Introduction
Lung cancer is the leading cause of death worldwide
(1). Radiotherapy has been applied to
treat advanced non-small-cell lung cancer (NSCLC) for the past two
decades. However, one major obstacle to successful radiotherapy is
the existence of radio-resistance (2). Tyrosine kinase inhibitors can improve
the survival of patients with radio-resistant lung adenocarcinoma.
However, few treatment targets have been identified for
treatment-resistant lung squamous carcinoma (3,4).
Therefore, it is important to explore more effective targeting
molecules for the radio-resistant lung squamous carcinoma.
Insulin-like growth factor 1 receptor (IGF-1R) is a
transmembrane receptor tyrosine kinase involved in the development
and progression of cancer, whereby receptor activation strongly
promotes cell growth and survival (5). IGF-1R is a heterotetrameric plasma
membrane glycoprotein. Once a ligand binds to the IGF-1R, it will
induce activation of the intrinsic tyrosine kinase of the
β-subunit, leading to receptor phosphorylation and activation of
protein kinases which are associated with carcinogenesis and tumor
progression (6,7). IGF-1R has been found to be
over-expressed in a variety of human malignancies including lung
cancer. One study showed that IGF-1R expression is significantly
associated a reduction in overall survival. Median survival in lung
cancer patients with high and low IGF-1R expression is 26.51 vs.
47.77 months (P=0.017), and disease-free survival is 17.44 vs.
37.65 months (P=0.045), respectively. IGF-1R is activated in NSCLC
patients, particularly in those with squamous cell carcinoma (SCC)
(8). IGF-1R plays an important role
in lung SCC. CP-751,871 is a fully human monoclonal antibody
against IGF-IR, which can increase the radio-sensitivity of NSCLC
cell lines and application of this antibody has been attributed to
inhibition of DNA repair and enhanced irradiation-induced apoptosis
(9). However, the mechanisms
underlying the association between IGF-1R over-expression and
radio-resistance in human lung squamous carcinoma cells are still
unknown.
Radiation induces DNA damage and destroys the
structure of DNA molecules, including nucleotide excision, single
strand breaks and double strand breaks (DSBs) (10,11). The
DNA lesions can be recognized by DNA checkpoints which can trigger
DNA repair pathways (12). However,
apoptosis will be initiated if DNA damage is not repaired. In the
current study, a lentiviral vector-based RNA interference
expression system was used to stably suppress the expression of
IGF-1R and the effects of IGF-1R knockdown on the radio-sensitivity
of lung SCC and the potential mechanisms were explored.
Materials and methods
Cell culture, materials and
reagents
The H520 human lung squamous carcinoma cells were
purchased from the Cancer Institute of Tongji University (Shanghai,
China). The cells were grown in Dulbecco's modified Eagle's medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and maintained in a humidified 5% CO2
atmosphere at 37°C. The major sources of materials and reagents
used in the present study are as follows: IGF-1R, phospho-IGF1R,
phospho-H2A histone family member X (H2AX), p53 binding protein 1
(53BP1), phospho-Akt, hypoxia inducible factor 1 α (HIF-1α), and
β-actin were supplied by Cell Signaling Technology, Inc., (Danvers,
MA, USA). Phosphorylated ataxia-telangiectasia mutated (p-ATM) was
purchased from Abcam (Cambridge, UK). An Annexin V-APC/PI apoptosis
detection kit was purchased from BioLegend, Inc., (San Diego, CA,
USA). TRIzol reagent was provided by Invitrogen; Thermo Fisher
Scientific, Inc. Cell Counting Kit-8 was purchased from Dojindo
Molecular Technologies, Inc., (Kumamoto, Japan).
X-ray irradiation
The H520 human lung squamous carcinoma cell line was
irradiated using a linear accelerator (Elekta Instrument AB,
Stockholm, Sweden) with an 8 MV X-ray at a dose rate of 500
cGy/min, and then cells were further incubated up to specified time
points, and harvested for the subsequent experiments.
CCK-8 assay
Cells were seeded into 96-well culture plates at a
density of 5,000 cells/well and allowed to adhere for 24 h. After
X-ray irradiation, the cells were then incubated for different h in
a humidified chamber at 37°C. Viable cells were evaluated with the
CCK-8 assay according to the manufacturer's instructions. CCK-8
solution was added to the cells in 96-well plates and then
incubated at 37°C for an additional 1 h, and the absorbance at 450
nm was determined at using a microplate reader (ELX800; BioTek
Instruments, Inc., Winooski, VT, USA).
