Introduction
As the majority of elderly patients with lung cancer
have a poor prognosis after treatment with either routine or
large-dose chemotherapy (1), it is
important to elucidate novel targeted therapies and immunotherapies
to improve quality of life and prolong survival (2). Previous studies have investigated the
signaling pathways that drive lung cancer progression and a number
of targeted therapeutic strategies have been proposed (3,4). However,
long-term effectiveness of lung cancer treatment is rare and
recurrence is promoted by various mechanisms, including
compensatory activation of cell survival signaling pathways
(5,6).
The phosphoinositide 3-kinase (PI3K) signaling
pathway serves an important role in the regulation of cell
proliferation, growth, survival and metabolism (7). Aberrant activation of the PI3K/AKT
pathway has been observed in several types of cancer, including
non-small cell lung cancer (NSCLC) (8,9).
Hyperactivation of the PI3K/AKT pathway may be involved in the
resistance to chemotherapeutic and targeted reagents in different
cancer types through anti-apoptotic functions (10), making it a therapeutic target of
cancer (11). Several small molecule
inhibitors of the PI3K/AKT signaling pathway have undergone
clinical trials in humans (12,13).
Despite the great potential of such targeted therapies in lung
cancer, a subset of patients exhibiting hyperactivation of PI3K/AKT
signaling do not respond to these inhibitor drugs (13,14).
In the present study, it was demonstrated that
inhibitors of the PI3K/AKT pathway activated the signal transducer
and activator of transcription 3 (STAT3) signaling pathway.
Previous studies have demonstrated that persistent activation of
the STAT3 signaling pathway is associated with various types of
solid cancer (15,16). While STAT3 hyperactivation has been
demonstrated to mediate drug resistance (17), inhibition of the STAT3 signaling
pathway has been demonstrated to reverse drug resistance (18). The aim of the present study was to
identify the molecular determinants driving the compensatory
activation of STAT3 after treatment with PI3K/AKT inhibitors, which
may limit the clinical efficacy of these compounds. The results
suggest novel cross-talk between the PI3K/AKT and STAT3 signaling
pathways, modulated by the MET proto-oncogene (MET). Targeting
MET/STAT3 signaling potentiates the antitumor activity of PI3K/AKT
inhibitors, and may be an effective therapeutic strategy for
NSCLC.
Materials and methods
Cell lines, reagents and
transfection
The NSCLC cell lines, H460 and H2126, were purchased
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The 2 cell lines were cultured in Dulbecco's modified Eagle's
medium (DMEM) with 10% heat-inactivated fetal bovine serum, 100
U/ml penicillin and 100 mg/ml streptomycin (all from HyClone; GE
Healthcare, Chicago, IL, USA) at 37°C with 5% CO2.
BKM120 (a selective PI3K inhibitor; S2247), LY294002 (a pan-PI3K
inhibitor; S1105), MK-2206 (an AKT allosteric inhibitor; S1078),
BEZ235 [a dual PI3K/mechanistic target of rapamycin (mTOR)
catalytic inhibitor; S1009], PF-2341066 (a MET inhibitor; S1068)
and stattic (a STAT3 inhibitor; S7024) were obtained from Selleck
Chemicals (Houston, TX, USA), and dissolved in DMSO. Specific small
interfering RNAs (siRNAs) for p110α (si-p110α; 100 nM; cat. no.
6359S), AKT (si-AKT; 100 nM; cat. no. 6211S) and MET (si-MET; 100
nM; cat. no. 6618S) were obtained from Cell Signaling Technology
Inc. (Danvers, MA, USA). Transfection was performed using
Lipofectamine 2000® in Opti-MEM (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Cells were transfected
with an individual siRNA or treated with an inhibitor for 24 h
prior to subsequent experimentation.
Western blotting
Cell lysates of H460 and H2126 cells were prepared
using radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and protease inhibitors. Total protein
concentration was determined by Pierce BCA Protein Assay Kit
(Thermo Fisher Scientific, Inc.). 12% Tricine-SDS-PAGE was used to
separate 10 µg protein. Subsequent to transfer to a nitrocellulose
membrane, the following primary antibodies were used:
Phosphorylated-STAT3 (p-STAT3, Y705; cat. no. 9145; dilution
1:1,000), p-extracellular regulation kinase 1/2 (p-ERK1/2,
T202/Y204; 4376; dilution 1:1,000), p-AKT (S473; cat. no. 4060;
dilution 1:1,000), p-AKT (T308; cat. no. 13038; dilution 1:1,000),
p-S6 (S240/244; cat. no. 5364; dilution 1:1,000), p-MET (Y1349;
cat. no. 3133; dilution 1:1,000), STAT3 (cat. no. 12604; dilution
1:1,000), ERK1/2 (cat. no. 4695; dilution 1:1,000), AKT (catalog
no., 4685; dilution, 1:1,000), S6 (cat. no. 2217; dilution 1:1,000)
and MET (cat. no. 8198; dilution 1:1,000) were obtained from Cell
Signaling Technology (Beverly, MA, USA), and incubation at 4°C with
gentle shaking overnight. A 10% (w/v) solution of bovine serum
albumin (Sigma-Aldrich; Merck KGaA) was used as blocking buffer (at
4°C with gentle shaking for 1 h). Blots were washed thrice in 1X
TBS with 0.1% Tween-20. HRP-linked anti-rabbit IgG secondary
antibody (Cell Signaling Technology; cat. no. 7074; dilution
1:2,000) incubated with the membranes at 4°C with gentle shaking
for 1 h. Anti-GAPDH was purchased from Beijing CoWin Biotech Co.,
Ltd. (Beijing, China) and used as a loading control. Blots were
washed thrice and exposed to the ECL Reagent substrate (Thermo
Fisher Scientific, Inc.). ImageJ (National Institutes of Health,
Bethesda, MD, USA) was used to compare the intensity of bands.
