Introduction
Pituicytomas are rare slow-growing tumours arising
from the glial cells of the neurohypophysis and infundibulum
(pituicytes). The histogenesis, natural history and prognosis of
these lesions still need to be elucidated, as data in the current
literature remain scarce.
Since the first reported case in 1955 (1), there has been a surge in the number of
case reports and small case series describing this rare entity.
There are no gender-specific differences in the incidence of
pituicytomas, and the average age of presentation is 50 years of
age (2). No specific risk factors
were identified. These neoplasms generally present with headaches,
visual disturbances and manifestations of hypopituitarism
(typically sexual dysfunction). The clinical presentation is almost
identical to other more frequent sellar lesions, therefore
correctly diagnosing these lesions can be challenging. Of about 80
cases described in the literature up until now (3), only one previous case reported an abrupt
onset of clinical features and this was due to an intraventricular
haemorrhage (4).
On imaging, even if pituicytomas present with
suspicious radiological peculiarities, their rarity often leads
them to be misdiagnosed for more common lesions. Surgical resection
represents the mainstay of treatment, and this is typically via an
endoscopic endonasal approach (5).
However, complete resection may be challenging due to the firm
consistency and highly vascularised nature of these tumours.
We report a novel case of a pituicytoma presenting
as pituitary apoplexy, with the aim to advance a hypothesis to
explain this phenomenon. To our knowledge this is the first
reported case in the literature that describes the development of
pituitary apoplexy in the context of a pituicytoma.
Case report
A 77-year-old man presented to the University
Hospital of Lausanne in October 2015 with fatigue and clinical
signs of hypogonadism. The clinical presentation was suggestive of
hypopituitarism and endocrine tests showed an anterior pituitary
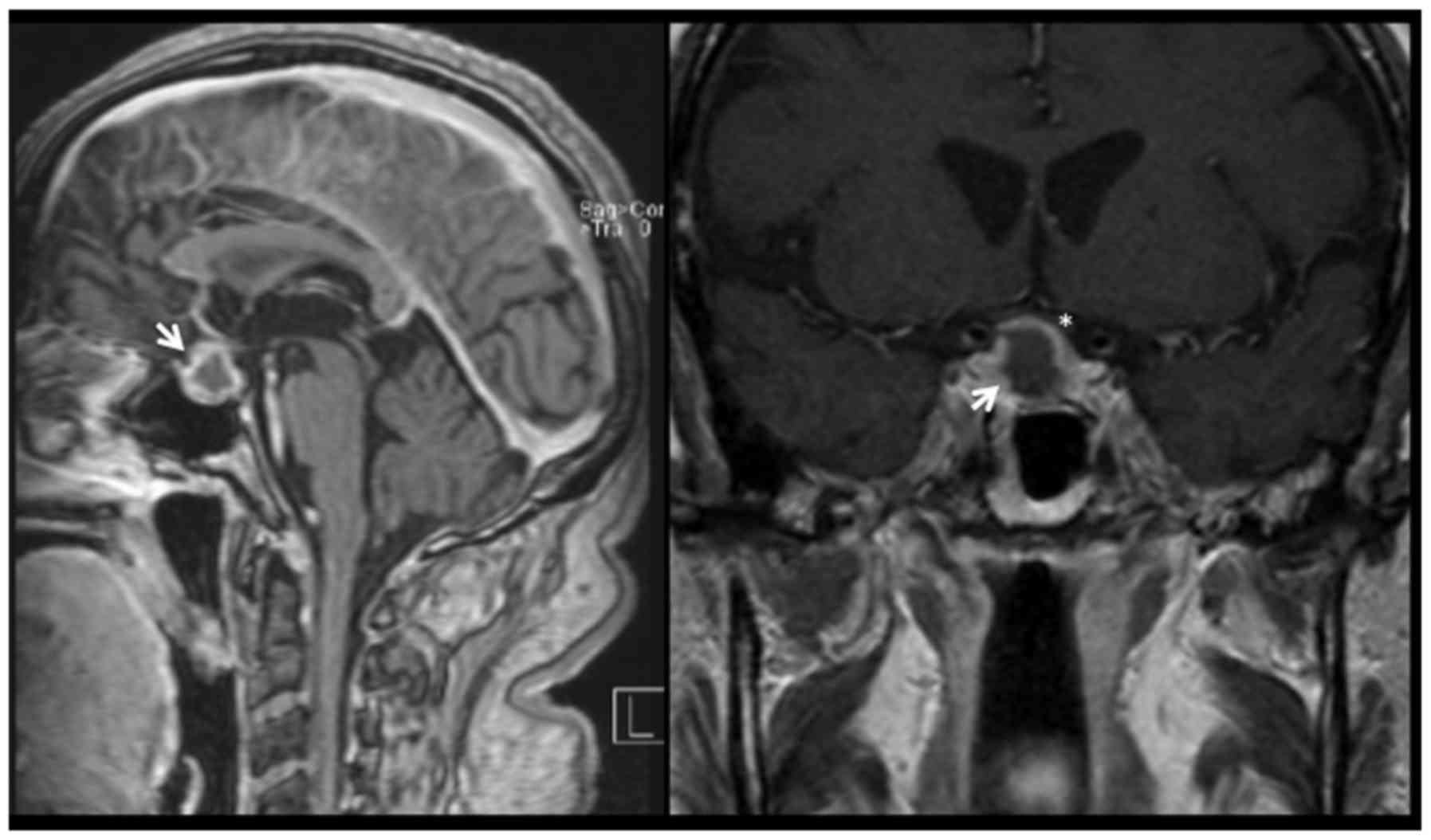