Clonogenic assay for
radio-sensitivity
Different numbers of cells (0 Gy with
1×103 cells, 2 Gy with 2×103 cells, 4 Gy with
4×103 cells, 6 Gy with 6×103 cells, 8 Gy with
8×103 cells, and 10 Gy with 1×104 cells) were
plated in 60-mm dishes depending on the dose of irradiation, and
cultured overnight. After X-Ray irradiation with 0, 2, 4, 6, 8, or
10 Gy, the cells were cultured for two weeks at 37°C. Cells were
washed three times using phosphate buffered saline (PBS) and fixed
using ice-cold methanol for 15 min, and then stained using 1%
crystal violet dye for 15 min and rinsed in distilled water to wash
away excess dye. Plates were allowed to dry before being scanned.
Only colonies consisting of 50 or more cells were counted.
Flow cytometry
The cells were cultured at a density of
5×104 cells per well in growth medium for 12 h in
24-well plates, and irradiated at the indicated doses. Apoptotic
cells were quantified using an Annexin-APC/PI apoptosis detection
kit and FACS Cali-bur flow cytometry, as described previously. The
cells were harvested through centrifugation after irradiation and
washed twice using cold PBS. Cells were then re-suspended in 100 µl
of Annexin-V binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM
Cacl2, pH 7.4), incubated with 5 µl of Annexin-V-APC for
15 min at room temperature, and counterstained using propidium
iodide (final concentration: 1 µg/ml). After the incubation period,
the cell suspension was diluted with 190 µl of Annexin-V binding
buffer. Ten thousand cells were acquired per sample, and data were
acquired using flow cytometry with a Becton-Dickinson FACS can flow
cytometer with Cell Quest software (BD Biosciences, Franklin Lakes,
NJ, USA). Cells in the early stages of apoptosis were
Annexin-positive, whereas cells that were Annexin-positive and
PI-positive were in the late stages of apoptosis.
Western blot analysis
The treated cells (1×107 cells/6 ml DMEM
with 10% FBS in a 90-mm dish) were collected and washed twice using
cold PBS. Cells were lysed in 200 µl of lysis buffer. The lysate
was incubated on ice for 30 min, vortexed and centrifuged at 14,000