Immunofluorescence (IF)
The adherent H460 cells were cultured on glass
coverslips and treated with 1 µM BKM120 for 12 h. Cells were fixed
with 2% buffered paraformaldehyde for 30 min on ice. The cells were
stained using p-MET (cat. no. 3077; dilution 1:50) and MET
antibodies (cat. no. 8198; dilution 1:50) for 1 h at 4°C.
Anti-rabbit IgG F (ab')2 Fragment (Alexa Fluor® 594
Conjugate; cat. no. 8889; Cell Signaling Technology) was incubated
with the cells for 30 min at 4°C. Fluorescence microscopy was used
to examine the expression of p-MET and MET.
STAT3 DNA-binding activity assay
H460 and H2126 cells were treated with 1 µM BKM120
for 12 h, and the Pierce LightShift Chemiluminescent EMSA kit
(Thermo Fisher Scientific, Inc.) was used to confirm that BKM120
increased STAT3 DNA-binding activity, as described previously
(19).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from H460 and H2126 cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
after 12 h of treatment with 0.1% dimethyl sulfoxide (DMSO), or 0.1
or 1 µM BKM120. The Maxima First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.) was used to obtain cDNA, which was used in
reaction with GoTaq qPCR Master Mix with SYBR-Green (Promega
Corporation, Madison, WI, USA) and primers specific for matrix
metallopeptidase 9 (MMP9), B-cell lymphoma 2 (bcl-2), survivin,
cyclin-D1, hepatocyte growth factor (HGF) and C-reactive protein
(CRP). PCR included a 12 min denaturation step at 94°C, followed by
38 cycles of 94°C for 10 sec, 60°C for 20 sec and 71°C for 10 sec.
GAPDH was used as endogenous control. Primers for these genes were
sourced from PrimerBank (http://pga.mgh.harvard.edu/primerbank/), and the
sequences are as follows: MMP9 forward, 5′-TGTGGGCATCAATGGATTTGG-3′
and reverse, 5′-ACACCATGTATTCCGGGTCAAT-3′; Bcl-2 forward,
5′-GGTGGGGTCATGTGTGTGG-3′ and reverse,
5′-CGGTTCAGGTACTCAGTCATCC-3′; surviving forward,
5′-AGGACCACCGCATCTCTACAT-3′ and reverse,
5′-AAGTCTGGCTCGTTCTCAGTG-3′; cyclin-D1 forward,
5′-GCTGCGAAGTGGAAACCATC-3′ and reverse,
5′-CCTCCTTCTGCACACATTTGAA-3′; HGF forward,
5′-GCTATCGGGGTAAAGACCTACA-3′ and reverse,
5′-CGTAGCGTACCTCTGGATTGC-3′; CRP forward,
5′-AACGAAGCCTCTCAAAGCCTT-3′ and reverse,
5′-CTCTTGGTGGCATACGAGAAAAT-3′. The 2-∆∆Cq method was
applied to present final results (20).
Receptor tyrosine kinase (RTK) array
analysis
After 24 h of cell starvation in serum-free medium,
the H460 cells were incubated for 12 h at 37°C in the absence
(DMSO) or presence of BKM120 (1 µM). The mixture of proteins was
prepared, and the phosphorylation status of each protein was
examined by RTK array analysis. Array 001 (R&D Systems, Inc.,
Minneapolis, MN, USA) was used to detect tyrosine-phosphorylated
RTKs, according to the manufacturer's instructions.
Cell apoptosis assay
H460 and H2126 cells were seeded at 2×105
cells/well in 24-well plates and left overnight prior to treatment
with DMSO [negative control (NC)], BKM120 (1 µM), PF-2341066 (1 µM)
or stattic (5 µM) for 60 h. Cells were harvested by centrifugation
(250 × g, 5 min, 4°C), washed once with PBS and resuspended in 1X
fluorescence-activated cell sorting buffer (BD Biosciences,
Franklin Lakes, NJ, USA) at 1×106 cells/ml. The
frequency of apoptosis was detected using the Alexa
Fluor® 488 Annexin V/Dead Cell Apoptosis kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Clonogenic assay
A total of 300 cells were seeded in 6-well plates
overnight and treated with DMSO (NC), BKM120 (1 µM), PF-2341066 (1
µM) or stattic (5 µM) and cultured for a further 14 days. The cell
clones were fixed and stained with 0.05% crystal violet at room
temperature for 15 min, and the number of clones was counted under
an optical microscope.
In vivo studies
5-week-old female BALB/c mice (Weitonglihua
Biotechnology, Beijing, China) weighing 17–20 g were maintained in
the pathogen-free animal facility under controlled conditions
(12:12 h light and dark cycle, 50% humidity and 22°C). These
animals were provided rodent chow and water. A total of
2×106 H460 cells were inoculated subcutaneously into the
flank of the mice. The mice were randomly divided into 6 groups
(n=10) when tumors reached a size of ~100 mm3. The mice
were treated with control [0.5% (w/v) aqueous
hydroxypropylmethylcellulose solution by oral gavage], BKM120 [15
mg/kg, in 0.5% (w/v) aqueous hydroxypropylmethylcellulose solution
by oral gavage], PF-2341066 [25 mg/kg, in 0.5% (w/v) aqueous
hydroxypropylmethylcellulose solution by oral gavage] or stattic [3
mg/kg, by intraperitoneal injection]. Each drug was administered
daily. The tumors were measured using calipers and volume was
calculated using the following formula: Length ×
width2/2. The procedures for care and use of animals
were approved by the Ethics Committee of the Nanjing Jiangbei
People's Hospital (approval no. IACUC/201606B03; Nanjing,
China).