dysfunction. Investigation with cerebral magnetic resonance imaging
(MRI) demonstrated a sellar lesion with suprasellar extension and
no compression of optic structures. He was admitted at the
neurosurgical department and initial management consisted of
hormonal replacement therapy and a ‘watch-and-wait’ approach. One
year after his initial presentation (October 2016), the patient
returned to the emergency department with a thunderclap headache. A
cerebral MRI demonstrated pituitary apoplexy (Fig. 1). There were no visual deficits on
clinical examination and we decided to continue the regular
monitoring and follow-up. Over 6 weeks of follow-up the patient
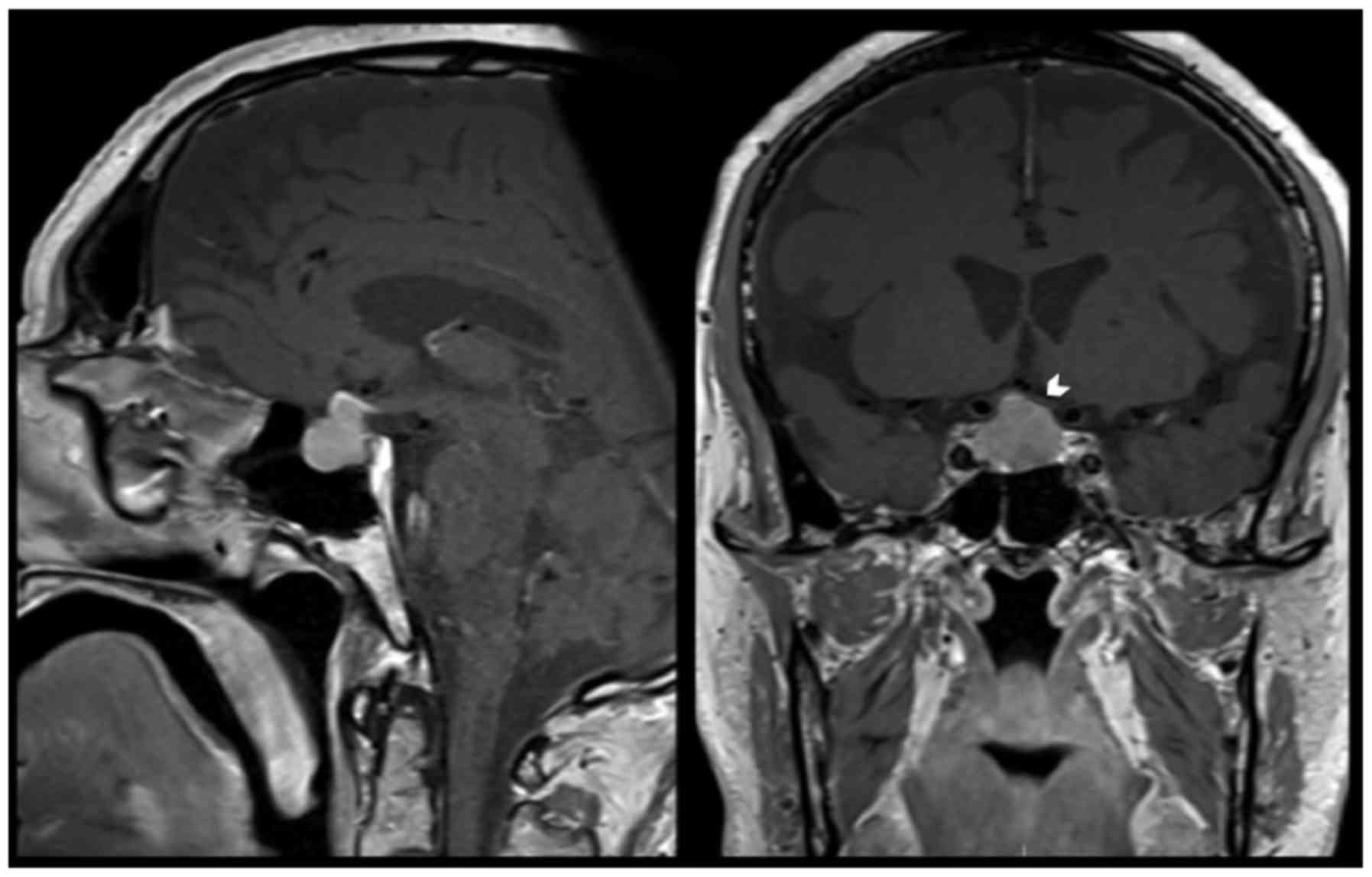
started complaining of visual symptoms, secondary to a progressive
growth of the pituitary lesion with compression of the optic chiasm
(Fig. 2). Ophthalmological
examination confirmed a right temporal quadrantanopia.
The patient underwent an uncomplicated endoscopic
endonasal transsphenoidal resection of the tumour. Post-operatively
