Introduction
Cervical cancer (CC) is a leading cause of
cancer-associated mortality in women globally, with 500,000 new
cases and 250,000 incidences of mortality annually in 2012
(1). Previous studies have reported
that human papillomavirus infection is a high risk factor for CC;
however, it is insufficient to initiate of malignancy alone
(2), and genetic alterations are
essential for the progression from precancerous disorder to
invasive cancer (3). Thus, it is
necessary to understand the pathogenic progresses that drive CC to
further prevent its development by dissecting the components
involved in the pathogenic process (4).
Clinically, early-stage and locally advanced CC may
be treated with standard radiotherapy and chemotherapy, or the two
treatments combined; however, patients with metastatic cancer types
and those with persistent or recurrent disease following
platinum-based chemoradiotherapy have limited options (5,6).
Furthermore, the clinical outcomes vary substantially and are
difficult to predict, owing to the lack of effective outcome
prediction models, which make it difficult to apply individualized
treatment protocols to patients with CC (7). With the development of gene
expression-associated analysis methods, target-gene treatments may
be applied to largely solve this problem and potentially improve
patient survival (8). Therefore, the
identification of target genes to aid the prediction of CC
prognosis is a necessary task.
MicroRNAs (miRNAs/miRs) are a family of small
non-coding RNA molecules (~22 nucleotides in length) that regulate
gene expression by promoting mRNA degradation and repressing
translation (9). miRNAs modulate the
expression of target mRNAs post-transcriptionally by base pairing
to complementary sequences in the 3′- and 5′-untranslated regions,
and occasionally the open-reading frames of mRNAs (10,11).
However, miRNA expression signatures have been revealed to be
promising potential biomarkers for the classification or outcome
prediction of a wide array of human cancer types (12), including lung cancer (13). miRNAs are involved in numerous
cancer-associated processes, including proliferation, metabolism,
differentiation, apoptosis, cellular signaling and cancer
development and progression (14).
Hence, the investigation of miRNA functions allows for the
elucidation of the complex pathological mechanisms underlying
malignant tumor types, and aids the design of drugs for the
treatment of malignant tumors.
However, to date, the prediction of miRNAs targets
in CC has rarely been investigated. Therefore, in the present
study, target miRNAs involved in CC were predicted utilizing an
ensemble method proposed by Le et al (15). The ensemble method integrated a
correlation method [Pearson's correlation coefficient (PCC)], a
causal inference method (IDA), and a regression method [least
absolute shrinkage and selection operator (Lasso)], which formed
the PCC (16), IDA (17,18) and
Lasso (19) (PIL) method, based on
the Borda count election method. The PIL approach may solve the
inconsistencies in results that result from individual methods as
it includes complementary results (20). Although there is not a full
understanding regarding miRNA target prediction, the ensemble
method may aid the identification of a number of confirmed
interactions that existing individual methods fail to discover
(15). Overall, using the PIL method,
more reliable results can be obtained compared with existing
individual methods (15).
To validate the activity of the predicted miRNA
targets in patients with CC, effective methods must be utilized.
Previous studies have proposed the use of a number of different
methods (21,22), including a semi-supervised method
(21). The semi-supervised method was
mainly dependent on a support vector machine model, which used the
experimentally confirmed database miRTarBase (23) as the control set and Tarbase (24) as the test set. Owing to the positive
classification performances, miRTarBase and Tarbase were utilized
in the present study, in addition to the other two commonly used
databases, miRecords (25) and
miRWalk (26), owing to the
sparseness of the number of confirmed interactions, for validation
of the miRNA targets.
Following the identification of miRNA targets using
the PIL method and validating them by matching them with confirmed
databases [miRTarBase (23), TarBase
(24), miRecords (25), and miRWalk (26)], Kyoto Encyclopedia of Genes and
Genomes (KEGG) (27) pathway
enrichment analysis was conducted for target genes in the 1,000
most frequently predicted miRNA-mRNA interactions to determine
target pathways associated with miRNA targets in CC. These targets
may be potential biomarkers for CC treatment, revealing the
pathological mechanism underlying this cancer.
Materials and methods
Collecting expression data
In the present study, miRNA and mRNA expression data
from patients with CC were downloaded from The Cancer Genome Atlas
(TCGA; http://cancergenome.nih.gov/). TCGA
is a comprehensive and coordinated effort to accelerate the current
understanding of the molecular basis of cancer through the
application of genome analysis technologies, including large-scale
genome sequencing (28). Owing to the
differing quantities and identities of miRNAs and mRNAs between
samples, only samples with common intersections were included as
study objects. A total of 309 samples were obtained.