Statistical analysis
Data are presented as the mean ± standard error
(SE). Multiple group comparisons of the means were performed by
one-way analysis of variance and post-hoc Student-Newman-Keuls
test. *P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibition of PI3K/AKT signaling
causes STAT3 activation
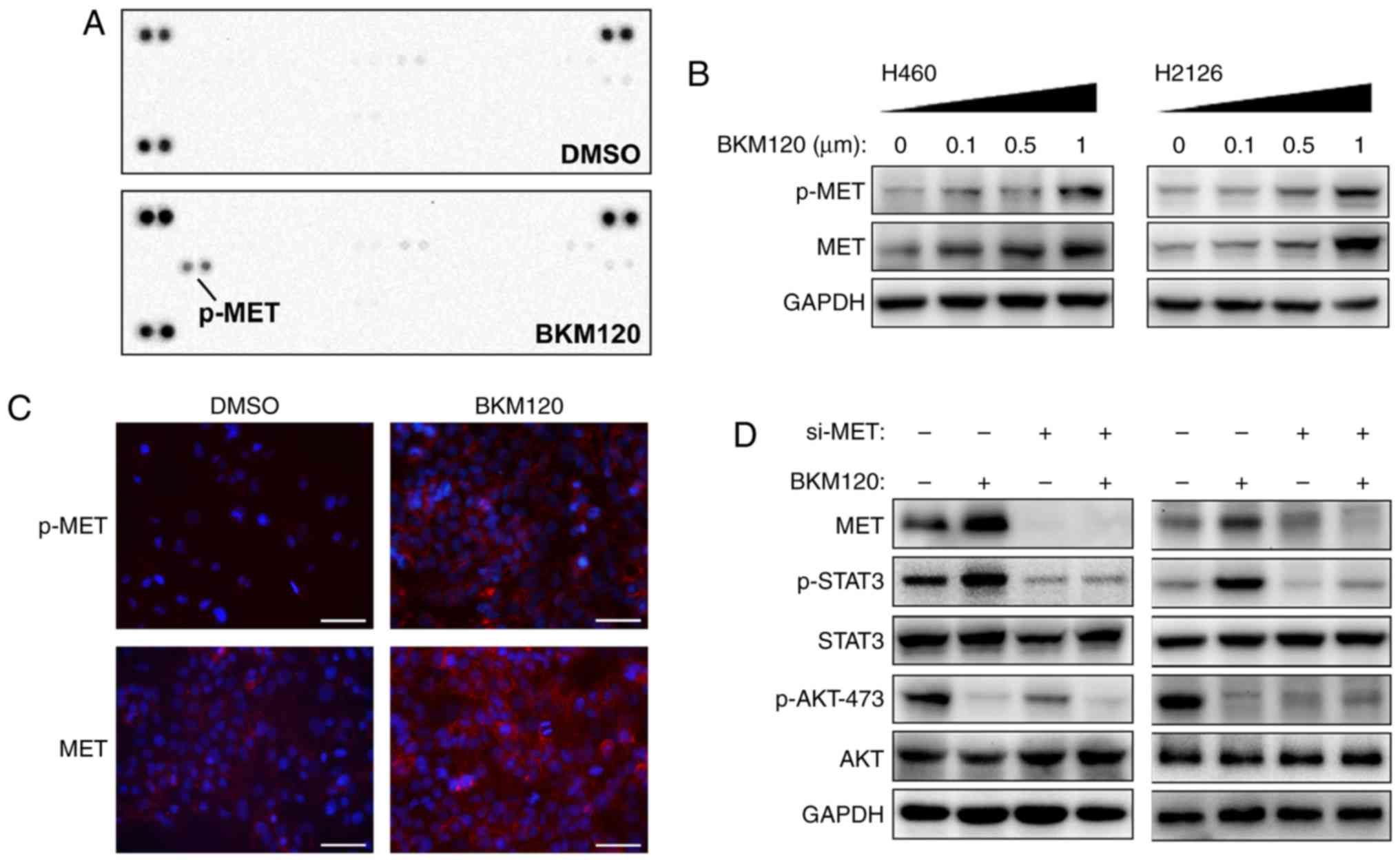
H460 and H2126 cells were treated with the selective
PI3K inhibitor, BKM120. BKM120 markedly inhibited p-AKT at Ser473
and Thr308, and a dose-dependent increase in p-STAT3 was observed,
whereas the phosphorylation of ERK1/2 remained unchanged (Fig. 1A). BKM120 also demonstrated mTORC1
inhibitory activity, which is evident from the reduction of p-S6
with the increasing concentration of BKM120.
 | Figure 1.Inhibition of PI3K/AKT leads to STAT3
activation. (A) Western blot analysis of H460 and H2126 cell
lysates using specific antibodies to phosphorylated or total
proteins of STAT3, ERK1/2, AKT, S6 or GAPDH (loading control). The
cells were treated with increasing concentrations of BKM120 for 24
h. (B) Western blot analysis of H460 cell lysates, following 24 h
cell culture in the absence (DMSO) or presence of LY294002 (10 µM),
MK2206 (10 µM) or BEZ235 (0.5 µM). (C) Western blotting analysis of
H460 cell proteins following transfection with negative control
siRNA (NC) or specific siRNAs for p110α (si-p110α) or AKT (si-AKT).