the patient recovered well, with the right temporal quadrantanopia
completely resolving. Endocrine function remained impaired, with a
persistent anterior hypopituitarism but no diabetes insipidus. A
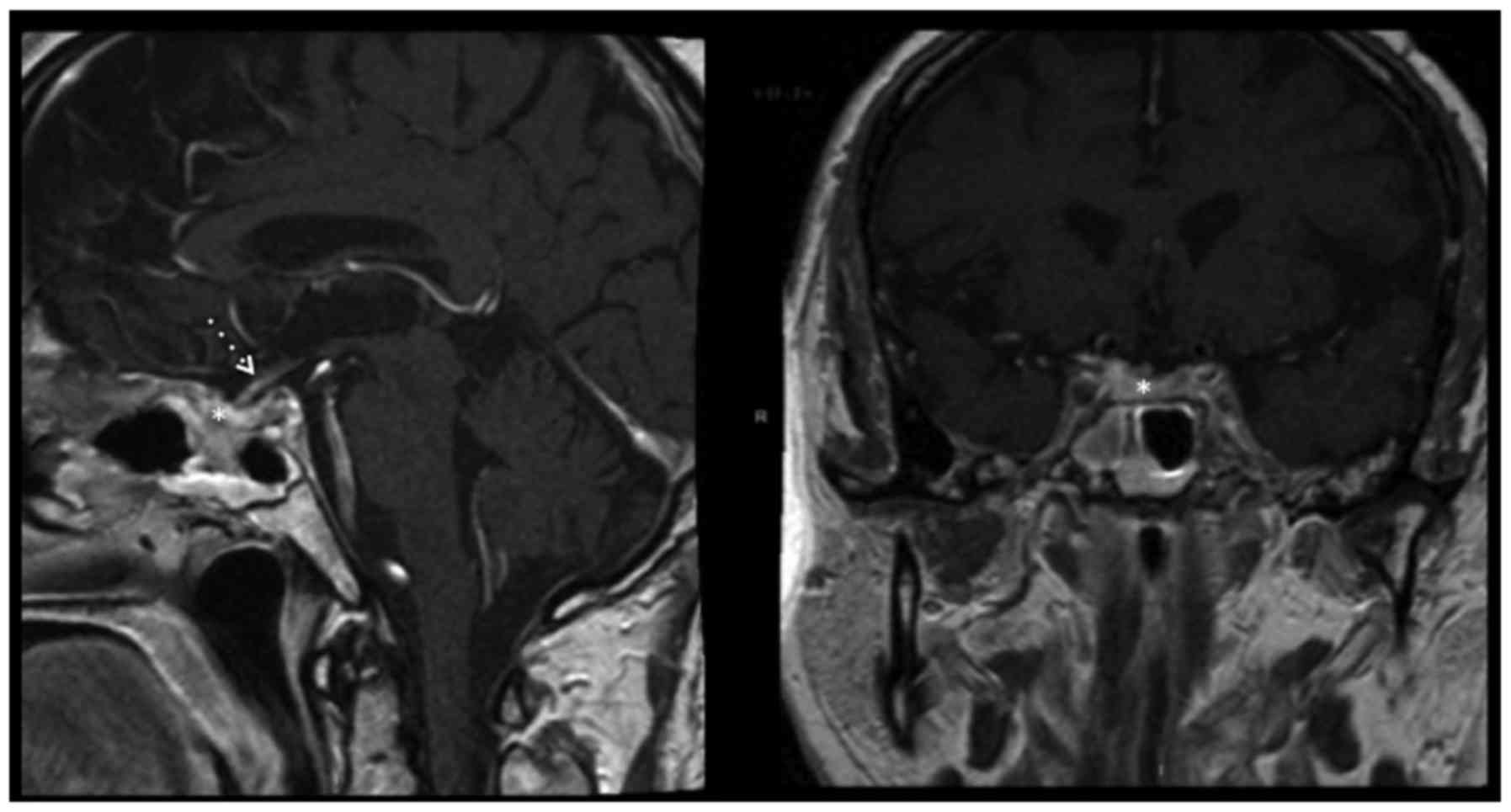
post-operative MRI confirmed complete excision of the lesion
(Fig. 3), and follow-up imaging at 15
months confirmed that the patient was still in remission.
The diagnosis of a pituicytoma was confirmed by
pathologic examination and immunohistochemistry performed on
four-micron thick, formalin fixed and paraffin embedded sections,
analysed with a Nikon Eclipse microscope and a Nikon DSFi1 camera
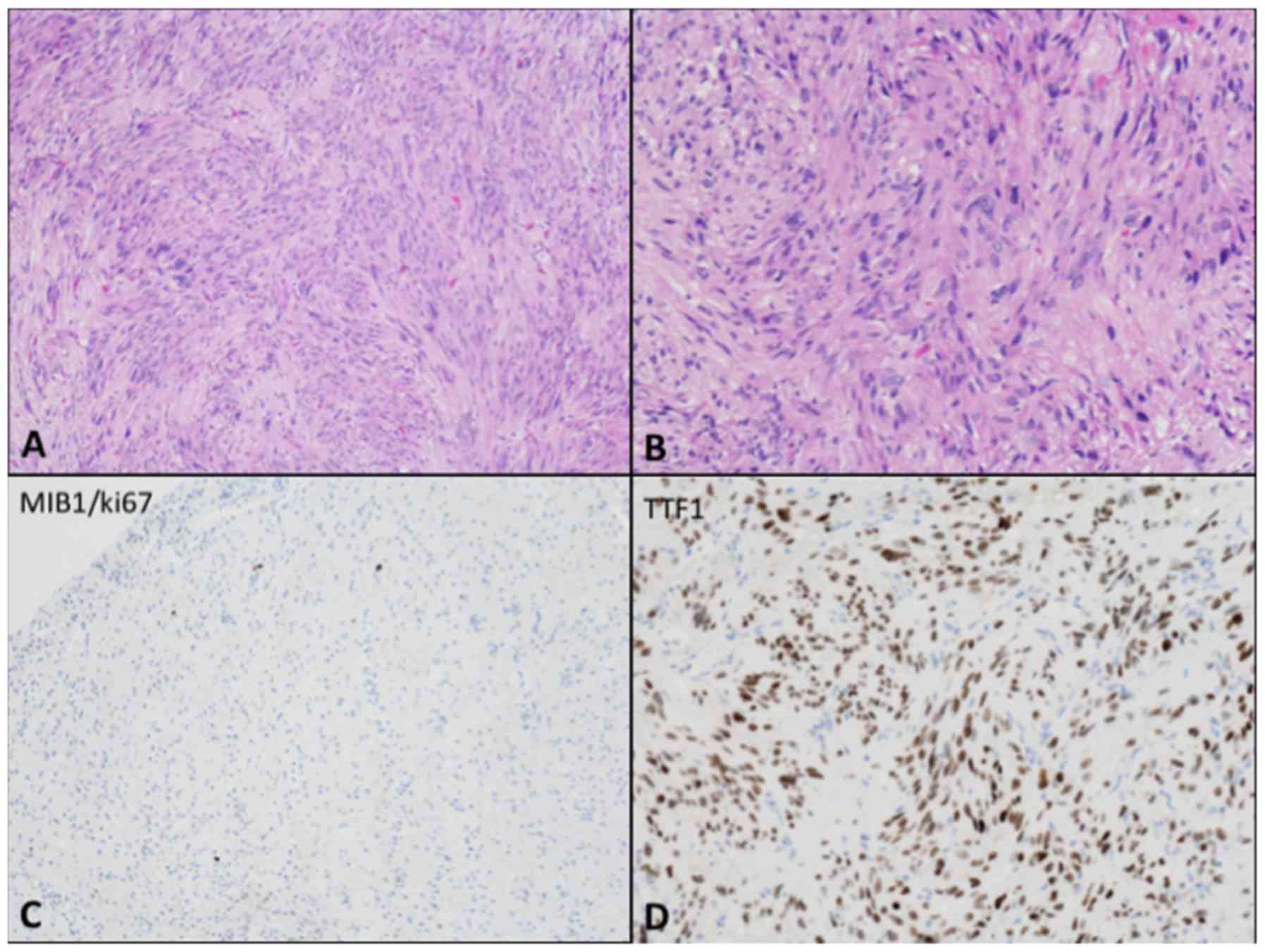
(both from Nikon Corporation, Tokyo, Japan). Spindle-shaped cells
with a fascicular growth pattern and heterogeneous nuclei were
identified with hematoxylin and eosin stain (Fig. 4A and B). There was no evidence of
atypical cellular features or mitotic figures and the detection of
proliferation markers was performed on paraffin sections with
anti-Ki67 (MIB-1; Immunotech; Beckman Coulter, Inc., Brea, CA, USA)
with the cell count performed at high power (×400). Cellular
proliferation resulted inferior to 1% (Fig. 4C). Immunohistochemical staining was
performed through the Ventana platform and it revealed tumour cells
positive for thyroid transcription factor-1 (TTF-1) (Fig. 4D), S-100, vimentin, B-cell lymphoma 2
(Bcl-2) and CD56. The cells were negative for synaptophysin, glial
fibrillary acidic protein (GFAP), epithelial membrane antigen
(EMA), signal transducer and activator of transcription (STAT)6,
cluster of differentiation 34 (CD34), Melan-A, human melanoma black
45 (HMB45), (tumor) protein 63 (P63), calretinin and neurofilament
(NF). The details for each antibody and for the methodology used to
perform immunohistochemistry were reported in Table I. The entire sections on the stained
slides were evaluated. Electron microscopy was not performed.
 | Table I.Antibodies used to perform the
immunohistochemistry with the Ventana platform. |
Table I.
Antibodies used to perform the
immunohistochemistry with the Ventana platform.