To control the quality of miRNA and mRNA in these
samples, standard pretreatments were performed. In the first step,
miRNAs or mRNAs with an expression value of zero were removed.
Secondly, the expression values were normalized and converted into
log2 forms, identifying 889 miRNAs and 20,104 mRNAs.
Thirdly, PCC, which evaluates the probability of two gene pairs
co-expressing (16), was used to
calculate the strength of the correlation between miRNAs and mRNAs.
Finally, if the absolute value of PCC for an interaction, denoted
as δ, met a threshold of δ≥0.70, the correlations were selected as
effective expression data. This resulted in the identification of
53 miRNAs and 216 mRNAs for further examination.
Predicting miRNA targets using the PIL
method
The PIL method is an ensemble method that integrates
three methods (PCC, IDA and Lasso) based on the Borda count
election method. This method mainly comprised three steps: Firstly,
for each miRNA, each of the individual methods (PCC, IDA and Lasso)
were utilized to produce rankings for each miRNA or to determine
the predicted targets of the miRNA, and the 1,000 most frequently
predicted performers in identifying miRNA targets were selected.
The second step was the application of the Borda rank election
method to the rankings for each miRNA to produce a single ranking
list of elected mRNAs with respect to the miRNA. Finally, the
highest-ranked genes from the list were extracted as the final
output, as the potential target genes for the given miRNA.
The Borda rank election method is an efficient
method to combine orderly appraising results from several separate
evaluating methods (29). Its
specific process is as follows: With an election consisting of a
set (V) of voters, each identified candidate is assigned a
preferential order, a strict, complete and transitive order on a
set of candidates (C). Subsequently, each of the candidates is
given ||C||-n points for each voter which ranked them in nth place
(for example, ||C||-1 points for first, ||C||-2 for second, and so
forth until the candidate voter ranked last receives no points).
Finally, the average point score of the candidate across all voters
was computed, and defined as the z-score. The higher the z-score,
the more significantly associated with CC the prediction results
were. Ranking the predicted miRNA targets according to their
z-scores, the 1,000 most frequently predicted ranked target genes
for CC were determined.
Validating predicted miRNA
targets
Validating computation results is difficult, as the
number of experimentally confirmed targets of miRNAs is limited and
there is no complete ground-truth for evaluating and comparing
different computational methods (30). In the present study, four databases,
miRTarBase v4.5 (23), TarBase v6.0
(24), miRecords v2013 (25) and miRWalk v2.0 (26), were combined to validate the
prediction of miRNA targets obtained from the PIL method.
miRTarbase provides the most up-to-date, comprehensive information
regarding experimentally validated miRNA-mRNA target interactions
(31). TarBase is the first resource
to provide experimentally verified miRNA target interactions by
surveying pertinent literature (32).
miRecords accumulates experimentally validated miRNA targets and
computationally predicted miRNA targets (25). miRWalk is a publicly available
comprehensive resource, hosting predicted and experimentally
validated miRNA target interaction pairs (26). There were 37,372 miRNA-mRNA
interactions with 576 miRNAs, 20,095 miRNA-mRNA interactions with
228 miRNAs, 21,590 miRNA-mRNA interactions with 195 miRNAs, and
1,710 miRNA-mRNA interactions with 226 miRNAs in the miRTarBase,
TarBase, miRecords and miRWalk databases, respectively. Following
the removal of the duplicated miRNA-mRNA interactions, a total of
62,858 interactions were retained for validation, termed background
interactions. If a miRNA target interaction was involved in
background interactions, the predicted miRNA target was
validated.
Pathway enrichment analysis
For the purpose of investigating functional
biological processes associated with target genes enriched in the
1,000 most frequently predicted miRNA-mRNA interactions for CC, the
Database for Annotation, Visualization, and Integrated Discovery
(DAVID) for KEGG pathway enrichment analysis were performed
(27). KEGG pathways with P<0.05
were selected based on an Expression Analysis Systematic Explored
(EASE) test applied in DAVID. EASE analysis of the regulated genes
indicated the molecular functions and biological processes unique
to each category (33). In functional
and pathway enrichment analysis, the threshold of the minimum
number of genes corresponded to ≥2, which was considered to be
significant for a category:
P=(a+ba)(c+dc)(na+c)
Where n = a′ + b + c + d was the number of
background genes; a′ was the gene number of one gene set in the
gene lists; a′ + b was the number of genes in the gene list
including at least one gene set; a′ + c was the gene number of one
gene list in the background genes; and a′ was replaced with a = a
′- 1 in EASE.