(D) The STAT3 DNA-binding activity of nuclear extracts from H460
and H2126 cells treated with 0.1, 0.5 or 1 µM BKM120 for 12 h. (E)
Reverse transcription quantitative-polymerase chain reaction
analysis of STAT3 downstream target genes after 24 h of BKM120
treatment. *P<0.05, **P<0.01 and ***P<0.001, compared with
the DMSO control group. PI3K, phosphoinositide 3-kinase; STAT3,
signal transducer and activator of transcription 3; ERK,
extracellular regulation kinase; DMSO, dimethyl sulfoxide; siRNA,
small interfering RNA; MMP9, matrix metallopeptidase 9; bcl-2,
B-cell lymphoma 2; HGF, hepatocyte growth factor; CRP, C-reactive
protein; Mcl-1, BCL2 family apoptosis regulator. |
A number of different PI3K/AKT inhibitors were used
to confirm the activation of STAT3 by pharmacological inhibition of
the PI3K/AKT pathway. Fig. 1B
demonstrates the elevated expression of p-STAT3 with the treatment
with all reagents, including LY294002 (pan-PI3K inhibitor), MK-2206
(AKT inhibitor) and BEZ235 (a dual p110/mTOR inhibitor).
To eliminate the possibility that these results were
due to off-target pharmacological effects of the inhibitors,
PI3K/AKT signaling was silenced via p110α or AKT using 2 different
target-specific siRNAs. Silencing of p110α or AKT resulted in
markedly enhanced activation of STAT3, and decreased AKT activity
in the si-AKT group (Fig. 1C).
The STAT3 DNA-binding activity increased in H460 and
H2126 cells with the concentration of BKM120 treatment (Fig. 1D). STAT3-dependent transcriptional
activity was examined by detecting the expression of STAT3
downstream target genes, including MMP9, bcl-2, survivin,
cyclin-D1, HGF, CRP and bcl-2 family apoptosis regulator (Mcl-1)
(21). As demonstrated in Fig. 1E, treatment with BKM120 increased the
expression of all genes, except Mcl-1. Taken together, these
results suggest that the pharmacological or genetic inhibition of
PI3K/AKT signaling triggered compensatory activation of STAT3 and
altered its downstream signaling events.
PI3K/AKT signaling inhibition induces
MET expression and activation
The regulatory mechanism behind the compensatory
activation of STAT3 induced by PI3K/AKT inhibition was further
investigated. Since multiple RTKs were involved in the activation
of STAT3 signaling, the phosphorylation level of RTKs was detected
in BKM120-treated H460 cells using an RTK array. Elevated p-MET was
observed in H460 cells with BKM120 treatment (Fig. 2A).
In consensus with these results, western blot
analysis demonstrated that BKM120 treatment upregulated the
expression levels of p-MET and total MET protein (Fig. 2B), which was further corroborated by
IF (Fig. 2C). In order to elucidate
the function of MET in regulating the activation of STAT3 signaling
following PI3K/AKT inhibition, MET expression was silenced by
