Introduction
As the most common malignancy in females with an
increasing rate of morbidity, breast cancer is a heterogeneous
disease with a high degree of diversity in histology, therapeutic
response and treatment outcomes (1).
Based on transcriptional profiling analysis, breast cancer is
reproducibly identified as one of the major intrinsic subtypes,
including normal breast-like, luminal A, luminal B, epidermal
growth factor receptor-2 (HER2)/Neu-enriched and basal-like breast
cancer (2,3).
Breast cancer can also be categorized on the basis
of expression of the estrogen receptor (ER), progesterone receptor
(PR) and human HER2 (4,5). Notably, basal-like breast cancer largely
overlaps with tumors lacking ER/PR and HER2 expression.
Triple-negative breast cancer (TNBC) is particularly
‘stem-cell-like’ as it adopts properties of stem cells, including
self-renewal (6). Cancer stem cells
(CSCs) have been considered as key contributors to the development
and progression of malignant tumors, including initiation,
sustenance, metastasis and recurrence, in addition to resistance to
conventional chemotherapy. Therefore, simultaneous targeting of
CSCs and non-CSCs holds great potential towards the development of
more efficient therapeutic methodologies for each type of cancer
(7).
Maternal embryonic leucine zipper kinase (MELK),
also known as murine protein serine threonine kinase 38 (MPK38), is
a member of AMPK/Snf1 family. MELK functions as a modulator of
intercellular signaling, including the apoptosis signal-regulating
kinase/Jun N-terminal kinase (JNK) pathway, p38 signaling (8) and NF-κB pathway (9), which affects various cellular and
biological processes. Currently, MELK has been demonstrated as a
key regulator in the malignancy and proliferation of CSCs, and
therefore it is considered as an attractive molecular target to
eliminate various CSCs (10–12). Previous data has documented that MELK
is an important contributor in the tumorigenesis of human mammary
epithelial cells. MELK is highly expressed in human breast cancer,
and its overexpression is strongly associated with poor disease
outcomes (13–20). In addition, MELK expression in breast
cancer has a significant inverse correlation with the expression of
luminal markers, including ER and PR (21). Therefore, MELK is aberrantly
overexpressed in ER/PR− tumors compared with tumors with
ER/PR+ status (21).
Indeed, MELK has been considered as an oncogenic kinase that is
essential for mitotic progression in basal-like breast cancer cells
(21). Specific targeting of MELK
enables induction of programmed cell death of specific breast
cancer cell lines, including TNBC MDA-MB-231, and BT-549, as
indicated by cleaved PARP (poly ADP-ribose polymerase) (21). PARP, a 116 kDa nuclear polymerase, is
a highly conserved nuclear enzyme implicated in DNA repair and in
the apoptotic response of cells. This protein can be cleaved by
numerous caspases in vitro and is one of the main cleavage
targets of caspase-3 in vivo. The cleavage occurs between
ASP214 and Gly 215, which separates PARP's N-terminal DNA binding
domain (24 kDa) from its C-terminal catalytic domain (89 kDa). It
has been demonstrated that cleavage of PARP facilitates cellular
disassembly and inhibition of PARP cleavage attenuates apoptosis
in vitro (22). Thus, MELK has
promising potential as a molecular target in breast cancer therapy,
and therefore it is warranted to extensive studies on the
mechanisms involved.
The present study reports that MELK expression does
not absolutely associate with ER expression. Although the knockdown
of MELK may lead to marked inhibition in the proliferation of TNBC
and non-TNBC cells, specific targeting of MELK did not result in
apoptosis in TNBC or HCC1143 cells. MELK exerts its effect on TNBC
and non-TNBC cells via inducing arrest at different phases of the
cell cycle and by different mediators. The ER signaling pathway may
participate in the regulation of MELK expression. When taking into
consideration with previous data, MELK may be used as a specific
target to control cell proliferation in MDA-MB-231 cells but not
all TNBC cells.
Materials and methods
Cell lines, antibodies and
reagents
Human mammary epithelial cell line MCF10A and
different breast cancer cell lines (T47D, HCC712, MCF7, ZR75-1,
MDA-MB-361, HCC1937, HCC1806 and MDA-MB-231) used in present study
were obtained from the American Type Culture Collection (Manassas,
VA, USA). DMEM/F12, RPMI 1640 and fetal bovine serum (FBS) were
purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Primary and secondary antibodies used for immunoblotting were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Other reagents including; EGF, insulin, hydrocortisone,
antibiotics, 50 µg/ml gentamycin, pyruvate, 10 mM Hepes, 4.5 g/l
glucose, 0.25% EDTA-containing trypsin, estradiol, dextran
charcoal-stripped bovine serum, MTT reagent, propidium iodide and
bovine serum albumin were products of Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Cell culture
Human mammary epithelial cells, MCF10A were
maintained in DMEM/F-12 supplemented with EGF (10 ng/ml), insulin
(10 µg/ml), and hydrocortisone (0.5 µg/ml) in a humidified
incubator with 5% CO2 at 37°C. All breast cancer cell
lines (T47D, HCC712, MCF7, ZR75-1, MDA-MB-361, HCC1937, HCC1806 and
MDA-MB-231) used in the present study were propagated in RPMI 1640
medium containing 10% FBS and antibiotics (penicillin and
streptomycin) and supplements (50 µg/ml gentamycin, pyruvate, 10 µM
Hepes and 4.5 g/l glucose) in a humidified 37°C incubator
containing 5% CO2.
Estrogen deprivation treatment
The wild-type MCF7 and ZR75-1 cells were cultured in
phenol red-free RPMI 1640 medium supplemented with 10% FBS and 1 nM
estradiol (E2) in a 37°C incubator for 1 week. For estradiol
deprivation treatment, cancer cells were cultured in phenol-free
RPMI medium in the absence of exogenous E2 and supplemented with
10% dextran charcoal-stripped bovine serum (DCC). The cells were
trypsinized using 0.25% EDTA-containing trypsin at base line,
1-week post estradiol deprivation (short-term estradiol
deprivation, STED) and at the point of resistance (long-term
estradiol deprivation, LTED) (23).