Results
Predicted miRNA targets
In the present study, based on the CC expression
data from TCGA database, a total of 53 miRNAs and 216 mRNAs were
obtained for further analysis subsequent to pretreatments. By
merging three methods on the basis of the Borda count election
algorithm, the PIL method was formed and was utilized to predict
miRNA targets. The miRNA targets were identified based on
miRNA-mRNA interactions, and the mRNAs or target genes were
additionally identified. During this process, each miRNA-mRNA
interaction was assigned a z-score, and all interactions were
ranked in a descending order of z-scores. The higher the z-score,
the more significantly associated with CC the prediction results.
Owing to the large number of miRNA targets, the 1,000 most
frequently predicted ranked interactions were selected, as they may
be more strongly associated with CC than other interactions.
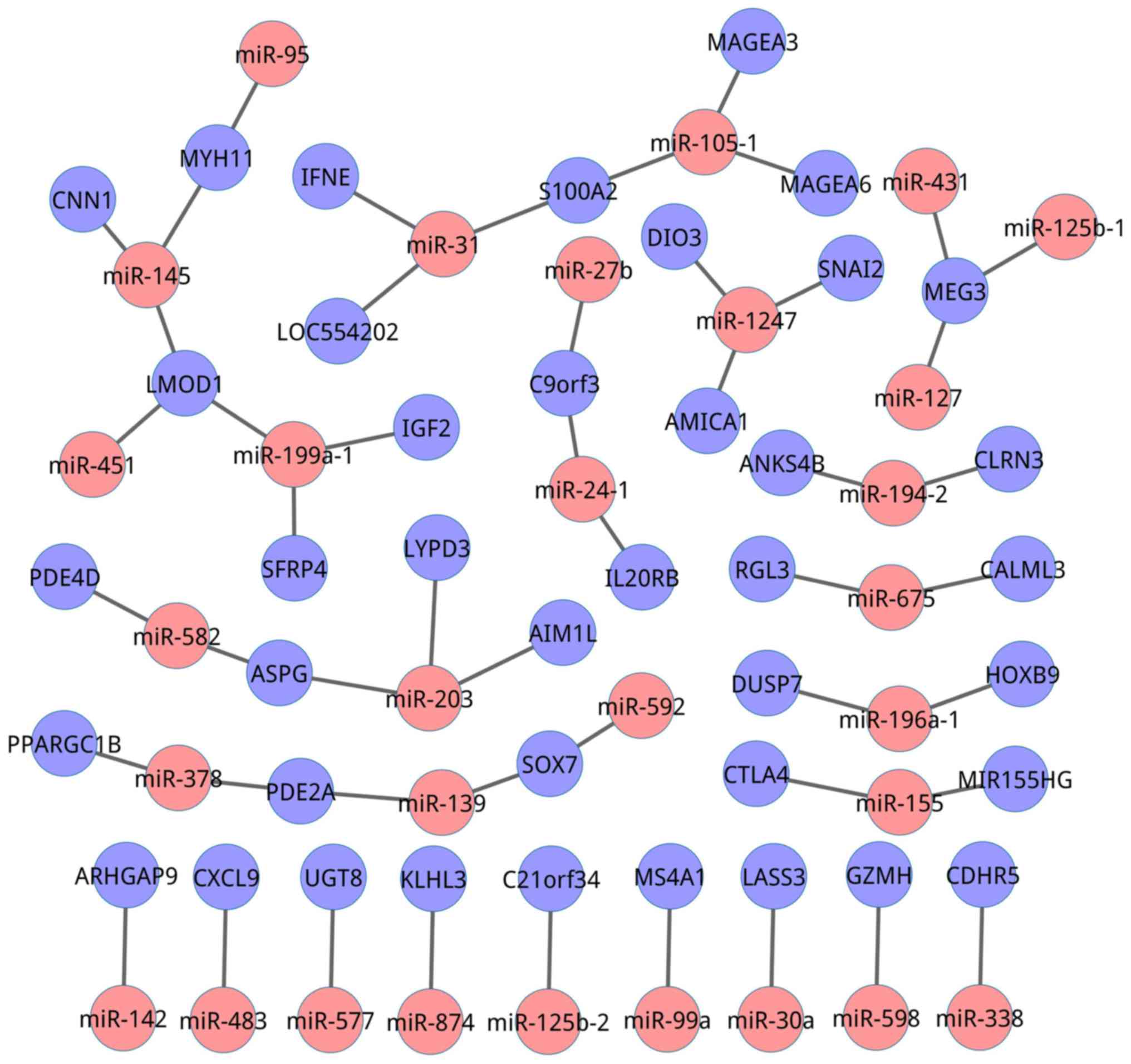
Table I displays the
50 most frequently predicted mRNA-miRNA interactions, and Fig. 1 represents the network of these
interactions. It was revealed that secreted frizzled-related
protein 4 (SFRP4; z-score=219.0) was the most frequently
predicted mRNA, and its corresponding miRNA was miR-199a-1. The
next four most frequent interactions were chromosome 9 open reading
frame 3 (z-score=218.5) with miR-24-1, iodothyronine deiodinase 3
(z-score=218.2) with miR-1247, maternally expressed 3 (MEG3;
z-score=217.8) with miR-431 and kelch-like family member 3
(z-score=216.9) with miR-874. Notably, among the 50 most frequently
predicted interactions, MEG3 was regulated by three miRNAs
(miR-431, miR-125b-1 and miR-127) simultaneously. miR-199a-1 may
regulate two mRNAs (SFRP4 and insulin like growth factor 2)
at the same time. Hence, a miRNA may regulate a plurality of genes,
and multiple miRNAs may alter the expression of a single gene.
 | Table I.All 50 most frequently predicted
mRNA-miRNA interactions. |
Table I.
All 50 most frequently predicted
mRNA-miRNA interactions.
| No. | mRNA | miRNA | z-score |
|---|
| 1 | SFRP4 | miR-199a-1 | 219.0 |