transfection with specific siRNA (si-MET). The results demonstrate
that the depletion of MET resulted in inhibition of p-STAT3, as
well as a modest suppression of p-AKT (Fig. 2D). Taken together, these results
suggest that PI3K/AKT inhibition may activate STAT3 signaling via
upregulating the expression and activation of MET.
MET/STAT3 inhibition promotes the
pro-apoptotic and anti-proliferative effects of PI3K/AKT
inhibitors
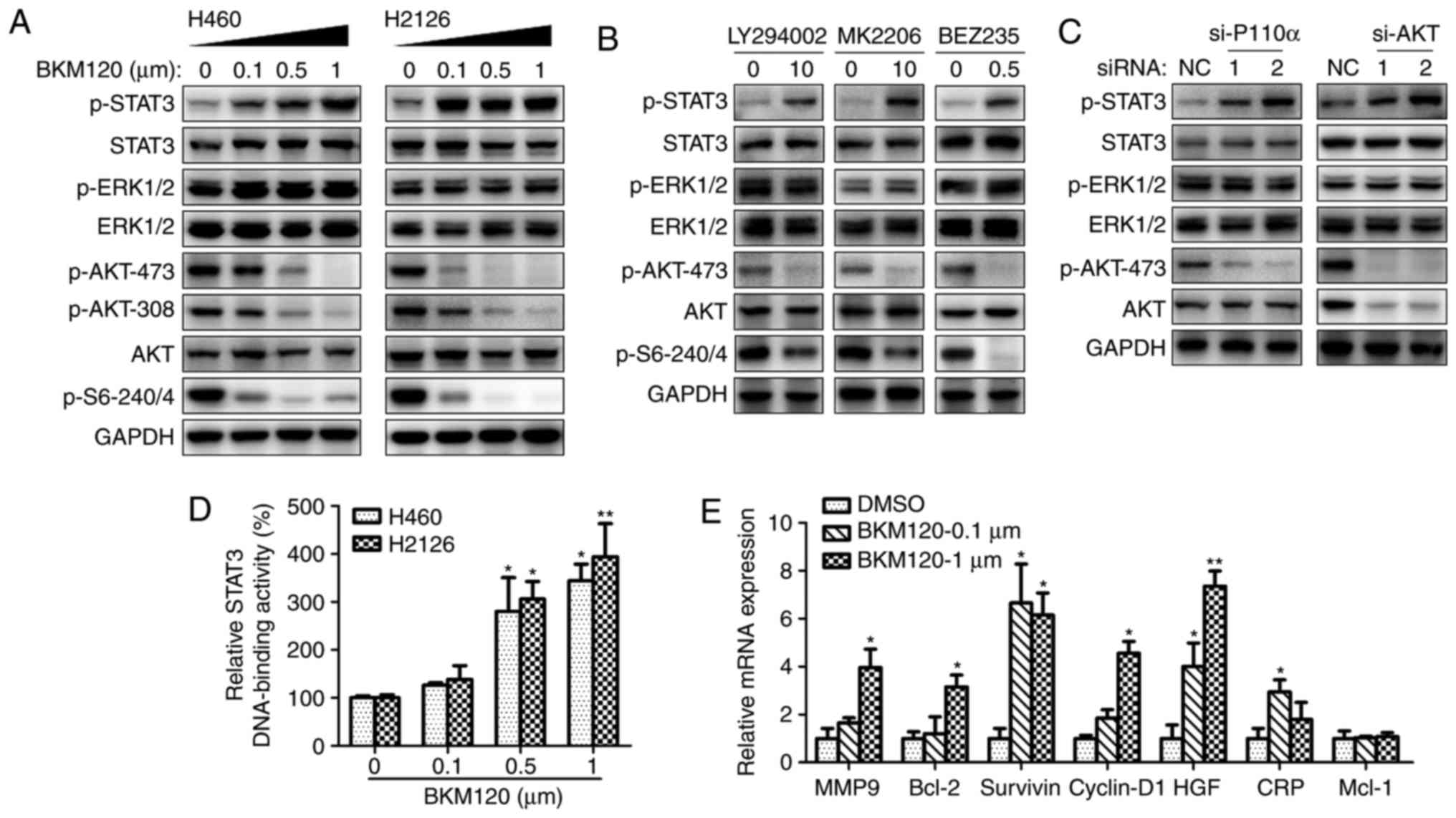
To investigate the compensatory activation of the
MET/STAT3 pathway as a resistance mechanism to PI3K/AKT targeted
therapy, the antitumor efficacy of PI3K/AKT inhibitors was
evaluated when MET/STAT3 signaling was inhibited. As demonstrated
in Fig. 3A, MET inhibition markedly
abrogated STAT3 phosphorylation and AKT activity, and STAT3
inhibition completely abrogated BKM120-induced activation of STAT3.
Antitumor efficacy was observed of BKM120 alone, and its potency
increased in combination with PF-2341066 or stattic. This was
indicated by increased apoptosis of tumor cells treated with the
inhibitor combination compared with those treated with each
inhibitor singly (Fig. 3B). A
significant decrease in colony number was also observed when cells
were treated with BKM120 in combination with PF-2341066 or stattic
compared with cells treated with a single inhibitor (Fig. 3C).
 | Figure 3.MET/STAT3 inhibition promotes the
pro-apoptotic and anti-proliferative effects of PI3K/AKT
inhibitors. (A) The expression of phosphorylated or total forms of
STAT3 and AKT were detected by western blotting following culture
of H460 cells with DMSO (NC), BKM120 (1 µM), PF-2341066 (1 µM) or
STAT3 inhibitor stattic (5 µM) for 60 h. (B) The rate of apoptosis
in H460 and H2126 cells treated with singular or combined
inhibitors was examined by flow cytometry. *P<0.05, **P<0.01
and ***P<0.001, compared with the NC group, (C) The number of
colonies formed after treatment with singular or combines
inhibitors. MET, MET proto-oncogene; STAT3, signal transducer and
activator of transcription 3; PI3K, phosphoinositide 3-kinase;
DMSO, dimethyl sulfoxide; NC, negative control (DMSO); B, BKM120;
P, PF-2341066; S, static. |
Targeting the MET/STAT3 signaling
pathway potentiates the antitumor efficacy of PI3K/AKT signaling
inhibitors in vivo
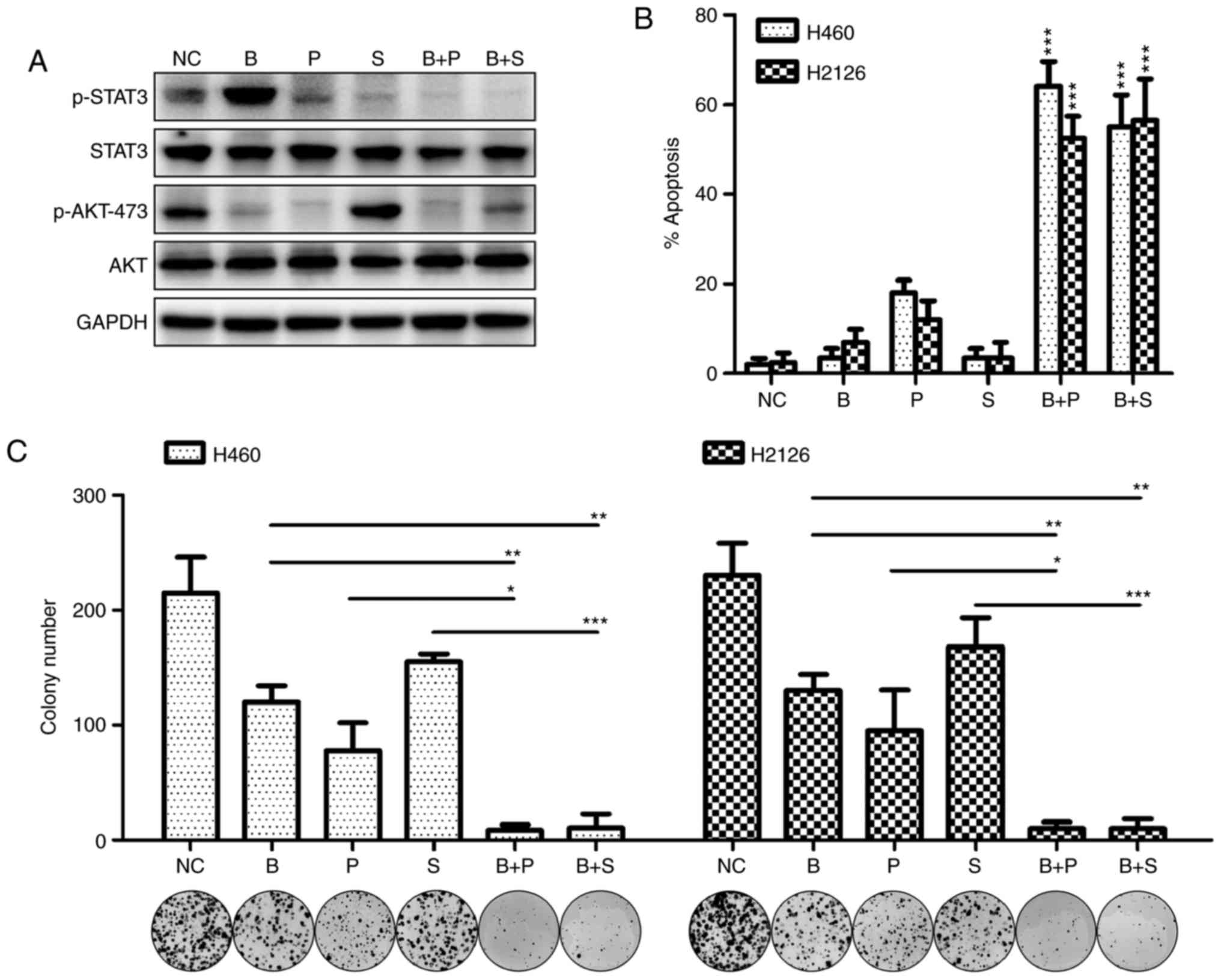
To further verify the antitumor efficacy of the
combination therapy in vivo, a subcutaneous xenotransplanted
tumor model of H460 cells was established in mice. As demonstrated
in Fig. 4A, combination treatment
with BKM120 and PF-2341066 inhibited tumor cell growth effectively
compared with treatment with each agent singly. The activity of
MET, STAT3 and AKT in tumor tissues was further investigated by
western blotting. In consensus with the in vitro