| Antibody | Clone | Fabricant | Reference | Dilution | Incubation |
|---|
| Antibodies that
stained positively |
|
|
|
|
|
|
TTF-1 | 8G7G3/1 | Invitrogen | 18–0221 | 1:30 | 32 min 37°C |
|
S-100 | Polyclonal | Novocastra
Laboratories, Ltd. | NCL-S100P | 1:400 | 24 min 37°C |
|
Vimentin | VIM3B4 | Progen Biotechnik
GmbH | 61013 | 1:3,200 | 32 min 37°C |
|
Bcl-2 | Bcl2/100/D5 | Novocastra
Laboratories, Ltd. | NCL-Bcl-2 | 1:30 | 92 min 37°C |
|
CD56/NCAM | CD564 | Novocastra
Laboratories, Ltd. | NCL-L-CD56-504 | 1:25 | 60 min 37°C |
| Antibodies that did
not stain tissue |
|
|
|
|
|
|
Synaptophysin | 27G12 | Novocastra
Laboratories, Ltd. | NCL-Synap-299 | 1:100 | 32 min 37°C |
| GFAP | G-A-5 | Sigma-Aldrich | G3893 | 1:15 | 92 min 37°C |
| EMA | E29 | DAKO | M0613 | 1:100 | 32 min 37°C |
| STAT-6
(S-20) | Polyclonal | Santa Cruz
Biotechnology, Inc. | SC-621 | 1:400 | 60 min 37°C |
| CD34 | Qbend-10 | DAKO | M7165 | 1:25 | 32 min 37°C |
|
Melan-A | A-103 | DAKO | M7196 | 1:50 | 32 min 37°C |
|
HMB45 | HMB45 | DAKO | M0634 | 1:50 | 32 min 37°C |
| P63 | DAC-P63 | DAKO | M7317 | 1:100 | 32 min 37°C |
|
Calretinin | CAL2 | Dianova GmbH | DIAL-CAL-250 | 1:20 | 32 min 37°C |
| NF | 2F11 | DAKO | M0762 | 1:1,000 | 37°C |
Discussion
Pituicytomas are defined as indolent World Health
Organization (WHO) grade 1 tumours involving the posterior lobe of
the pituitary and/or the pituitary stalk. They are solid tumours,
generally well circumscribed and composed of spindle cells derived
from pituicytes (6,7).
Pituicytomas are slow growing and they may often go
unnoticed until clinical signs and symptoms secondary to mass
effect start to develop. These lesions are known to compress the
pituitary gland and the optic system, thus provoking more
frequently hypopituitarism and visual disturbance. We describe here
the first reported case of pituitary apoplexy secondary to a
pituicytoma. The primary goal of the management was to mitigate the
consequences of apoplexy through urgent hormonal substitution and
to monitor for visual deficits. Surgical management was decided
upon following the development of a visual deficit.
Pituicytomas have similar radiological features to
pituitary adenomas, thus distinguishing these diagnoses from one
another based on radiological imaging remains challenging. They are
often described as sellar lesions with frequent suprasellar
extension and sellar enlargement (and/or bone remodelling). They
generally present as isointense lesions on T1-weighted MRI and the
posterior lobe of the pituitary gland is generally not
identifiable, while they appear as hyperintense lesions on
T2-weighted images (2,8). T1-weighted MRI sequences with gadolinium
contrast enhancement may prove useful in diagnosis, as pituicytomas
may appear brighter than classical adenomas due to their dense
vascularisation (4). A recent paper
reports the usefulness of flow voids at the MRI to differentiate
pituicytomas from pituitary adenomas. Flow voids represent signal
loss due to rapidly flowing blood, and thus indirectly equate to
increased vascularity (9).
Angiography may also be a useful tool, with a vascular blush
typical of pituicytomas (2). Despite
these differences, radiological distinction from other more common
sellar lesions, such as pituitary adenomas and meningiomas, is not
always easy.
Considering this, the diagnosis of a pituicytoma
strongly relies on pathological analysis and immunohistochemical
investigations, as pituicytomas demonstrate a characteristic
immunohistochemical profile with a strong positive staining for
S-100 and TTF-1, a focal positivity for GFAP, and an absence of
staining for synaptophysin, p53 or pericellular reticulin (6,10,11).
The mainstay of treatment for symptomatic
pituicytomas is surgery, with gross total resection considered
curative. Unfortunately, due to the dense vascularity and the firm
consistency of these tumours, the extent of resection is often
limited. Precise surgical planning is necessary, with a focus on
the extension of the tumour itself and its radiological
characteristics. The development of endoscopic surgery has led to
an endonasal approach being the preferred procedure.
Our case had a peculiar clinical presentation and we
suggest that apoplexy was most likely due to the haemorrhagic
transformation or to the intralesional ischemia secondary to the
rupture or the occlusion of pathological vessels arising directly
from the internal carotid artery or from its branches. We put forth
the hypothesis that the incidence of pituicytoma apoplexy might
actually be underestimated, as with pituitary apoplexy, surgery and
tissue diagnosis is performed only in cases complicated by visual
disturbance.