| 2 | C9orf3 | miR-24-1 | 218.5 |
| 3 | DIO3 | miR-1247 | 218.2 |
| 4 | MEG3 | miR-431 | 217.8 |
| 5 | KLHL3 | miR-874 | 216.9 |
| 6 |
LOC554202 | miR-31 | 216.8 |
| 7 | MAGEA6 | miR-105-1 | 216.8 |
| 8 |
MIR155HG | miR-155 | 216.5 |
| 9 | MYH11 | miR-145 | 216.2 |
| 10 | PDE2A | miR-139 | 216.1 |
| 11 | PDE4D | miR-582 | 216.1 |
| 12 |
PPARGC1B | miR-378 | 216.1 |
| 13 |
C21orf34 | miR-125b-2 | 216.0 |
| 14 | UGT8 | miR-577 | 216.0 |
| 15 | CALML3 | miR-675 | 130.0 |
| 16 | HOXB9 | miR-196a-1 | 129.6 |
| 17 | CNN1 | miR-145 | 128.7 |
| 18 | CXCL9 | miR-483 | 123.1 |
| 19 | IFNE | miR-31 | 122.2 |
| 20 | MAGEA3 | miR-105-1 | 118.4 |
| 21 | SOX7 | miR-139 | 108.9 |
| 22 | CLRN3 | miR-194-2 | 102.6 |
| 23 | LMOD1 | miR-451 | 97.5 |
| 24 | C9orf3 | miR-27b | 85.6 |
| 25 | LMOD1 | miR-145 | 81.8 |
| 26 | MEG3 | miR-125b-1 | 77.7 |
| 27 | S100A2 | miR-31 | 72.3 |
| 28 | RGL3 | miR-675 | 64.8 |
| 29 | AIM1L | miR-203 | 62.9 |
| 30 | ARHGAP9 | miR-142 | 60.9 |
| 31 | ASPG | miR-582 | 59.7 |
| 32 | CDHR5 | miR-338 | 59.1 |
| 33 | IL20RB | miR-24-1 | 58.9 |
| 34 | SNAI2 | miR-1247 | 57.9 |
| 35 | LMOD1 | miR-199a-1 | 55.3 |
| 36 | S100A2 | miR-105-1 | 54.0 |
| 37 | ANKS4B | miR-194-2 | 52.6 |
| 38 | ASPG | miR-203 | 51.9 |
| 39 | GZMH | miR-598 | 49.4 |
| 40 | LYPD3 | miR-203 | 49.1 |
| 41 | MEG3 | miR-127 | 48.3 |
| 42 | MYH11 | miR-95 | 47.8 |
| 43 | AMICA1 | miR-1247 | 47.3 |
| 44 | CTLA4 | miR-155 | 46. 9 |
| 45 | DUSP7 | miR-196a-1 | 46.7 |
| 46 | PDE2A | miR-378 | 46.3 |
| 47 | SOX7 | miR-592 | 45.3 |
| 48 | IGF2 | miR-199a-1 | 44.2 |
| 49 | LASS3 | miR-30a | 44.2 |
| 50 | MS4A1 | miR-99a | 43.2 |
For the specific 1,000 miRNA targets, genes which
were regulated by a greater number of miRNAs or which were
predicted more frequently, may have a stronger association with CC
compared with those only predicted once. This may offer another way
to evaluate the importance of one gene in certain tumor. Therefore,
the predicted frequency for mRNAs were computed, and the targets
which were predicted >5 times amongst the 1,000 miRNA-mRNA
interactions were listed (Table II).