experimental results, the protein expression levels of p-MET,
p-STAT3 and p-AKT were all reduced with combination therapy
compared with BKM120 or PF-2341066 treatment alone, which only
inhibited AKT or MET/STAT3 phosphorylation respectively (Fig. 4B). Similar effects were achieved when
BKM120 was applied in combination with stattic (Fig. 4C). The combined effect of the reagents
was more effective than each single agent in repressing tumor
growth and the expression of proteins of the STAT3 signaling
pathway (Fig. 4D). Fig. 4E illustrates a schematic presentation
of the potential molecular mechanism behind these effects.
 | Figure 4.Targeting the MET/STAT3 pathway
potentiates the antitumor efficacy of PI3K/AKT inhibitors in
vivo. (A) BALB/c nude/nude mice (n=10 per group) bearing H460
xenografts were treated with DMSO, BKM120 (15 mg/kg), PF-2,341066
(25 mg/kg), or BKM120 and PF-2341066 and the tumor volumes were
determined over time. (B) Western blot analysis of the changes in
phosphorylated or total forms of MET, STAT3 and AKT protein
expression in the xenograft tumors. (C) The rate of tumor growth of
H460-derived xenografts treated with DMSO, BKM120 (15 mg/kg),
stattic (3 mg/kg) or both. (D) Western blot analysis of protein
expression of members of the STAT3/AKT pathway in mouse tumor
samples. (E) Schematic representation of the PI3K/AKT and MET/STAT3
pathways. Inhibition of PI3K/AKT caused activation of the MET/STAT3
signaling pathway, leading to cell survival. Dual inhibition of the
two pathways by combination targeted therapy enhanced the antitumor
efficacy in non-small cell lung cancer. MET, MET proto-oncogene;
STAT3, signal transducer and activator of transcription 3; p-,
phosphorylated; PI3K, phosphoinositide 3-kinase; DMSO, dimethyl
sulfoxide; NC, negative control (DMSO); B, BKM120; P, PF-2341066;
S, static; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PDK1, pyruvate
dehydrogenase kinase isozyme 1; mTORC1, mammalian target of
rapamycin complex-1; S6K, ribosomal protein S6 kinase; 4EBP1,
eukaryotic translation initiation factor 4E-binding protein 1. |
Discussion
PI3K/AKT signaling is a core regulatory mechanism in
cancer cells. Hyperactivation of the signaling pathway is evident
in NSCLC, and is therefore a therapeutic target for the disease
(22,23). Small molecule inhibitors of this
pathway are a focus in current research, and demonstrate potential
applications in targeted therapy (24). However, the compensatory activation of
other pathways is a major limitation in the feasibility and
effectiveness of these small-molecule inhibitors. It has been
reported that inhibition of the PI3K signaling pathway leads to
enhanced ERK signaling in human epidermal growth factor receptor 2
(HER2)-positive breast cancer (25).