× g for 15 min at 4°C. The supernatant was collected and protein
concentration was determined using the Bradford Assay. After the
addition of sample loading buffer, the protein samples underwent
electrophoresis using a 10% SDS-polyacrylamide gel and were then
transferred to a polyvinylidene fluoride membrane (EMD Millipore,
Billerica, MA, USA). After blocking for 4 h at room temperature in
a solution of 5% non-fat dry milk in Tris-buffered saline
containing 0.1% Tween-20, the membranes were incubated overnight at
4°C with primary antibody (against phospho-IGF-1R, IGF-1R,
phospho-ATM, phospho-AKT, phospho-H2AX, 53BP1, HIF-1α, matrix
metallopeptidase 9 (MMP-9) and VEGFA) at a concentration of 1:1,000
in Tris-buffered saline with 0.1% Tween-20 containing 5% non-fat
dry milk. After washes four times, the membranes were incubated
with a horseradish peroxidase-conjugated secondary anti-rabbit IgG
anti-body (1:5,000 dilution) at room temperature for 1 h and were
then washed six times. The blots were developed using an Immobilon™
Western Chemiluminescent detection reagent (EMD Millipore) and the
results were recorded using the ChemiDox™ XRS+ system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Short hairpin RNA (shRNA)
infection
The lentiviral vectors expressing shRNA were
successfully constructed by Shanghai GenePharma Co., Ltd, Shanghai,
China. Cells were seeded at 2×105 per well in 6-well
plates and incubated overnight, and the cells were then infected
using the following sequences: IGF-1R shRNA-1
(5′-GCAACCTGAGTTACTACATTG-3′), IGF-1R shRNA-2
(5′-GCACCATCTTCAAGGGCAATT-3′), IGF-1R shRNA-3
(5′-GCCCAACACCTACAGGTTTGA-3′), IGF-1R shRNA-4
(5′-GCAAAGTCTTTGAGAATTTCC-3′) or negative control shRNA
(5′-TTCTCCGAACGTGTCACGT-3′). Then the cells were selected using
Puromycin (2 µg/ml), whereby puromycin-resistant colonies were
picked, expanded, and analyzed separately.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Cells were seeded in 6-well plates and allowed to
grow in DMEM supplemented with 10% FBS until semi-confluent, before