Small-interfering RNA (siRNA)
treatment
For knockdown experiments, breast cancer cell lines
(HIM3, HCC1806, MDA-MB-231, HCC1143, BT549, HCC1937, SKBR3, T47D,
MCF7 and HCC712) and human mammary epithelial cell MCF10 were
transiently transfected with 200 pmol oligo siRNA using
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
siRNA targeting MELK (siMELK, 5′-GACAUCCUAUCUAGCUGCA-3′) and
scrambled negative control (5′-GUGGGCAACAUUCUUCGAATT-3′) were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Subsequent experimentation was conducted 3 days following
transfection.
Cell proliferation assay
The cells treated with siMELK or negative control
(50 nM) were seeded at a density of 1×104 cells/well in
96-well plates. Cell proliferation was quantified by MTT reduction
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide).
Formazan salt was dissolved in acid isopropanol, and absorbance was
assessed at 570 and 630 nm on a microplate reader. The results are
expressed as increases in absorbance (570–630 nm). The experiments
were performed at least three times in triplicate.
Immunoblotting
The cells were lysed at 4°C for 15 min with RIPA
buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet p-40, 0.5%
sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing
protease cocktail inhibitors (Roche Applied Science, Penzberg,
Germany) and phosphatase inhibitor cocktail (Thermo Fisher
Scientific, Inc.). Protein quantification was performed using a BCA
Protein assay (Pierce Biotechnology; Thermo Fisher Scientific,
Inc.), and aliquots of 20 µg total protein was resolved on SDS-PAGE
(12% gel) and transferred onto a nitrocellulose membrane.
Subsequently, the membrane was blocked with 5% non-fat milk
(dissolved in TBST; incubation for 1 h at room temperature) and was
then incubated with the following primary antibodies: Anti human
β-actin (monoclonal; cat. no. 3700; 1:1,000), anti human MELK
(polyclonal; cat. no. 2274; 1:1,000), anti human p-JNK (monoclonal;
cat. no. 9255; 1:2,000), anti human p-p38 (polyclonal; cat. no.
4511; 1:1,000), anti human p-ERK1/2 (polyclonal; cat. no. 4370;
1:2,000), anti human p27 (polyclonal; cat. no. 3686; 1:1,000), anti
human p21 (polyclonal; cat. no. 2947; 1:1,000), anti human p53
(monoclonal; cat. no. 2524; 1:1,000), anti human cyclin B
(monoclonal; cat. no. 4135; 1:2,000), anti human p-cdc2
(polyclonal; cat. no. 9111; 1:1,000), anti human cyclin A
(monoclonal; cat. no. 4656; 1:2,000), anti human cyclin E
(polyclonal; cat. no. 20808; 1:1,000), anti human cyclin D1
(polyclonal; cat. no. 2978; 1:1,000), anti human CDK2 (polyclonal;
cat. no. 2546; 1:1,000), anti human caspase-3 (polyclonal; cat. no.
9662; 1:1,000) overnight at 4°C. After washing with TBST, the
membrane was incubated with horseradish peroxidase-conjugated
secondary antibodies including: Anti-mouse immunoglobulin G (cat.
no. 7076; 1:1,000) and anti-rabbit immunoglobulin G (cat. no. 7074;
1:1,000) at room temperature for 2 h. The target signals were
visualized and semi-quantified as the ratio of target protein
relative to β-actin.
Cell cycle analysis
The cells were trypsinized and repeatedly pipetted
into single-cell suspension. Following centrifugation at 600 × g
for 5 min at 4°C, the cells were fixed by adding 70% ethanol
(−20°C) in drops while vortexing. Subsequently, the cells were
stained with 50 µg/ml propidium iodide solution containing 50 µg/ml
DNase-free RNase A and 0.5% bovine serum albumin (BSA). Following
incubation for 30 min, the cells were washed and resuspended in
0.5% BSA. Cell cycle analysis was performed on a LSRFortessa (BD
Biosciences, San Jose, CA, USA) at the DFCI flow cytometry core
facility. Single cells were gated by plotting FL3-A to FL3-H in
order to exclude cell debris and doublets. A minimum of
1×104 single cells was collected from each sample.
Statistical analysis
The data are presented as the mean ± standard
deviation, and analyzed using SPSS 17.0 (SPSS, Chicago, IL, USA).