The WNT/β-catenin pathway, neurogenic locus notch homolog protein 1
and eukaryotic translation initiation factor 4E have been
demonstrated to mediate PI3K inhibitor resistance (26–28). In
the present study, enhanced activation of MET/STAT3 signaling
following PI3K/AKT inhibition was demonstrated in NSCLC.
Persistent STAT3 signaling has been demonstrated to
promote cancer progression through assisting proliferation,
metastasis, angiogenesis and resistance to various drugs (17,18). In
the present study, multiple PI3K/AKT inhibitors induced the
activation of the STAT3 signaling pathway, including BKM120 (an
inhibitor of PI3K), LY294002 (a pan-PI3K inhibitor), MK2206 (an AKT
allosteric inhibitor) and BEZ235 (a dual PI3K/mTOR catalytic
inhibitor) (Fig. 1A and B). Knocked
down expression of PI3K subunit p110α and AKT by siRNA increased
the protein expression level of p-STAT3 (Fig. 1C). Acquired ERK dependency has been
reported in HER2-overexpressing breast cancer following PI3K
inhibition (25). However, no
difference in the phosphorylation of ERK1/2 was observed after
PI3K/AKT inhibition in the present study. PI3K/AKT inhibition did
result in the overexpression of several STAT3 target genes,
including MMP9, bcl-2, survivin, cyclin-D1, HGF and CRP (Fig. 1E). Collectively, these results suggest
novel cross-talk between the PI3K/AKT and STAT3 pathways, and the
compensatory activation of STAT3 may limit the clinical application
of PI3K/AKT inhibitors in cancer treatment.
Activation of RTKs is a key event in signal
transduction and regulates multiple pathways, including PI3K/AKT
and STAT3 signaling (29). An RTK
array was used to study the effects of BKM120 administration on the
activation status of RTKs, and indicated a high level of tyrosine
phosphorylation of MET subsequent to PI3K/AKT signaling inhibition
(Fig. 2A). These results were further
confirmed by western blotting and IF (Fig. 2B and C). Genetic or pharmacological
inhibition of MET disrupted PI3K inhibition-induced STAT3
activation (Fig. 2D), indicating that
STAT3 signaling was regulated by MET. MET encodes an RTK and has
been indicated to function as an oncogene (30). Amplification of MET has been
demonstrated to contribute to tumorigenesis in multiple types of
human cancer (31,32). Numerous studies have demonstrated that
MET-mediated STAT3 signaling is involved in tumor cell growth and
survival, as well as in tumor metastasis (33,34). A
recent study demonstrated that MET-STAT3 signaling was a potential
mechanism of resistance to MEK1/2 inhibitors in colorectal cancer
originating from KRAS mutations (35).
Since compensatory activation of the MET/STAT3
signaling pathway may be involve in the failure of PI3K/AKT
inhibitor treatment in clinical trials, alternative therapeutic
strategies against NSCLC were also explored in the present study.
MET and STAT3 inhibitors not only inhibited STAT3 phosphorylation
but also increased the pro-apoptotic and anti-proliferative effects
of PI3K inhibitors in vitro (Fig.
3). These results were also obtainable in lung cancer-bearing
nude mice (Fig. 4). This suggests the
administration of PI3K inhibitors in combination with either MET or
STAT3 inhibitors as a potential therapeutic strategy to replace
monotherapy (Fig. 4E).
In conclusion, the present study highlights the role
of MET/STAT3 signaling as a compensatory response to PI3K/AKT
blockade, suggesting dual inhibition of PI3K/AKT and MET/STAT3
pathways as an effective NSCLC therapy. Further studies will be
required to validate these results in clinical tumor samples. It
may be a useful to consider the MET/STAT3 activation state in
targeted therapy against NSCLC.
References
|
1
|
Blanco R, Maestu I, de la Torre MG,
Cassinello A and Nunez I: A review of the management of elderly
patients with non-small-cell lung cancer. Ann Oncol. 26:451–463.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Naylor EC, Desani JK and Chung PK:
Targeted therapy and immunotherapy for lung cancer. Surg Oncol Clin
N Am. 25:601–609. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang L, Li G, Zhao L, Pan F, Qiang J and
Han S: Blocking the PI3K pathway enhances the efficacy of
ALK-targeted therapy in EML4-ALK-positive nonsmall-cell lung
cancer. Tumour Biol. 35:9759–9767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gadepalli VS, Deb SP, Deb S and Rao RR:
Lung cancer stem cells, p53 mutations and MDM2. Subcell Biochem.
85:359–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bild AH, Yao G, Chang JT, Wang Q, Potti A,
Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al:
Oncogenic pathway signatures in human cancers as a guide to
targeted therapies. Nature. 439:353–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baselga J: Targeting the
phosphoinositide-3 (PI3) kinase pathway in breast cancer.