being irradiated. After irradiation, total RNA was extracted using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the recommendations provided by the manufacturer. cDNA
was generated from sample RNA using the RevertAid First strand cDNA
Cynthesis kit (no. 1622). Subsequently, 2 µl of cDNA and Taq PCR
Master Mix (Qiagen GmbH, Hilden, Germany) were used for PCR. The
primer sequences used for the RT-qPCR were as follows: IGF-1R
forward primer: CCTGAAAGGAAGCGGAGAG, reverse primer:
GGGTCGGTGATGTTGTAGGT; GAPDH forward primer: ATGACATCAAGAAGGTGGTG,
reverse primer: CATACCAGGAAATGAGCTTG. PCR amplifications were
performed as follows: 5 min at 94°C followed by 25 cycles of [30
sec at 94°C, 30 sec at 55°C, 30 sec at 72°C] and a final extension
at 72°C for 5 min.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Each experiment was performed three times. All data are
expressed as mean ± standard deviation (SD) unless otherwise
specified. Comparison of data between two groups was statistically
analyzed using a two-tailed Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Levels of statistical significance are denoted as *P<0.05,
**P<0.01 and OL-0-0-8705P<0.001, as shown in the
Figures.
Results
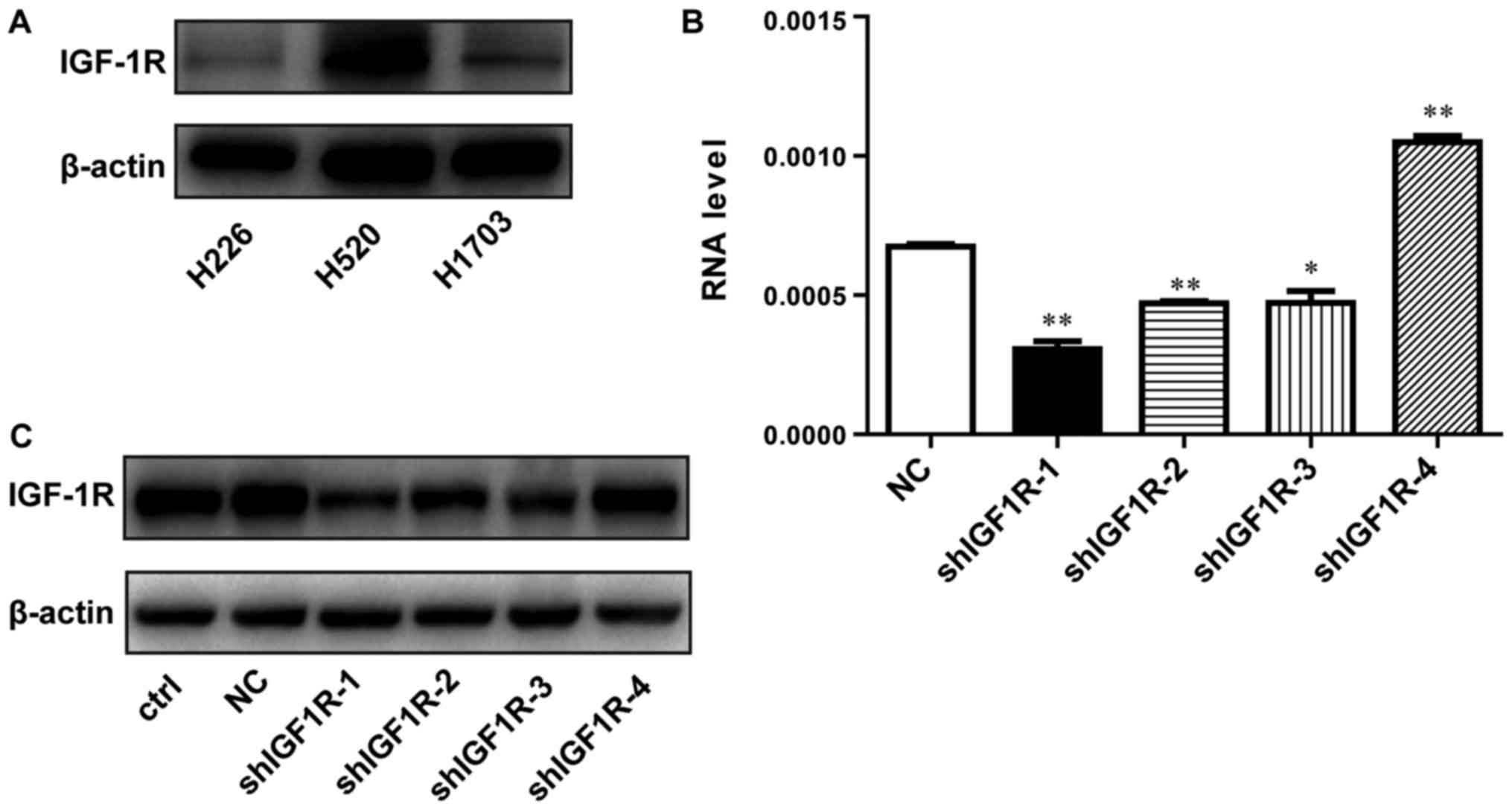
Lentivirus-mediated shRNA efficiently
inhibited the expression of IGF-1R in H520 lung squamous carcinoma
cells
To examine whether IGF-1R expression is associated
with the radio-resistance of SCC cells, we detected the expression
of IGF-1R in several SCC cells by western blotting. As shown in
Fig. 1A, the level of IGF-1R was
relatively higher in H520 cells compared with H1703 and H226 cells.
Therefore, H520 cells was selected for the following experiments.
Lentivirus-mediated shRNA was applied to knockdown the IGF-1R gene.
The shRNA infection efficiency was evaluated using RT-qPCR and
western blotting. The IGF-1R mRNA levels were reduced by 55% in
shIGF1R-1-treated cells compared with cells in the negative control
(NC) group, in which no gene was targeted (Fig. 1B). As shown in Fig. 1C, western blotting was used to examine