Two-tailed unpaired Student's t-test was used to analyze difference
between two groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
High MELK protein expression is
observed in MDA-MB-231 cells
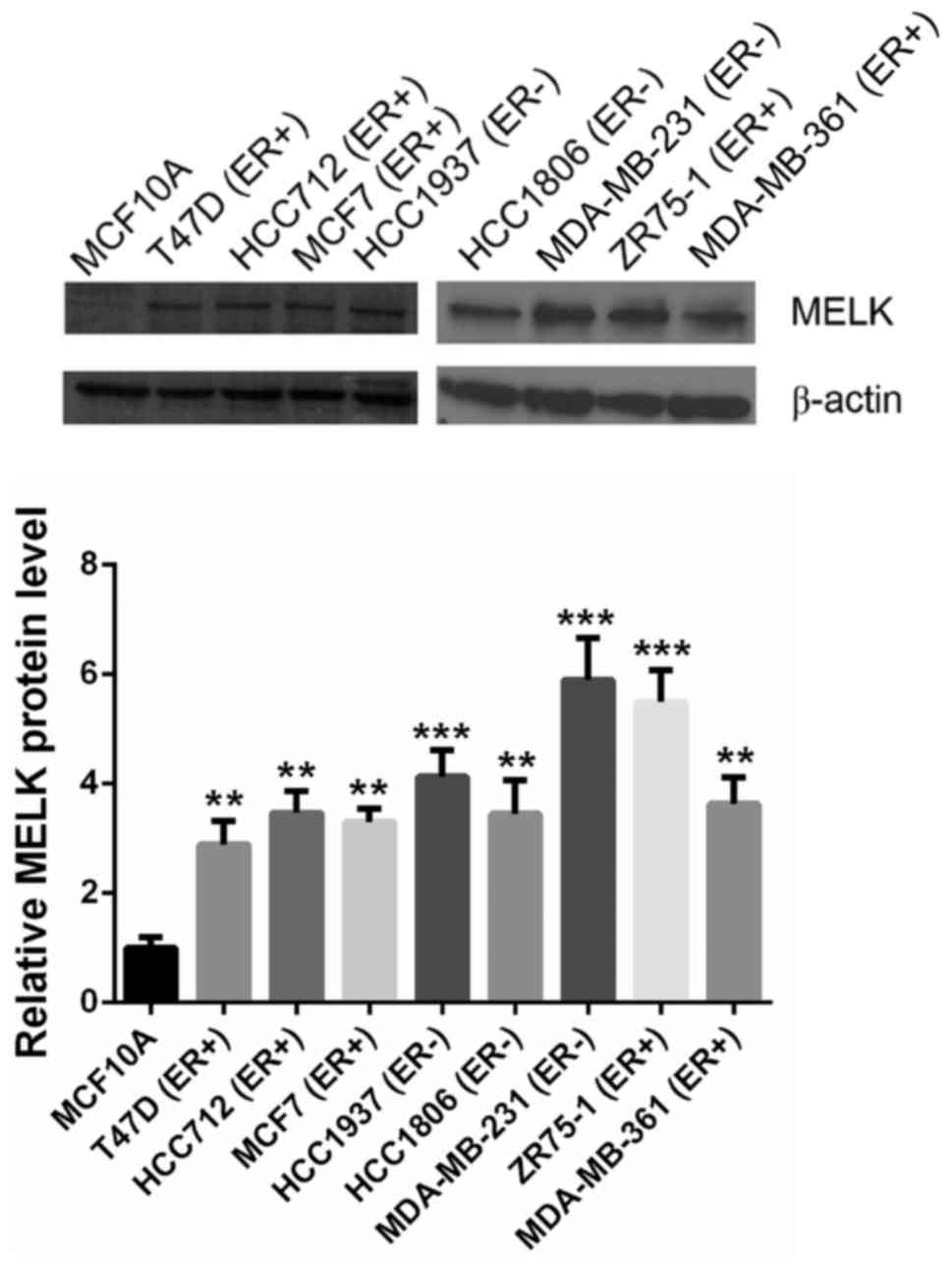
To validate the expression patterns of MELK protein,
human breast epithelial MCF10A cell line and eight breast cancer
lines, including ER+ and ER− breast cancer
cell lines, were subjected to semi-quantification analysis. MELK
was hardly detectable in human breast epithelial MCF10A cells, but
aberrantly overexpressed in ER+ and ER− human
breast cancer cells (Fig. 1).
Treatment with MELK siRNA suppresses
the proliferation of TNBC and non-TNBC cells
Following the confirmation of MELK overexpression in
breast cancer cell lines, MELK siRNA and scrambled siRNA (as a
negative control) was used to treat human breast epithelial MCF10A
cells and several ER+ and ER− human breast
cancer cell lines. Following 5 days of incubation, cell
proliferation was analyzed. As indicated in Fig. 2, the silencing of MELK resulted
in a small decrease in the proliferation rate of human breast
epithelial cells MCF10A, which is as hypothesized as MELK
expression was barely detectable in this cell line. By contrast,
the knockdown of MELK in breast cancer cell lines (duration
of incubation, 5 days) resulted in marked decreases in
proliferation compared with the cells that were transfected with
negative control. Notably, the biggest decrease in proliferation
was observed in the siRNA-transfected HCC1143 TNBC cells. By
contrast, there was only a small decrease in proliferation in
siRNA-transfected HCC712 cells, which are
ER+/PR+/HER−. Furthermore,
specific targeting of MELK results in inhibitory effects on
MDA-MB-231 cells, which was comparable to the effects observed on
MCF-7 and T47D cells.
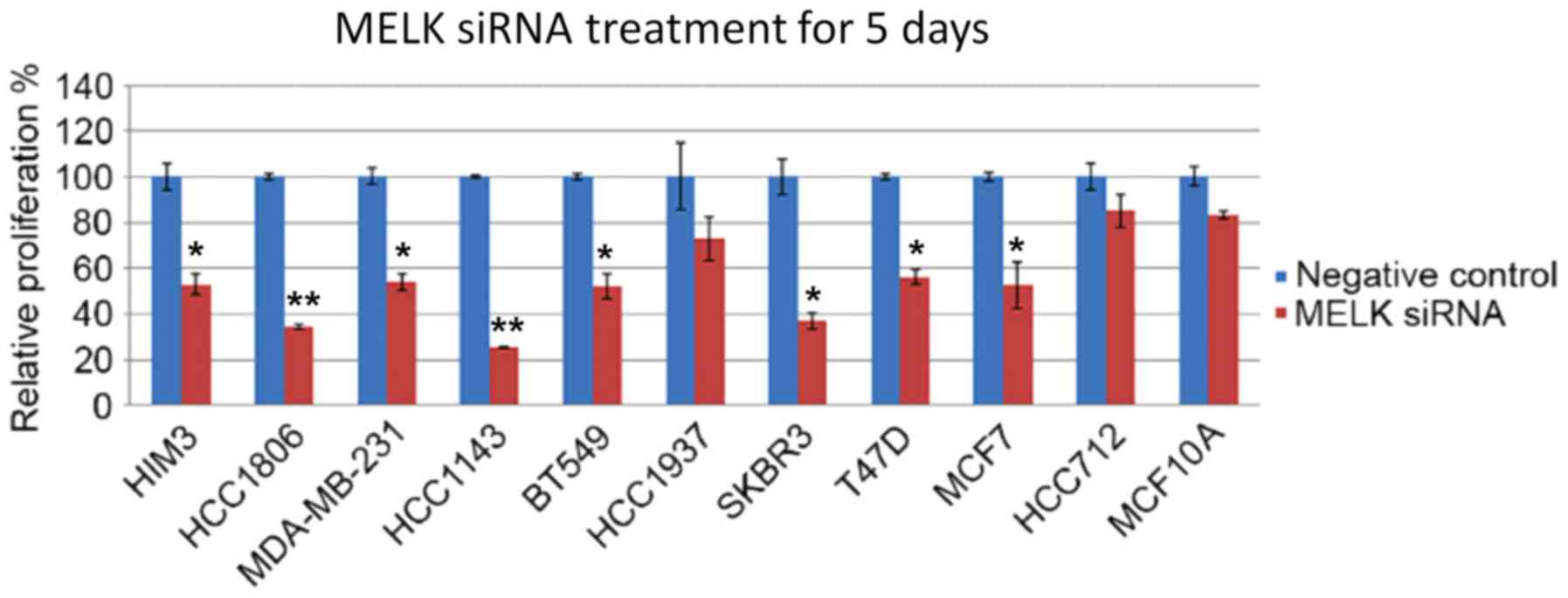 | Figure 2.Effects of MELK siRNA treatment on the
proliferation of human mammary epithelial cells and various TNBC
and non-TNBC cell lines. Human mammary epithelial cells are MCF10.