SFRP4 and MEG3 were predicted 8 times. A total of 7
miRNAs co-regulated NIPA like domain containing 4, whilst guanylate
binding protein family member 6, interferon gamma inducible protein
6, family with sequence similarity 83 member C, serpin family B
member 5, adhesion G protein-coupled receptor F4, calponin 1 and
leiomodin 1 were all regulated by 6 miRNAs simultaneously.
| Table II.mRNA targets predicted >5
times. |
Table II.
mRNA targets predicted >5
times.
| Target | Predictions, n | miRNAs |
|---|
| SFRP4 | 8 | miR-105-2,
miR-125b-2, miR-127, miR-199a-1, miR-335, miR-451, miR-95,
miR-99a |
| MEG3 | 8 | miR-1247,
miR-125b-1, miR-127, miR-199a-1, miR-431, miR-483, miR-493,
miR-874 |
| NIPAL4 | 7 | miR-1287, miR-144,
miR-27b, miR-451, miR-592, miR-675, miR-95 |
| GBP6 | 6 | let-7c, miR-105-2,
miR-125b-2, miR-30a, miR-582, miR-592 |
| IFI16 | 6 | let-7c, miR-105-2,
miR-196a-2, miR-24-1, miR-452, miR-95 |
| FAM83C | 6 | let-7c, miR-127,
miR-30a, miR-378, miR-452, miR-582 |
|
SERPINB5 | 6 | miR-105-2, miR-152,
miR-23b, miR-449a, miR-584, miR-708 |
| GPR115 | 6 | miR-1287, miR-452,
miR-584, miR-592, miR-874, miR-944 |
| CNN1 | 6 | miR-139, miR-144,
miR-145, miR-23b, miR-431, miR-95 |
| LMOD1 | 6 | miR-144, miR-145,
miR-199a-1, miR-31, miR-451, miR-95 |
Validation of miRNA targets
To validate the prediction of miRNA targets
identified by the PIL method, the miRTarBase, TarBase, miRecords
and miRWalk databases were used. By removing duplicated
interactions, 62,858 interactions were obtained, which were denoted
as background interactions. Selecting intersections between
background interactions and all predicted miRNA-mRNA interactions,
105 intersected interactions were detected. The results indicated
the feasibility and stability of the PIL method.
Enriched pathways for target
genes
KEGG pathway enrichment analysis was conducted for
genes identified within the 1,000 miRNA-mRNA interactions (Table III). With the threshold set at
P<0.05 and target genes count ≥2, a total of 17 pathways were
identified, which were termed ‘target pathways’. In addition, 5 out
of the 17 target pathways were signaling pathways. The five target
pathways with the highest significance were cytokine-cytokine
receptor interactions (P=8.91×10−7), the chemokine
signaling pathway (P=1.55×10−5), cell adhesion molecules
(CAMs) (P=1.37×10−4), the T-cell receptor signaling
pathway (P=1.58×10−4) and primary immunodeficiency
(P=1.75×10−4).
| Table III.Kyoto Encyclopedia of Genes and
Genomes pathways enriched in cervical cancer. |
Table III.
