoux D,
Chaigneau T, Saffar L and Gattegno L: The shedding of syndecan-4
and syndecan-1 from HeLa cells and human primary macrophages is
accelerated by SDF-1/CXCL12 and mediated by the matrix
metalloproteinase-9. Glycobiology. 16:488–501. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Asuthkar S, Velpula KK, Nalla AK, Gogineni
VR, Gondi CS and Rao JS: Irradiation-induced angiogenesis is
associated with an MMP-9-miR-494-syndecan-1 regulatory loop in
medulloblastoma cells. Oncogene. 33:1922–1933. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wen N, Wang Y, Wen L, Zhao SH, Ai ZH, Wang
Y, Wu B, Lu HX, Yang H, Liu WC and Li Y: Overexpression of FOXM1
predicts poor prognosis and promotes cancer cell proliferation,
migration and invasion in epithelial ovarian cancer. J Transl Med.
12:1342014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tzeng HT, Tsai CH, Yen YT, Cheng HC, Chen
YC, Pu SW, Wang YS, Shan YS, Tseng YL, Su WC, et al: Dysregulation
of Rab37-mediated cross-talk between cancer cells and endothelial
cells via thrombospondin-1 promotes tumor neovasculature and
metastasis. Clin Cancer Res. 23:2335–2345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Limoge M, Safina A, Beattie A, Kapus L,
Truskinovsky AM and Bakin AV: Tumor-fibroblast interactions
stimulate tumor vascularization by enhancing cytokine-driven
production of MMP9 by tumor cells. Oncotarget. 8:35592–35608. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lorenc Z, Waniczek D, Lorenc-Podgórska K,
Krawczyk W, Domagała M, Majewski M and Mazurek U: Profile of
expression of genes encoding matrix metallopeptidase 9 (MMP9),
matrix metallopeptidase 28 (MMP28) and TIMP metallopeptidase
inhibitor 1 (TIMP1) in colorectal cancer: Assessment of the role in
diagnosis and prognostication. Med Sci Monit. 23:1305–1311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roomi MW, Kalinovsky T, Niedzwiecki A and
Rath M: Modulation of u-PA, MMPs and their inhibitors by a novel
nutrient mixture in adult human sarcoma cell lines. Int J Oncol.
43:39–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lusby K, Savannah KB, Demicco EG, Zhang Y,
Ghadimi MP, Young ED, Colombo C, Lam R, Dogan TE, Hornick JL, et
al: Uterine leiomyosarcoma management, outcome, and associated
molecular biomarkers: A single institution's experience. Ann Surg
Oncol. 20:2364–2372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu XQ, Shi YF, Cheng XD and Wu YZ: The
differential diagnosis between uterine leiomyosarcoma and the
special subtypes of leiomyoma. Zhonghua Yi Xue Za Zhi.
83:1419–1421. 2003.(In Chinese). PubMed/NCBI
|
|
20
|
Koivisto-Korander R, Butzow R, Koivisto AM
and Leminen A: Immunohistochemical studies on uterine
carcinosarcoma, leiomyosarcoma, and endometrial stromal sarcoma:
Expression and prognostic importance of ten different markers.
Tumour Biol. 32:451–459. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garcia C, Kubat JS, Fulton RS, Anthony AT,
Combs M, Powell CB and Littell RD: Clinical outcomes and prognostic
markers in uterine leiomyosarcoma: A population-based cohort. Int J
Gynecol Cancer. 25:622–628. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
George S, Feng Y, Manola J, Nucci MR,
Butrynski JE, Morgan JA, Ramaiya N, Quek R, Penson RT, Wagner AJ,
et al: Phase 2 trial of aromatase inhibition with letrozole in
patients with uterine leiomyosarcomas expressing estrogen and/or
progesterone receptors. Cancer. 120:738–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho YL, Bae S, Koo MS, Kim KM, Chun HJ,
Kim CK, Ro DY, Kim JH, Lee CH, Kim YW and Ahn WS: Array comparative
genomic hybridization analysis of uterine leiomyosarcoma. Gynecol
Oncol. 99:545–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Uluer ET, Inan S, Ozbilgin K, Karaca F,
Dicle N and Sanci M: The role of hypoxia related angiogenesis in
uterine smooth muscle tumors. Biotech Histochem. 90:102–110. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bodner-Adler B, Nather A, Bodner K,
Czerwenka K, Kimberger O, Leodolter S and Mayerhofer K: Expression
of thrombospondin 1 (TSP 1) in patients with uterine smooth muscle
tumors: An immunohistochemical study. Gynecol Oncol. 103:186–189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D
and Li W: Beta-adrenergic signaling promotes tumor angiogenesis and
prostate cancer progression through HDAC2-mediated suppression of
thrombospondin-1. Oncogene. 36:1525–1536. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adams JC, Kureishy N and Taylor AL: A role
for syndecan-1 in coupling fascin spike formation by
thrombospondin-1. J Cell Biol. 152:1169–1182. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alexander CM, Reichsman F, Hinkes MT,