TNBC cell lines are as follows: HIM3, HCC1806, MDA-MB-231,
HCC-1143, BT-549. Non-TNBC cell lines are as follows: HCC1937,
SKBR3, T47D, MCF7A HCC712. Data is presented as the mean ± standard
deviation. *P<0.05, **P<0.01 compared with negative siRNA
treatment controls. MELK, maternal embryonic leucine zipper kinase;
TNBC, triple-negative breast cancer; HIM, human in mouse. |
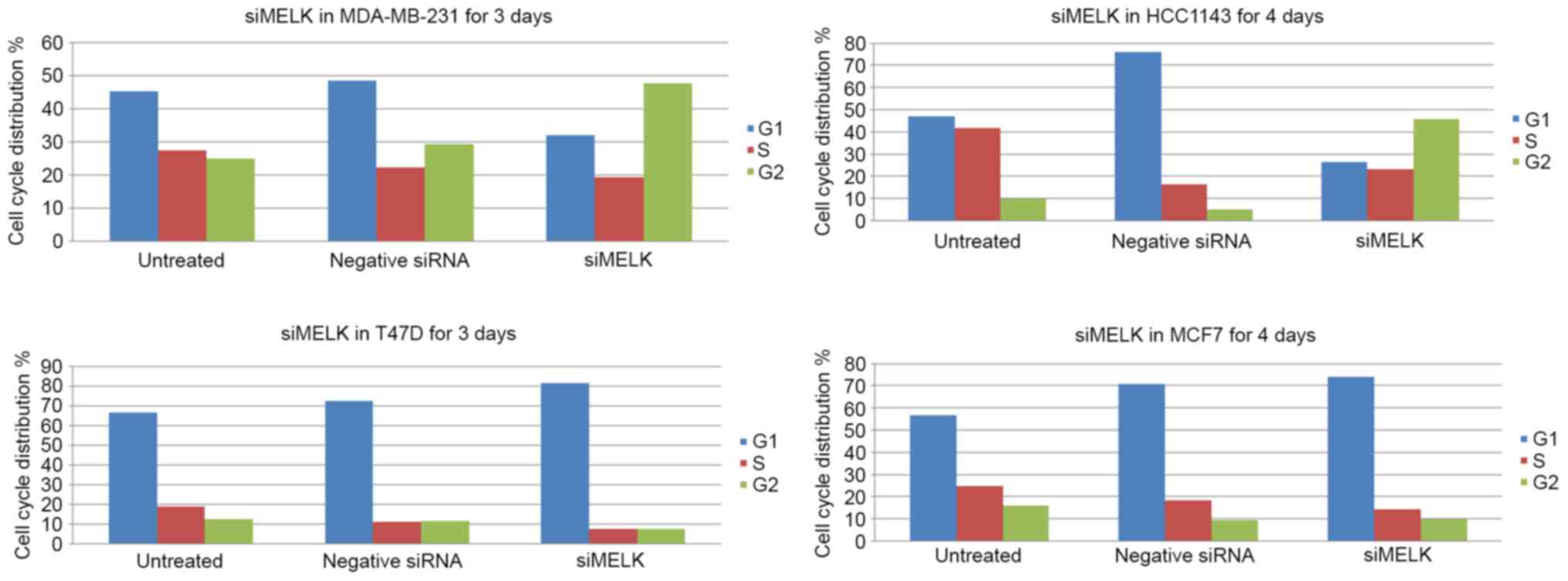
Treatment with MELK siRNA induces G2
arrest in TNBC cell line and G1 arrest in non-TNBC cell line
In order to determine whether MELK affects the
proliferation of breast cancer cells, which may be attributable to
its regulation of the cell cycle, two TNBCs lines (MDA-MB-231 and
HCC1143) and two non-TNBC breast cancer lines (T47D and MCF7) were
analyzed for cell cycle distribution. Compared with the data from
MDA-MB-231 cells that were treated with scrambled siRNA, specific
silencing of MELK in MDA-MB-231 cells for 4 days produced a
decrease in the number of cells in the G1 phase, and markedly
increased the number of cells in the G2 phase (Fig. 3). Notably, the number of
si-MELK-treated HCC1143 cells in the S phase was markedly increased
compared with scrambled siRNA treated cells. Additionally, non-TNBC
T47D cells that were untreated or treated with scrambled siRNA were
predominantly distributed in the G1 phase, and a small proportion
of the cells were distributed in the G2 phase. Specific silencing
of MELK for 3 days resulted in a decreased number of cells
in the G2 phase, and the number of cells in the G1 phase increased.