Oncologist. 16 Suppl 1:S12–S19. 2011. View Article : Google Scholar
|
|
9
|
Engelman JA: The role of phosphoinositide
3-kinase pathway inhibitors in the treatment of lung cancer. Clin
Cancer Res. 13:s4637–s4640. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li
JH, Xu XM, Liu S, Chen J, Liu F, et al: Phosphoinositide
3-kinase/Akt pathway plays an important role in chemoresistance of
gastric cancer cells against etoposide and doxorubicin induced cell
death. Int J Cancer. 122:433–443. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brana I and Siu LL: Clinical development
of phosphatidylinositol 3-kinase inhibitors for cancer treatment.
BMC Med. 10:1612012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Seront E, Rottey S, Filleul B, Glorieux P,
Goeminne JC, Verschaeve V, Vandenbulcke JM, Sautois B, Boegner P,
Gillain A, et al: Phase II study of dual phosphoinositol-3-kinase
(PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in
patients with locally advanced or metastatic transitional cell
carcinoma. BJU Int. 118:408–415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bedard PL, Tabernero J, Janku F, Wainberg
ZA, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Pérez-García J,
Stathis A, Britten CD, et al: A phase Ib dose-escalation study of
the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with
the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with
selected advanced solid tumors. Clin Cancer Res. 21:730–738. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen CL, Loy A, Cen L, Chan C, Hsieh FC,
Cheng G, Wu B, Qualman SJ, Kunisada K, Yamauchi-Takihara K and Lin
J: Signal transducer and activator of transcription 3 is involved
in cell growth and survival of human rhabdomyosarcoma and
osteosarcoma cells. BMC Cancer. 7:1112007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang C, Wang L, Yang X, Lai L, Chen D and
Duan C: Expression of activated signal transducer and activator of
transcription-3 as a predictive and prognostic marker in advanced
esophageal squamous cell carcinoma. World J Surg Oncol. 13:3142015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gritsko T, Williams A, Turkson J, Kaneko
S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D, et al:
Persistent activation of stat3 signaling induces survivin gene
expression and confers resistance to apoptosis in human breast
cancer cells. Clin Cancer Res. 12:11–19. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masciocchi D, Gelain A, Villa S,
Meneghetti F and Barlocco D: Signal transducer and activator of
transcription 3 (STAT3): A promising target for anticancer therapy.
Future Med Chem. 3:567–597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fossey SL, Liao AT, McCleese JK, Bear MD,
Lin J, Li PK, Kisseberth WC and London CA: Characterization of
STAT3 activation and expression in canine and human osteosarcoma.
BMC Cancer. 9:812009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carpenter RL and Lo HW: STAT3 target genes
relevant to human cancers. Cancers (Basel). 6:897–925. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martinez-Marti A and Felip E: PI3K pathway
in NSCLC. Front Oncol. 1:552011.PubMed/NCBI
|
|
23
|
Heavey S, O'Byrne KJ and Gately K:
Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC.
Cancer Treat Rev. 40:445–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ando Y, Inada-Inoue M, Mitsuma A, Yoshino
T, Ohtsu A, Suenaga N, Sato M, Kakizume T, Robson M, Quadt C and
Doi T: Phase I dose-escalation study of buparlisib (BKM120), an
oral pan-class I PI3K inhibitor, in Japanese patients with advanced
solid tumors. Cancer Sci. 105:347–353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Serra V, Scaltriti M, Prudkin L, Eichhorn
PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M,
Rodriguez S, et al: PI3K inhibition results in enhanced HER
signaling and acquired ERK dependency in HER2-overexpressing breast
cancer. Oncogene. 30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ilic N, Utermark T, Widlund HR and Roberts
TM: PI3K-targeted therapy can be evaded by gene amplification along
the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis.
Proc Natl Acad Sci USA. 108:E699–E708. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tenbaum SP, Ordóñez-Morán P, Puig I,
Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert
JD, Mendizabal L, et al: β-catenin confers resistance to PI3K and
AKT inhibitors and subverts FOXO3a to promote metastasis in colon
cancer. Nat Med. 18:892–901. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu P, Cheng H, Santiago S, Raeder M,
Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA, et al:
Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K
pathway-dependent and PI3K pathway-independent mechanisms. Nat Med.
17:1116–1120. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
He L and Hristova K: Quantification of the
effects of mutations on receptor tyrosine kinase (RTK) activation
in mammalian cells. Methods Mol Biol. 1233:81–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kubo T, Yamamoto H, Lockwood WW, Valencia
I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, et al: MET
gene amplification or EGFR mutation activate MET in lung cancers
untreated with EGFR tyrosine kinase inhibitors. Int J Cancer.
124:1778–1784. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Janbabai G, Oladi Z, Farazmandfar T,
Taghvaei T and Naghshvar F: The prognostic impact of EGFR, ErbB2
and MET gene amplification in human gastric carcinomas as measured
by quantitative Real-Time PCR. J Cancer Res Clin Oncol.
141:1945–1952. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Syed ZA, Yin W, Hughes K, Gill JN, Shi R
and Clifford JL: HGF/c-met/Stat3 signaling during skin tumor cell
invasion: Indications for a positive feedback loop. BMC Cancer.
11:1802011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang YW, Wang LM, Jove R and Vande Woude
GF: Requirement of Stat3 signaling for HGF/SF-Met mediated
tumorigenesis. Oncogene. 21:217–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Van Schaeybroeck S, Kalimutho M, Dunne PD,
Carson R, Allen W, Jithesh PV, Redmond KL, Sasazuki T, Shirasawa S,
Blayney J, et al: ADAM17-dependent c-MET-STAT3 signaling mediates
resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell
Rep. 7:1940–1955. 2014. View Article : Google Scholar : PubMed/NCBI
|