Lincecum J, Becker KA, Cumberledge S and Bernfield M: Syndecan-1 is
required for Wnt-1-induced mammary tumorigenesis in mice. Nat
Genet. 25:329–332. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marinkovich MP: Tumor microenvironment:
Laminin 332 in squamous-cell carcinoma. Nat Rev Cancer. 7:370–380.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jary M, Lecomte T, Bouché O, Kim S, Dobi
E, Queiroz L, Ghiringhelli F, Etienne H, Léger J, Godet Y, et al:
Prognostic value of baseline seric Syndecan-1 in initially
unresectable metastatic colorectal cancer patients: A simple
biological score. Int J Cancer. 139:2325–2335. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wilsie LC, Gonzales AM and Orlando RA:
Syndecan-1 mediates internalization of apoE-VLDL through a low
density lipoprotein receptor-related protein (LRP)-independent,
non-clathrin-mediated pathway. Lipids Health Dis. 5:232006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang YA, Zhou B, Wernig M and Südhof TC:
ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription
and Aβ secretion. Cell. 168:427–441.e21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo J, Song J, Feng P, Wang Y, Long W, Liu
M and Li L: Elevated serum apolipoprotein E is associated with
metastasis and poor prognosis of non-small cell lung cancer. Tumour
Biol. 37:10715–10721. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu X, Wan J, Yuan L, Ba J, Feng P, Long W,
Huang H, Liu P, Cai Y, Liu M, et al: Serum levels of apolipoprotein
E correlates with disease progression and poor prognosis in breast
cancer. Tumour Biol. Oct 5–2016.(Epub ahead of print). View Article : Google Scholar
|
|
35
|
Shi X, Xu J, Wang J, Cui M, Gao Y, Niu H
and Jin H: Expression analysis of apolipoprotein E and its
associated genes in gastric cancer. Oncol Lett. 10:1309–1314. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Roy I, Zimmerman NP, Mackinnon AC, Tsai S,
Evans DB and Dwinell MB: CXCL12 chemokine expression suppresses
human pancreatic cancer growth and metastasis. PLoS One.
9:e904002014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang YP and Wu XH: Correlations of
chemokine CXCL12 and its receptor to tumor metastasis. Ai Zheng.
26:220–224. 2007.(In Chinese). PubMed/NCBI
|
|
38
|
Wu Y, Guo X, Brandt Y, Hathaway HJ and
Hartley RS: Three-dimensional collagen represses cyclin E1 via β1
integrin in invasive breast cancer cells. Breast Cancer Res Treat.
127:397–406. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakayama N, Nakayama K, Shamima Y,
Ishikawa M, Katagiri A, Iida K and Miyazaki K: Gene amplification
CCNE1 is related to poor survival and potential therapeutic target
in ovarian cancer. Cancer. 116:2621–2634. 2010.PubMed/NCBI
|
|
40
|
Kanska J, Zakhour M, Taylor-Harding B,
Karlan BY and Wiedemeyer WR: Cyclin E as a potential therapeutic
target in high grade serous ovarian cancer. Gynecol Oncol.
143:152–158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin SC, Kao CY, Lee HJ, Creighton CJ,
Ittmann MM, Tsai SJ, Tsai SY and Tsai MJ: Dysregulation of
miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of
prostate cancer. Nat Commun. 7:114182016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gormally MV, Dexheimer TS, Marsico G,
Sanders DA, Lowe C, Matak-Vinković D, Michael S, Jadhav A, Rai G,
Maloney DJ, et al: Suppression of the FOXM1 transcriptional
programme via novel small molecule inhibition. Nat Commun.
5:51652014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leitao MM, Soslow RA, Nonaka D, Olshen AB,
Aghajanian C, Sabbatini P, Dupont J, Hensley M, Sonoda Y, Barakat
RR and Anderson S: Tissue microarray immunohistochemical expression
of estrogen, progesterone, and androgen receptors in uterine
leiomyomata and leiomyosarcoma. Cancer. 101:1455–1462. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nachtigal MW, Hirokawa Y,
Enyeart-VanHouten DL, Flanagan JN, Hammer GD and Ingraham HA:
Wilms' tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1
in sex-specific gene expression. Cell. 93:445–454. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Patil DT, Laskin WB, Fetsch JF and
Miettinen M: Inguinal smooth muscle tumors in women-a dichotomous
group consisting of Müllerian-type leiomyomas and soft tissue
leiomyosarcomas: An analysis of 55 cases. Am J Surg Pathol.
35:315–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Coosemans A, Van Calster B, Verbist G,
Moerman P, Vergote I, Van Gool SW and Amant F: Wilms tumor gene 1
(WT1) is a prognostic marker in high-grade uterine sarcoma. Int J
Gynecol Cancer. 21:302–308. 2011.PubMed/NCBI
|