Kyoto Encyclopedia of Genes and
Genomes pathways enriched in cervical cancer.
| Rank | Pathway | P-value |
|---|
| 1 | Cytokine-cytokine
receptor interaction |
8.91×10−7 |
| 2 | Chemokine signaling
pathway |
1.55×10−5 |
| 3 | Cell adhesion
molecules |
1.37×10−4 |
| 4 | T-cell receptor
signaling pathway |
1.58×10−4 |
| 5 | Primary
immunodeficiency |
1.75×10−4 |
| 6 | Chagas disease
(American trypanosomiasis) |
6.09×10−4 |
| 7 | Hematopoietic cell
lineage |
9.24×10−3 |
| 8 | Rheumatoid
arthritis |
9.34×10−3 |
| 9 | Natural killer cell
mediated cytotoxicity | 1.02×10-2 |
| 10 | Toll-like receptor
signaling pathway | 1.21×10-2 |
| 11 | Prion diseases |
1.25×10−2 |
| 12 | Adherens
junction |
1.54×10−2 |
| 13 | Type I diabetes
mellitus |
1.87×10−2 |
| 14 | Malaria |
2.73×10−2 |
| 15 | Staphylococcus
aureus infection |
3.11×10−2 |
| 16 | Maturity onset
diabetes of the young |
3.75×10−2 |
| 17 | Tumor protein p53
signaling pathway |
4.70×10−2 |
Discussion
A number of differing computational methods have
been proposed to identify miRNA targets from expression data,
including PCC (16), IDA (17,18) and
Lasso (19). PCC is commonly used to
measure the strength of associations between a pair of variables
(16). PCC is used to rank data in
descending order of absolute PCC values, and may result in negative
miRNA-mRNA correlations being highly ranked, as miRNAs mainly
downregulate mRNAs (15).
Additionally, the practicability of PCC would be substantially
reduced if the correlations were non-linear (34). IDA, a causal inference method,
evaluates the causal effect between two variables (17,18). Le
et al found that miRNA-mRNA causal regulatory associations
revealed by IDA overlapped substantially with the results of
follow-up gene-knockdown experiments (35). Lasso, a regression method, minimizes
the usual sum of squared errors, with a limit on the sum of the
absolute values of the coefficients (19). Similar to the PCC method, the
miRNA-mRNA pairs with limitations are ranked highly to favor
downregulation. Therefore, the Borda count election method was used
to integrate the aforementioned three methods together, giving the
PIL method, and validated by identifying intersections between
predicted miRNA-mRNA interactions and background interactions.
Using the PIL method, predicted miRNA targets for
patients with CC were ranked according to their z-scores and the
1,000 highest ranked interactions were obtained. For specific
interactions, target genes that were predicted multiple times were
identified. Notably, SFRP4 was the most frequently predicted
gene during the prediction process. SFRP4 is a member of the
SFRP family that contains a cysteine-rich domain homologous
to the putative Wnt-binding site of Frizzled proteins (36); it serves notable functions in tumor
progress through antagonizing Wnt signaling (37). Hypermethylation of the SFRP4
promoter was associated with CC and may have utility for the
molecular screening of cervical neoplasia (38). Brebi et al (39) revealed that SFRP4 may be used
as a potential biomarker for CC diagnosis. Therefore, SFRP4
was identified to be closely associated with CC.
For the purpose of investigating functional gene
sets involved with miRNA-mRNA targets, pathway enrichment analysis
was conducted. A total of 17 target pathways were identified for
genes in the 1,000 miRNA-mRNA interactions most notably associated
with CC based on KEGG pathway enrichment analysis, of which
cytokine-cytokine receptor interactions (P=8.91×10−7)
was the most significantly associated with CC. Cytokines are
soluble extracellular proteins, usually secreted in response to an
activating stimulus, which induce responses through binding to
specific receptors on the surface of target cells (40). Cytokines can be grouped by structure
into different families, as can and their receptors. It had been
reported that the cytokine-cytokine receptor interaction gene set
may induce cancer (41) and was
upregulated in cancer cachexia (42);
these changes should result in the development of markers for early
diagnosis and a better understanding of the conditions of a tumor.
Mak et al (43) revealed that
the marked upregulation of genes involved in cytokine-cytokine
receptor interactions were consistently detected in tumor cell
lines. Signaling pathway impact analysis implicated that this
pathway was commonly altered in triple-negative breast cancer
(44). Therefore, it can be inferred
that cytokine-cytokine receptor interactions additionally serve
notable functions in the progression of CC, and, to the best of our
knowledge, the present study is the first to reveal the correlation
between cytokine-cytokine receptor interactions and CC.