the interference efficiency of different shRNA sequences. Compared
with the NC group, IGF-1R expression was also significantly
decreased by approximately 69% in shIGF1R-1-treated H520 cells. The
knockdown IGF1R-1 cell line was called shIGF-1R H520 cells.
Therefore, this sequence (shIGF1R-1) was selected for its effective
knockdown of IGF-1R expression and was used for subsequent
experiments.
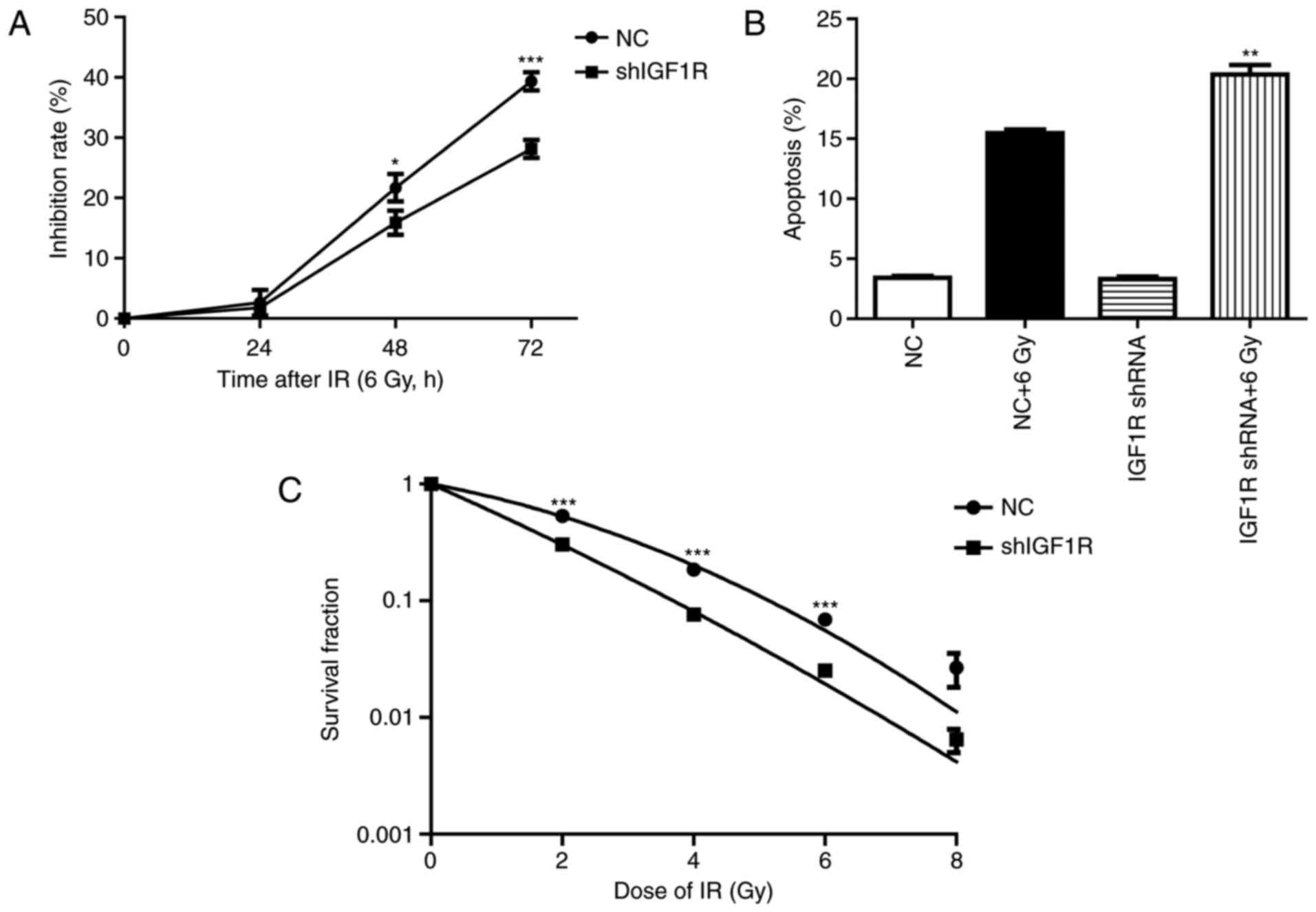
Knockdown of IGF-1R suppression cell
growth, increased apoptosis and enhanced the radio-sensitivity of
H520 cells
To investigated the effect of IGF1R knockdown on the
H520 cells after irradiation. Cell growth was assessed by CCK-8
assay (13). As shown in Fig. 2A, the Inhibition rate of IGF1R
knockdown cells at 48 h were (21.7±1.3) and (15.89±1.2%) in the NC
group cells (P<0.05, n=3). The Inhibition rate of IGF1R
knockdown cells at 72 h were (39.3±0.9) and (28.14±0.9%) in the NC
group cells (P<0.001, n=3). Though the proliferation inhibition
rate were no significant difference between IGF1R knockdown cells
and NC cells after irradiation for 24 h, it has shown increase at
48 h and obviously increased at 72 h in the IGF1R knockdown cells
compared with the NC cells.
To test the effect of IGF-1R knockdown on the
apoptosis induced by irradiation, flow cytometry was used to
investigate the percentage of apoptotic cells. After an irradiation
of 6 Gy for 72 h, the percentage of early apoptosis increased by
78% in the NC group. However, apoptosis increased by 84% after
irradiation in the shIGF-1R H520 group. This result indicates that
knockdown of IGF-1R increased the early apoptosis of H520 cells
after irradiation (P<0.01, n=3; Fig.
2B).
The radio-sensitivity of H520 cells after IGF-1R
knockdown was investigated by clone formation assay (12). Compared with the negative control
group, IGF-1R knockdown significantly enhanced inhibition of clone
formation in H520 cells, which was dependent on the dose of
delivered radiation. These survival fraction results illustrate
that IGF-1R knockdown increased the radio-sensitivity of H520 cells
(P<0.05, compared with NC group; Fig.
2C).