In TNBC MCF7 cells, specific silencing of MELK for 4 days
led to an increase in the proportion of cells in the G1 phase,
accompanied with a marked decrease in cells in the S and G2 phases
compared with untreated cells and negative siRNA-transfected with
(Fig. 3).
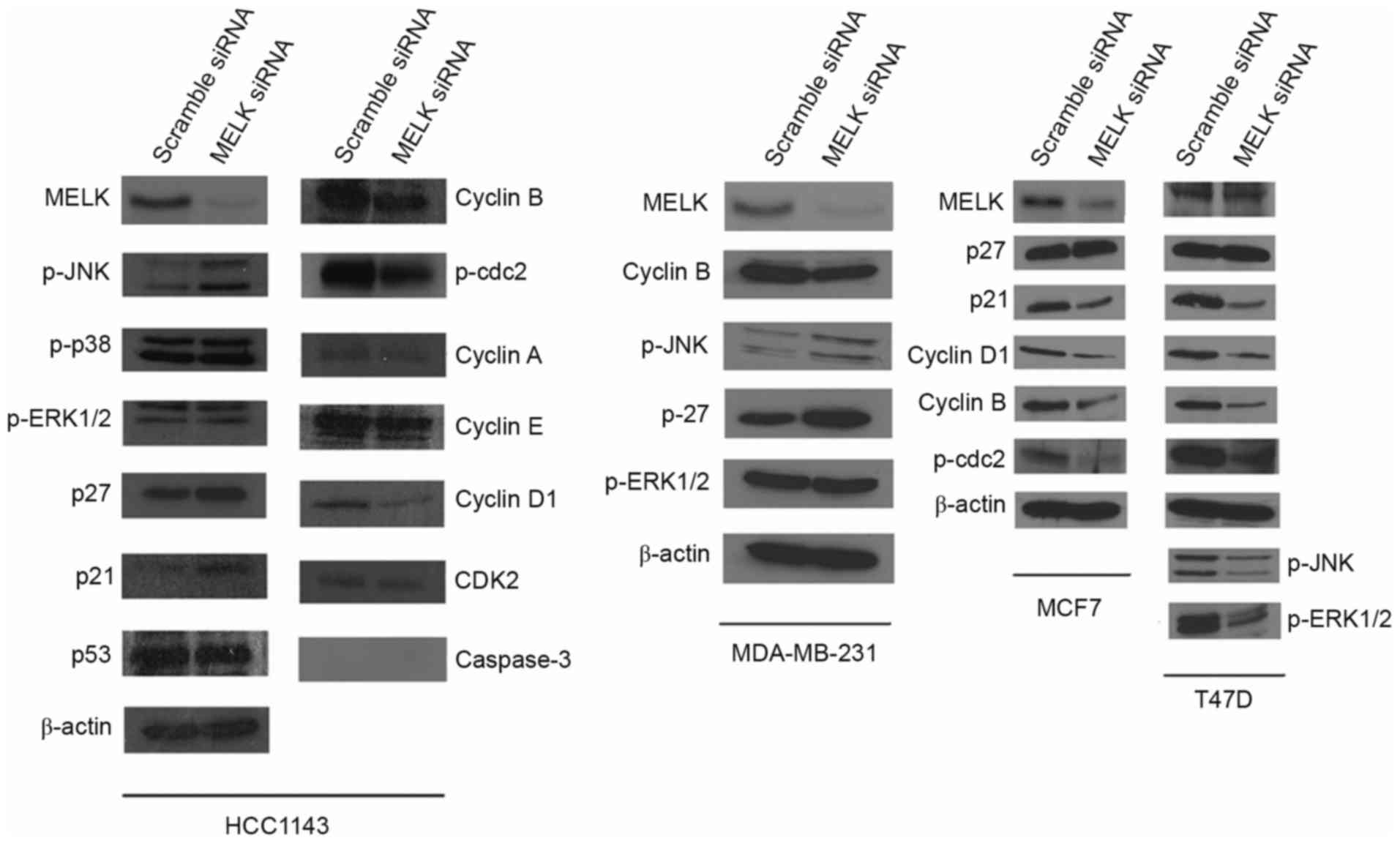
Treatment with MELK siRNA induces
differential expression patterns in cell cycle-regulatory
proteins
To investigate the molecular mechanisms of MELK on
the proliferation of breast cancer cells, the levels of cell
cycle-regulatory proteins were assessed in TNBC cells and non-TNBC
cells. It was demonstrated that specific silencing of MELK
resulted in marked decrease in the levels of MELK protein in
HCC1143 cells. Accompanied with this inhibition, specific targeting
of MELK by siRNA for 2 days resulted in the inhibition of
cyclin B, cyclin D1 and phosphorylated (p)-cdc 2 expression, as
well as the promotion of p-c-JNK, p27 and p21 in HCC1143 cells.
Furthermore, detectable changes in p-p38, p53, and CDK2 expression
were not observed in HCC1143 cells. Notably, caspase-3 was not
detected in cells that were treated with siRNA-MELK or scrambled
siRNA in HCC1143 cells. The increase in p27 and p-JNK expression as
a consequence of MELK knockdown was also revealed in TNBC
MDA-MB-231 cells, compared with scrambled siRNA. Additionally, a
significant decrease in cyclin B expression was revealed in
MDA-MB-231 cells as a result of specific silencing of MELK.
As for non-TNBC MCF7 and T47D cells, specific silencing targeting
MELK for 2 days resulted in marked decrease in cyclin D1,
cyclin B, p-cdc2, p-extracellular signal-regulated kinase (ERK)
1/2, p-JNK and p21. There was no significant difference in p27
expression following the silencing of MELK (Fig. 4).