In conclusion, the present study predicted target
genes and pathways for patients with CC based on miRNA expression
data, the PIL method and pathway analysis. The results of the
present study may provide insights into the pathological mechanism
underlying CC, and provide potential biomarkers for the diagnosis
and treatment of this tumor type. However, these biomarkers are yet
to be validated, and the relevant validations should be performed
in future studies.
References
|
1
|
Creasman WT and Miller DS: Adenocarcinoma
of the uterine corpusClinical Gynecologic Oncology. Elsevier;
Philadelphia, PA: pp. 141–174. 2012, View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tjalma WA, Van Waes TR, Van den Eeden LE
and Bogers JJ: Role of human papillomavirus in the carcinogenesis
of squamous cell carcinoma and adenocarcinoma of the cervix. Best
Pract Res Clin Obstet Gynaecol. 19:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin CM, Astbury K, McEvoy L, O'Toole S,
Sheils O and O'Leary JJ: Gene expression profiling in cervical
cancer: Identification of novel markers for disease diagnosis and
therapy. Methods Mol Biol. 511:333–359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang YX and Zhao YL: Pathogenic Network
analysis predicts candidate genes for cervical cancer. Comput Math
Methods Med. 2016:31860512016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tewari KS: PRO: Patients with
metastatic/recurrent cervical cancer should be treated with
cisplatin plus paclitaxel. Clinical Ovarian Cancer. 4:90–93. 2011.
View Article : Google Scholar
|
|
6
|
Moore DH, Tian C, Monk BJ, Long HJ, Omura
GA and Bloss JD: Prognostic factors for response to cisplatin-based
chemotherapy in advanced cervical carcinoma: A Gynecologic Oncology
Group Study. Gynecol Oncol. 116:44–49. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu X, Schwarz JK, Lewis JS Jr, Huettner
PC, Rader JS, Deasy JO, Grigsby PW and Wang X: A microRNA
expression signature for cervical cancer prognosis. Cancer Res.
70:1441–1448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Itahana Y, Han R, Barbier S, Lei Z, Rozen
S and Itahana K: The uric acid transporter SLC2A9 is a direct
target gene of the tumor suppressor p53 contributing to antioxidant
defense. Oncogene. 34:1799–1810. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berezikov E, Cuppen E and Plasterk RH:
Approaches to microRNA discovery. Nat Genet. 38 Suppl:S2–S7. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jay C, Nemunaitis J, Chen P, Fulgham P and
Tong AW: miRNA profiling for diagnosis and prognosis of human
cancer. DNA Cell Biol. 26:293–300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu SL, Chen HY, Chang GC, Chen CY, Chen
HW, Singh S, Cheng CL, Yu CJ, Lee YC, Chen HS, et al: MicroRNA
signature predicts survival and relapse in lung cancer. Cancer
Cell. 13:48–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le TD, Zhang J, Liu L and Li J: Ensemble
methods for miRNA target prediction from expression data. PLoS One.
10:e01316272015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nahler G: Pearson correlation coefficient.
Dictionary of Pharmaceutical Med. 132:2009.
|
|
17
|
Maathuis MH, Kalisch M and Bühlmann P:
Estimating high-dimensional intervention effects from observational
data. Annals Statist. 37:3133–3164. 2009. View Article : Google Scholar
|
|
18
|
Maathuis MH, Colombo D, Kalisch M and
Bühlmann P: Predicting causal effects in large-scale systems from
observational data. Nat Methods. 7:247–248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Friedman J, Hastie T and Tibshirani R:
Glmnet: Lasso and Elastic-Net Regularized Generalized Linear
Models. R package version 1.9–5R Foundation for Statistical
Computing. Vienna, Austria: 2013
|
|
20
|
Marbach D, Costello JC, Küffner R, Vega
NM, Prill RJ, Camacho DM and Allison KR: DREAM5 Consortium, Kellis
M, Collins JJ and Stolovitzky G: Wisdom of crowds for robust gene
network inference. Nat Methods. 9:796–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pio G, Malerba D, D'Elia D and Ceci M:
Integrating microRNA target predictions for the discovery of gene
regulatory networks: A semi-supervised ensemble learning approach.