Irradiation induced the activation of
IGF-1R signaling pathways in H520 cells
To study the relationship between IGF-1R and
irradiation, the effect of irradiation on the activation of IGF-1R
and total levels of IGF-1R in H520 cells was assessed. Cells were
treated with irradiation of 6 Gy and harvested at different times
after irradiation (0, 6, 12, 24, 48, 72 and 96 h). As shown in
Fig. 3A, increased levels of
phosphorylated IGF-1R were apparent at 12 h and this was sustained
to 72 h and increased further at 96 h. Meanwhile, the total levels
of IGF-1R were increased at 12 h and this was sustained to 96 h.
This finding further indicated that IGF-1R signaling pathways may
affect radio-sensitivity in H520 cells.
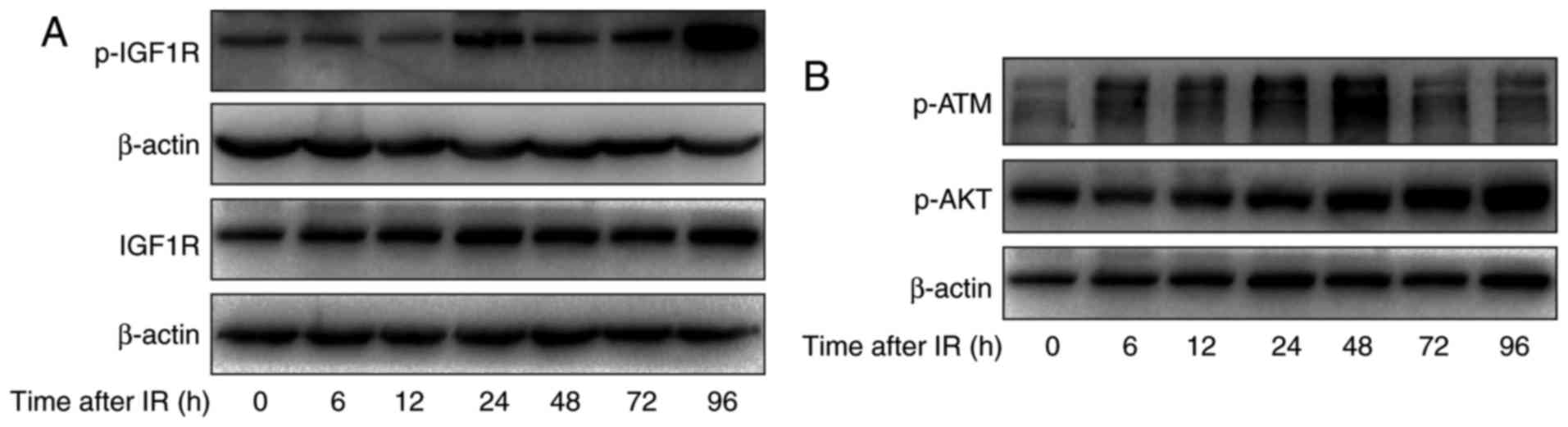 | Figure 3.The effect of irradiation on the
IGF-1R downstream signaling pathway in H520 cells. (A) H520 cells
were irradiated at 6 Gy for 0, 6, 12, 24, 48, 72 and 96 h, and
their protein content was harvested. Western blot analysis of the
expression of p-IGF1R and IGF1R was conducted. (B) H520 cells were
irradiated at 6 Gy for 0, 6, 12, 24, 48, 72 and 96 h, and their
protein content was harvested. Western blot analysis of the
expression of p-ATM and p-AKT was conducted. IGF-1R, insulin-like
growth factor-1 receptor. |
To explore the mechanism of IGF-1R in the regulation
of apoptosis, first, apoptosis-related signaling pathways were
investigated using western blotting. As shown in Fig. 3B, irradiation induced the activation
of ATM in H520 cells at 6 h and this was sustained to 48 h.
Moreover, the phosphorylation of AKT was significantly increased
after irradiation, in a time-dependent manner.
Irradiation induced DNA damage
repair-related protein expression in H520 cells
To further study the effect of irradiation on DNA
damage repair in H520 cells after irradiation with 6 Gy, the levels
of γ-H2AX, the phosphorylated form of H2AX, which can indicate DNA
damage repair, were investigated. As shown in Fig. 4A, after short durations of irradiation
(0–6 h), the levels of γ-H2AX were first increased at 0.5 h, were
reduced substantially by 2 h and were reduced further by 6 h.