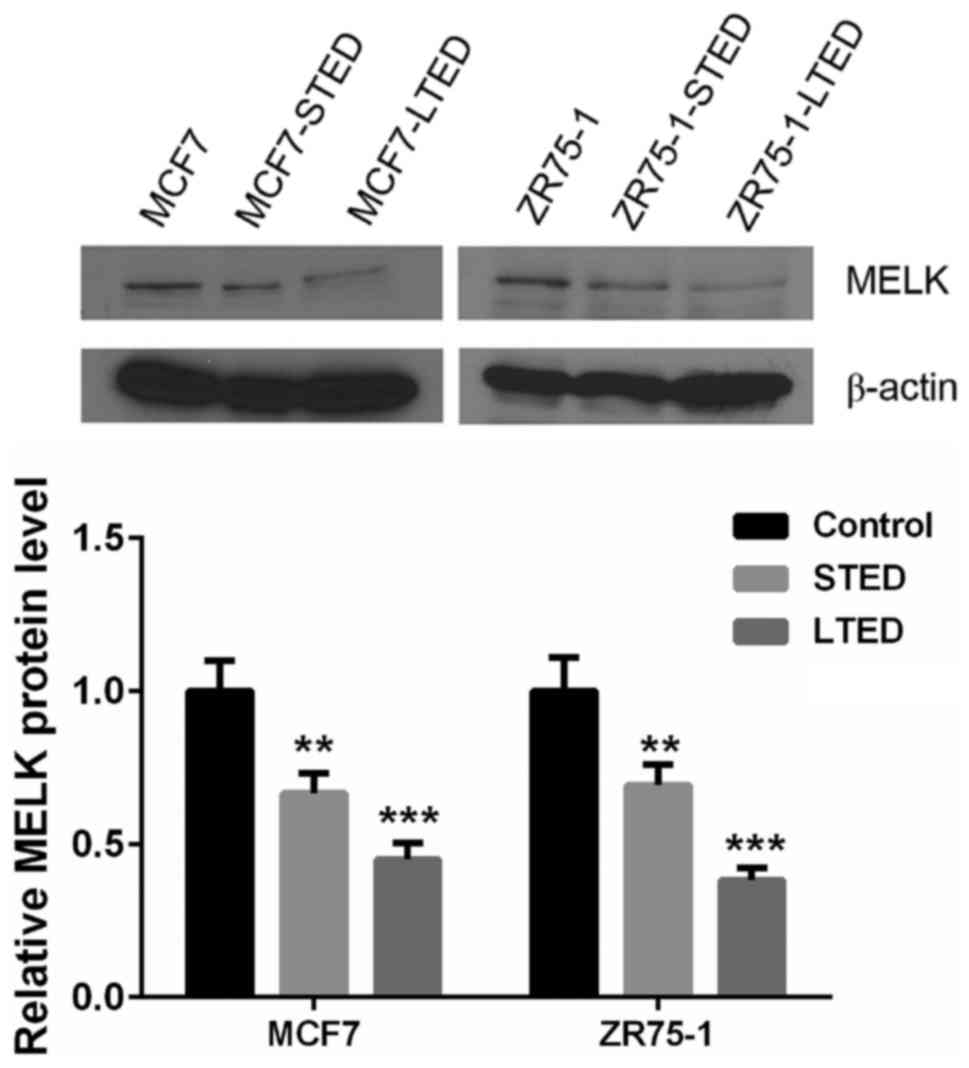
MELK protein is downregulated in
response to estrogen deprivation in ER+ MCF-7 and ZR75-1
cells
To investigate the role of estrogen on MELK
expression, ER+ breast cancer lines, MCF7 and ZR75-1
underwent estradiol deprivation treatment. In the presence of E2,
there was a low expression of MELK in ER+ breast cancer
MCF7 and ZR75-1 cells. MELK expression was significantly suppressed
in breast cancer cell lines that were cultured in STED medium
compared with untreated cells, and hardly detectable in the LTED
medium (P<0.01; Fig. 5).
Discussion
Despite marked advances in breast cancer therapy,
TNBCs remain a challenge in the clinic, attributable to stem
cell-like characteristics and a relative unresponsiveness to
targeted therapies. MELK is a key regulator of malignancy and
proliferation of CSCs and an oncogenic kinase that is essential for
mitotic progression in basal-like breast cancer cells (21). A previous study has documented that
targeting MELK resulted in cell death of TNBC MDA-MB-231 cells
(24). However, the molecular mech
TNBC anism underlying this effect is still unknown (24).
In the present study, it was observed that MELK
protein was aberrantly expressed in 8 ER− and
ER+ breast cancer cell lines. MELK is highly expressed
in MDA-MB-231 cells revealed in the present study may provide new
data that MELK could be used a specific target to eliminate
MDA-MB-231 cells. Furthermore, it was indicated in the present
study that the silencing of MELK induced a marked decrease in the
proliferation of MDA-MB-231 cells, which was in accordance with the
findings in a previous study where the loss of MELK promoted
programmed cell death of MDA-MB-231 cells (21). It was further observed that the
biggest decrease in proliferation was observed in the
siRNA-transfected HCC1143 TNBC cells. In addition, inhibition in
the viability of non-TNBC T47D and MCF7 cells was also observed,
which was comparable to the effects observed in MDA-MB-231 cells.
Therefore, the four cell lines (HCC1143, T47D, MCF7 and MDA-MB-231)
were employed for subsequent analysis.
In order to identify whether MELK is a cell cycle
regulator, which participates in mediating inhibitory effects on
breast cancer cells, the aforementioned four cell lines were
employed for cell cycle analysis. The results revealed that
specific targeting of MELK caused G2 arrest in TNBC lines (HCC1143
and MDA-MB-231), and G1 arrest in non-TNBC lines (T47D and MCF7),
suggesting that different molecules mediate the specific targeting
MELK on the proliferation of TNBC and non-TNBC cells. It is notable
that specific targeting of MELK resulted in weak downregulation in
the level of MELK protein in TNBC HCC1143, non-TNBC T47D and MCF7
cells. The sensitivity to specific siRNA targeting MELK in various
breast cancer cell lines should be investigated individually
(21). Protein quantification
revealed that caspase-3 was undetectable in HCC1143 cells treated
with MELK siRNA or scrambled siRNA, suggesting that genetic
knockdown of MELK did not promote apoptosis of HCC1143 cells. In
addition, cyclin B and cyclin D1 in TNBC and non-TNBC cells were
markedly downregulated in response to silencing of MELK.