We assert that pathologists, endocrinologists and
neurosurgeons should be mindful of pituicytoma as a possible
differential diagnosis when dealing with pituitary apoplexy.
Recommendations and future directions
Over the last few years there has been substantial
advancement regarding the definition and classification of
pituicytomas (7). However, the
histogenesis of these lesions remains controversial, and the
natural history of progression of this disease is yet to be
elucidated. Understanding the natural history of these tumours is
difficult, because all of the cases described so far have been
diagnosed retrospectively following surgical management and
pathological confirmation.
Treatment is mainly empirical, and when gross total
resection is possible, management is relatively straightforward. In
cases where only subtotal resection is possible, the use of
adjuvant therapies is highly debated, and there is no existing
evidence to recommend specific management.
Further work to clarify the optimal management of
patients with pituicytomas still needs to be undertaken, and we
hope that future studies will help to standardise treatment
strategies.
To conclude, pituicytomas are rare and indolent
lesions. However, atypical presentations may be challenging and
should not be underestimated. Due to the vascularised nature of
these lesions, haemorrhagic or ischaemic events are potential
serious consequences. We assert that a wider range of differential
diagnoses should be considered when clinicians encounter pituitary
apoplexy in the context of hypervascularised pituitary lesions not
associated with abnormal hormonal secretion.
Acknowledgments
We would like to thank Mr. Michael Nohrenberg from
the Medical University of Melbourne for his help with revising the
English of the article.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GC, JD, RTD and MM contributed to the design and
conception of the study. GC and JD proceeded with the acquisition
of data, GC performed a literature review and JPB performed the
pathological analysis and provided the necessary material. All the
authors contributed substantially to the drafting of the manuscript
and revised it critically for important intellectual content. They
all gave final approval of the version to be published.
Ethics approval and consent to
participate
Informed consent was obtained from the patient.
Consent for publicationv
The patient has consented to the submission of this
case report for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Scothorne CM: A glioma of the posterior
lobe of the pituitary gland. J Pathol Bacteriol. 69:109–112. 1955.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolfe SQ, Bruce J and Morcos JJ:
Pituicytoma: Case report. Neurosurgery. 63:E173–E174; discussion
E174. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang X, Liu X, Li W and Chen D:
Pituicytoma: A report of three cases and literature review. Oncol
Lett. 12:3417–3422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benveniste RJ, Purohit D and Byun H:
Pituicytoma presenting with spontaneous hemorrhage. Pituitary.
9:53–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Messerer M, De Battista JC, Raverot G,
Kassis S, Dubourg J, Lapras V, Trouillas J, Perrin G and Jouanneau
E: Evidence of improved surgical outcome following endoscopy for
nonfunctioning pituitary adenoma removal. Neurosurg Focus.
30:E112011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brat DJ, Scheithauer BW, Fuller GN and
Tihan T: Newly codified glial neoplasms of the 2007 WHO
classification of tumours of the central nervous system:
Angiocentric glioma, pilomyxoid astrocytoma and pituicytoma. Brain
Pathol. 17:319–324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mete O and Lopes MB: Overview of the 2017
WHO classification of pituitary tumors. Endocr Pathol. 28:228–243.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hurley TR, D'Angelo CM, Clasen RA,
Wilkinson SB and Passavoy RD: Magnetic resonance imaging and
pathological analysis of a pituicytoma: Case report. Neurosurgery.
35:314–317; discussion 317. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Law-Ye B, Cholet C and Leclercq D: First
depiction of flow voids to differentiate pituicytomas from giant
adenomas. World Neurosurg. 109:304–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Figarella-Branger D, Dufour H, Fernandez
C, Bouvier-Labit C, Grisoli F and Pellissier JF: Pituicytomas, a
mis-diagnosed benign tumor of the neurohypophysis: Report of three
cases. Acta Neuropathol. 104:313–319. 2002.PubMed/NCBI
|
|
11
|
Ulm AJ, Yachnis AT, Brat DJ and Rhoton AL
Jr: Pituicytoma: Report of two cases and clues regarding
histogenesis. Neurosurgery. 54:753–757; discussion 757–758. 2004.
View Article : Google Scholar : PubMed/NCBI
|