BMC Bioinformatics. 15 Suppl 1:S42014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y and Verbeek FJ: Comparison and
integration of target prediction algorithms for microRNA studies. J
Integr Bioinform. 7:2010.doi: 10.2390/biecoll-jib-2010-127.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin
YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al: miRTarBase
2016: Updates to the experimentally validated miRNA-target
interactions database. Nucleic Acids Res. 44:D239–D247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vergoulis T, Vlachos IS, Alexiou P,
Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N,
Dalamagas T and Hatzigeorgiou AG: TarBase 6.0: Capturing the
exponential growth of miRNA targets with experimental support.
Nucleic Acids Res. 40:(Database issue). D222–D229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:(Database issue). D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dweep H, Gretz N and Sticht C: miRWalk
database for miRNA-target interactions. Methods Mol Biol.
1182:289–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu Y, Qiu P and Ji Y: TCGA-assembler:
Open-source software for retrieving and processing TCGA data. Nat
Methods. 11:599–600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Russell N: Complexity of control of Borda
count elections. Rochester Institute of Technology. 2007.http://scholarworks.rit.edu/theses/332
|
|
30
|
Le TD, Liu L, Zhang J, Liu B and Li J:
From miRNA regulation to miRNA-TF co-regulation: Computational
approaches and challenges. Brief Bioinform. 16:475–496. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hsu SD, Tseng YT, Shrestha S, Lin YL,
Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, et al:
miRTarBase update 2014: An information resource for experimentally
validated miRNA-target interactions. Nucleic Acids Res.
42:(Database issue). D78–D85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: A functional update of TarBase. Nucleic Acids
Res. 37:(Database issue). D155–D158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ford G, Xu Z, Gates A, Jiang J and Ford
BD: Expression analysis systematic explorer (EASE) analysis reveals
differential gene expression in permanent and transient focal
stroke rat models. Brain Res. 1071:226–236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Speed T: Mathematics. A correlation for
the 21st century. Science. 334:1502–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Le TD, Liu L, Tsykin A, Goodall GJ, Liu B,
Sun BY and Li J: Inferring microRNA-mRNA causal regulatory
relationships from expression data. Bioinformatics. 29:765–771.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Drake JM, Friis RR and Dharmarajan AM: The
role of sFRP4, a secreted frizzled-related protein, in ovulation.
Apoptosis. 8:389–397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang D, Yu B, Deng Y, Sheng W, Peng Z,
Qin W and Du X: SFRP4 was overexpressed in colorectal carcinoma. J
Cancer Res Clin Oncol. 136:395–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chung MT, Sytwu HK, Yan MD, Shih YL, Chang
CC, Yu MH, Chu TY, Lai HC and Lin YW: Promoter methylation of SFRPs
gene family in cervical cancer. Gynecol Oncol. 112:301–306. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brebi P, Hoffstetter R, Andana A, Ili CG,
Saavedra K, Viscarra T, Retamal J, Sanchez R and Roa JC: Evaluation
of ZAR1 and SFRP4 methylation status as potentials biomarkers for
diagnosis in cervical cancer: Exploratory study phase I.
Biomarkers. 19:181–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Turrin NP and Plata-Salamán CR:
Cytokine-cytokine interactions and the brain. Brain Res Bull.
51:3–9. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
El Hassan Abou M, Huang K, Eswara MB, Zhao
M, Song L, Yu T, Liu Y, Liu JC, McCurdy S, Ma A, et al: Cancer
cells Hijack PRC2 to modify multiple cytokine pathways. PLoS One.
10:e01264662015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iman Al-Azwani Bs, Al Haddad A, Mohamoud
Y, Farouk S, Haytham S, Joel M and Thomas A: Rna-seq study of
muscle and adipose tissue in cancer patients with early cachexia.
Qatar Foundation Annual Research Conference, HBPP0326.
2014.https://doi.org/10.5339/qfarc.2014.HBPP0326
|
|
43
|
Mak CK, Chung GTY, Yip KYL, Ken KYT,
Sau-Dan L, Siu-Tim C, Sai-Wah T, Pierre B, Ka-Fai T and Kwok-Wai L:
Abstract 3425: Whole-transcriptome analyses of EBV-associated
nasopharyngeal carcinoma using next-generation transcriptome
sequencing. Cancer Res. 74:3425. 2014. View Article : Google Scholar
|
|
44
|
Thompson KJ, Tang X, Sun Z, Sinnwell JP,
Sicotte H, Mahoney DW, Hart S, Vedell PT, Barman B, Passow JEE, et
al: Molecular classification of triple negative breast cancer via
RNA-sequencing data (Abstract). Cancer Res. 74:55922014. View Article : Google Scholar
|