Additionally, silencing of MELK in HCC1143 TNBC cells resulted in
upregulation of p27 and p21, and downregulation of p21 in non-TNBC
cells (MCF7 and T47D). Notably, p-JNK was upregulated in TNBC cells
and downregulated in non-TNBC T47D cells as a consequence of
silencing of MELK. In response to silencing of MELK, the level of
p-ERK1/2 protein was decreased in T47D cells.
The effects of MELK silencing are mediated through
substrates. According to previous data, MELK is able to
phosphorylate CDC25B on Ser323 in vitro (25), which is a critical 14-3-3 binding site
(26). The 14-3-3 binding to the
Ser323 site on CDC25B, blocks access of the substrate, cyclin/cdk,
to the catalytic site of the enzyme and therefore directly inhibit
the activity of CDC25B, which initially results in arrest at G2
(27). Additionally, MELK
phosphorylates zinc finger-like protein 9 (ZPR9) and promotes its
nuclear localization (28), therefore
ZPR9 interacts with and increases the transcriptional activity of
Myb-like protein 2 (29). This has
been shown to promote DNA replication and transcriptional
activation of genes, including cyclin B1, which is essential for
G2/M phase progression (30,31).
In addition to substrates, molecules regulating MELK
expression should be also considered. As demonstrated in the
present study, estrogen deprivation led to a marked suppression in
MELK protein expression in non-TNBC MCF7 and ZR75-1 cells,
indicating that estrogen may be required to maintain MELK
expression. It has been well documented that the ER signaling
pathway crosstalks with the phosphatidylinositol 3-kinase/protein
kinase B/mechanistic target of rapamycin (PI3K/Akt/mTOR) signaling
pathway. Furthermore, protein kinase B (PKB)/Akt is able to
phosphorylate Forkhead box protein O1 (FOXO) transcription factors
and create docking sites for 14-3-3 (32). The binding of 14-3-3 to FOXO excludes
FOXO from the nucleus, therefore the transcriptional activity of
FOX is inhibited. By contrast, c-Jun N-terminal kinases (JNKs) and
MST1 are activated by stimuli, which results in the phosphorylation
of FOXOs at two different sites. The phosphorylation by JNK or MST1
promotes the nuclear localization of FOXO despite phosphorylation
by Akt, thus various genes are targeted, including p27 and p21
(32,33).
It has been documented that FOXOs have a major role
in G1 arrest by upregulating cell cycle inhibitors (p21 and p27),
and the consequent attenuation of cell cycle promotes CDKs.
Furthermore, the activation of FOXO3 is sufficient to elevate p27
mRNA and protein levels and to induce apoptosis (32–34). The
data from a previous study on glioma stem cells indicated that JNK
signaling regulates MELK expression and forms a complex with
oncoprotein c-JUN in glioma stem cells (35), suggesting that JNK may have a dual
role in regulating FOXOs and MELK expression. However, the
hypothesis described here should be further validated by future
studies.
In conclusion, MELK expression does not absolutely
associate with ER expression in breast cancer tissues. Although the
sensitivity of MELK for specific siRNA varies in TNBC and non-TNBC
cells, the genetic knockdown of MELK resulted in a marked
decrease in the proliferation of TNBC and non-TNBC cells. The
silencing of MELK did not result in apoptosis in HCC1143 cells,
which is indicated by the lack of caspase-3 expression. The
specific targeting of MELK on TNBC and non-TNBC cells induces cell
cycle arrest and different stages, for example, induces G2 arrest
in TNBC cell lines and G1 arrest in non-TNBC cell lines, due to
causing a decrease in cyclin B1 and an increase in p27 and p-JNK in
TNBC cell lines; and a decrease in p21, cyclin B1, cyclin D1 and
p-cdc2 in non-TNBC cell lines. In addition to ER66, other ER
isoforms may participate in the regulation of MELK expression
(36). The expression of JNK and p27
varied in response to silencing of MELK, therefore it is warranted
to further investigate the role of JNK and p27 in mediating the
inhibitory effect of MELK siRNA. Taken together, the results from
the present study provide evidence that MELK may have potential as
a specific target in MDA-MB-231 cells.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rakha EA, Reis-Filho JS and Ellis IO:
Basal-like breast cancer: A critical review. J Clin Oncol.
26:2568–2581. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jamdade VS, Sethi N, Mundhe NA, Kumar P,
Lahkar M and Sinha N: Therapeutic targets of triple-negative breast
cancer: A review. Br J Pharmacol. 172:4228–4237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soady KJ, Kendrick H, Gao Q, Tutt A,
Zvelebil M, Ordonez LD, Quist J, Tan DW, Isacke CM, Grigoriadis A
and Smalley MJ: Mouse mammary stem cells express prognostic markers
for triple-negative breast cancer. Breast Cancer Res. 17:312015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chiotaki R, Polioudaki H and
Theodoropoulos PA: Stem cell technology in breast cancer: Current
status and potential applications. Stem Cells Cloning. 9:17–29.
2016.PubMed/NCBI
|
|
8
|
Jiang P and Zhang D: Maternal embryonic
leucine zipper kinase (MELK): A novel regulator in cell cycle
control, embryonic development, and cancer. Int J Mol Sci.
14:21551–21560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janostiak R, Rauniyar N, Lam TT, Ou J, Zhu
LJ, Green MR and Wajapeyee N: MELK promotes melanoma growth by
stimulating the NF-κB pathway. Cell Rep. 21:2829–2841. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakano I, Masterman-Smith M, Saigusa K,
Paucar AA, Horvath S, Shoemaker L, Watanabe M, Negro A, Bajpai R,
Howes A, et al: Maternal embryonic leucine zipper kinase is a key
regulator of the proliferation of malignant brain tumors, including
brain tumor stem cells. J Neurosci Res. 86:48–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoshimoto K, Ma X, Guan Y, Mizoguchi M,
Nakamizo A, Amano T, Hata N, Kuga D and Sasaki T: Expression of
stem cell marker and receptor kinase genes in glioblastoma tissue
quantified by real-time RT-PCR. Brain Tumor Pathol. 28:291–296.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakano I, Paucar AA, Bajpai R, Dougherty
JD, Zewail A, Kelly TK, Kim KJ, Ou J, Groszer M, Imura T, et al:
Maternal embryonic leucine zipper kinase (MELK) regulates
multipotent neural progenitor proliferation. J Cell Biol.
170:413–427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma XJ, Dahiya S, Richardson E, Erlander M
and Sgroi DC: Gene expression profiling of the tumor
microenvironment during breast cancer progression. Breast Cancer
Res. 11:R72009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richardson AL, Wang ZC, De Nicolo A, Lu X,
Brown M, Miron A, Liao X, Iglehart JD, Livingston DM and Ganesan S:
X chromosomal abnormalities in basal-like human breast cancer.
Cancer Cell. 9:121–132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Desmedt C, Piette F, Loi S, Wang Y,
Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y,
d'Assignies MS, et al: Strong time dependence of the 76-gene
prognostic signature for node-negative breast cancer patients in
the TRANSBIG multicenter independent validation series. Clin Cancer
Res. 13:3207–3214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hatzis C, Pusztai L, Valero V, Booser DJ,
Esserman L, Lluch A, Vidaurre T, Holmes F, Souchon E, Wang H, et
al: A genomic predictor of response and survival following
taxane-anthracycline chemotherapy for invasive breast cancer. JAMA.
305:1873–1881. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmidt M, Böhm D, von Törne C, Steiner E,
Puhl A, Pilch H, Lehr HA, Hengstler JG, Kölbl H and Gehrmann M: The
humoral immune system has a key prognostic impact in node-negative
breast cancer. Cancer Res. 68:5405–5413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM,
Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu
J, et al: Gene-expression profiles to predict distant metastasis of
lymph-node-negative primary breast cancer. Lancet. 365:671–679.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Esserman LJ, Berry DA, Cheang MC, Yau C,
Perou CM, Carey L, DeMichele A, Gray JW, Conway-Dorsey K, Lenburg
ME, et al: Chemotherapy response and recurrence-free survival in
neoadjuvant breast cancer depends on biomarker profiles: Results
from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast
Cancer Res Treat. 132:1049–1062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Lee YM, Baitsch L, Huang A, Xiang
Y, Tong H, Lako A, Von T, Choi C, Lim E, et al: MELK is an
oncogenic kinase essential for mitotic progression in basal-like
breast cancer cells. Elife. 3:e017632014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soldani C and Scovassi AI:
Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update.
Apoptosis. 7:321–328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murphy CS, Meisner LF, Wu SQ and Jordan
VC: Short- and long-term estrogen deprivation of T47D human breast
cancer cells in culture. Eur J Cancer Clin Oncol. 25:1777–1788.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gray D, Jubb AM, Hogue D, Dowd P, Kljavin
N, Yi S, Bai W, Frantz G, Zhang Z, Koeppen H, et al: Maternal
embryonic leucine zipper kinase/murine protein serine-threonine
kinase 38 is a promising therapeutic target for multiple cancers.
Cancer Res. 65:9751–9761. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davezac N, Baldin V, Blot J, Ducommun B
and Tassan JP: Human pEg3 kinase associates with and phosphorylates
CDC25B phosphatase: a potential role for pEg3 in cell cycle
regulation. Oncogene. 21:7630–7641. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mils V, Baldin V, Goubin F, Pinta I, Papin
C, Waye M, Eychene A and Ducommun B: Specific interaction between
14-3-3 isoforms and the human CDC25B phosphatase. Oncogene.
19:1257–1265. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Forrest A and Gabrielli B: Cdc25B activity
is regulated by 14-3-3. Oncogene. 20:4393–4401. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Seong HA, Gil M, Kim KT, Kim SJ and Ha H:
Phosphorylation of a novel zinc-finger-like protein, ZPR9, by
murine protein serine/threonine kinase 38 (MPK38). Biochem J.
361:597–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seong HA, Kim KT and Ha H: Enhancement of
B-MYB transcriptional activity by ZPR9, a novel zinc finger
protein. J Biol Chem. 278:9655–9662. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Joaquin M and Watson RJ: Cell cycle
regulation by the B-Myb transcription factor. Cell Mol Life Sci.
60:2389–2401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tao D, Pan Y, Lu H, Zheng S, Lin H, Fang H
and Cao F: B-myb is a gene implicated in cell cycle and
proliferation of breast cancer. Int J Clin Exp Pathol. 7:5819–5827.
2014.PubMed/NCBI
|
|
32
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hay N: Interplay between FOXO, TOR, and
Akt. Biochim Biophys Acta. 1813:1965–1970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Medema RH, Kops GJ, Bos JL and Burgering
BM: AFX-like Forkhead transcription factors mediate cell-cycle
regulation by Ras and PKB through p27kip1. Nature. 404:782–787.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gu C, Banasavadi-Siddegowda YK, Joshi K,
Nakamura Y, Kurt H, Gupta S and Nakano I: Tumor-specific activation
of the C-JUN/MELK pathway regulates glioma stem cell growth in a
p53-dependent manner. Stem Cells. 31:870–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim KH, Young BD and Bender JR:
Endothelial estrogen receptor isoforms and cardiovascular disease.
Mol Cell Endocrinol. 389:65–70. 2014. View Article : Google Scholar : PubMed/NCBI
|