However, after irradiation for longer durations (0–96 h), the
γ-H2AX levels were substantially increased from 24 to 96 h, and
remained high at 48 to 96 h. Meanwhile, the levels of
phosphorylation ATM were obviously increase at 30 min after
irradiation. In addition, the levels of 53BP1, another protein
associated with the DNA damage response were also determined, and
the expression of 53BP1 increased at 0.5 h and returned to basal
levels at 6 h after irradiation in H520 cells, which is similar to
the trend of H2AX activation after irradiation. These results
suggest that irradiation induced DNA damage repair at 30 min after
irradiation in H520 cells by inducing phosphorylation of H2AX and
recruiting 53BP1 to the DNA damage sites.
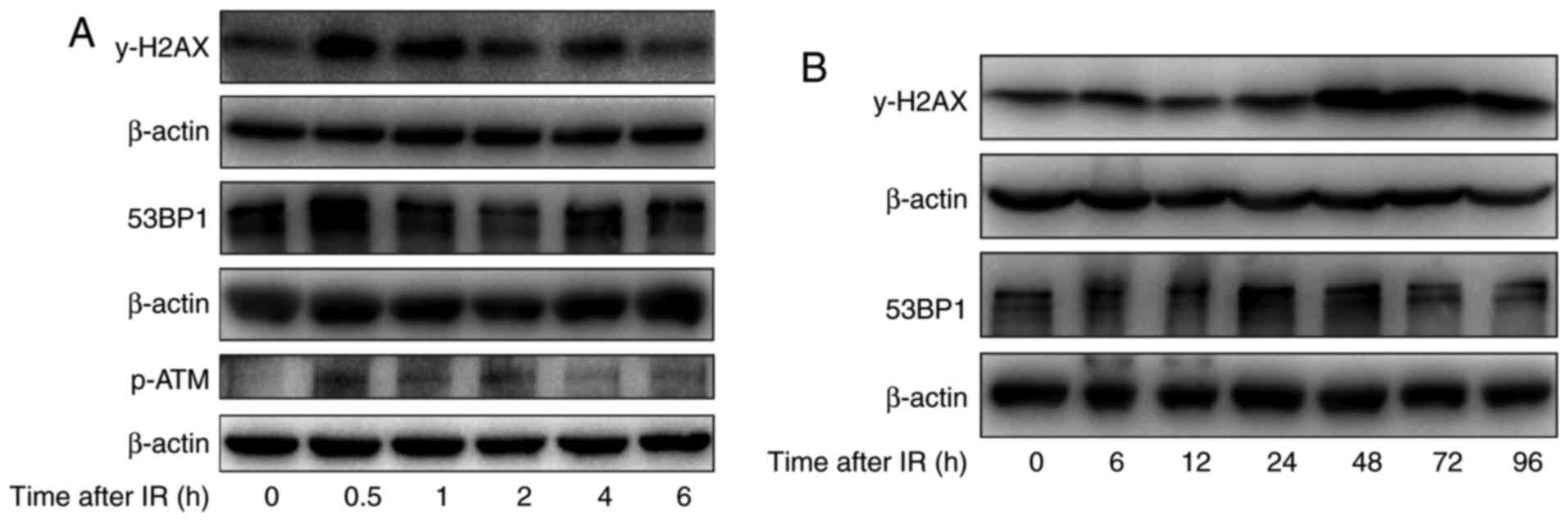 | Figure 4.The effect of irradiation on DNA
damage repair-related proteins in H520 cells. (A) H520 cells were
irradiated at 6 Gy for 0, 0.5, 1, 2, 4 and 6 h, and their protein
content was harvested. Western blot analysis of the expression of
53BP1, p-ATM and γ-H2AX was conducted. (B) H520 cells were
irradiated at 6 Gy for 0, 6, 12, 24, 48, 72 and 96 h, and their
protein content was harvested. Western blot analysis of the
expression of 53BP1 and γ-H2AX was conducted. p-ATM, phosphorylated
ataxia-telangiectasia mutated. |
In addition to its role in DNA damage repair after a
short duration of irradiation, H2AX is also associated with
apoptosis after exposure to irradiation. The western blotting
analysis indicates that irradiation induced the phosphorylation of
H2AX in a time-dependent manner, especially at 48 and 72 h
(Fig. 4B).
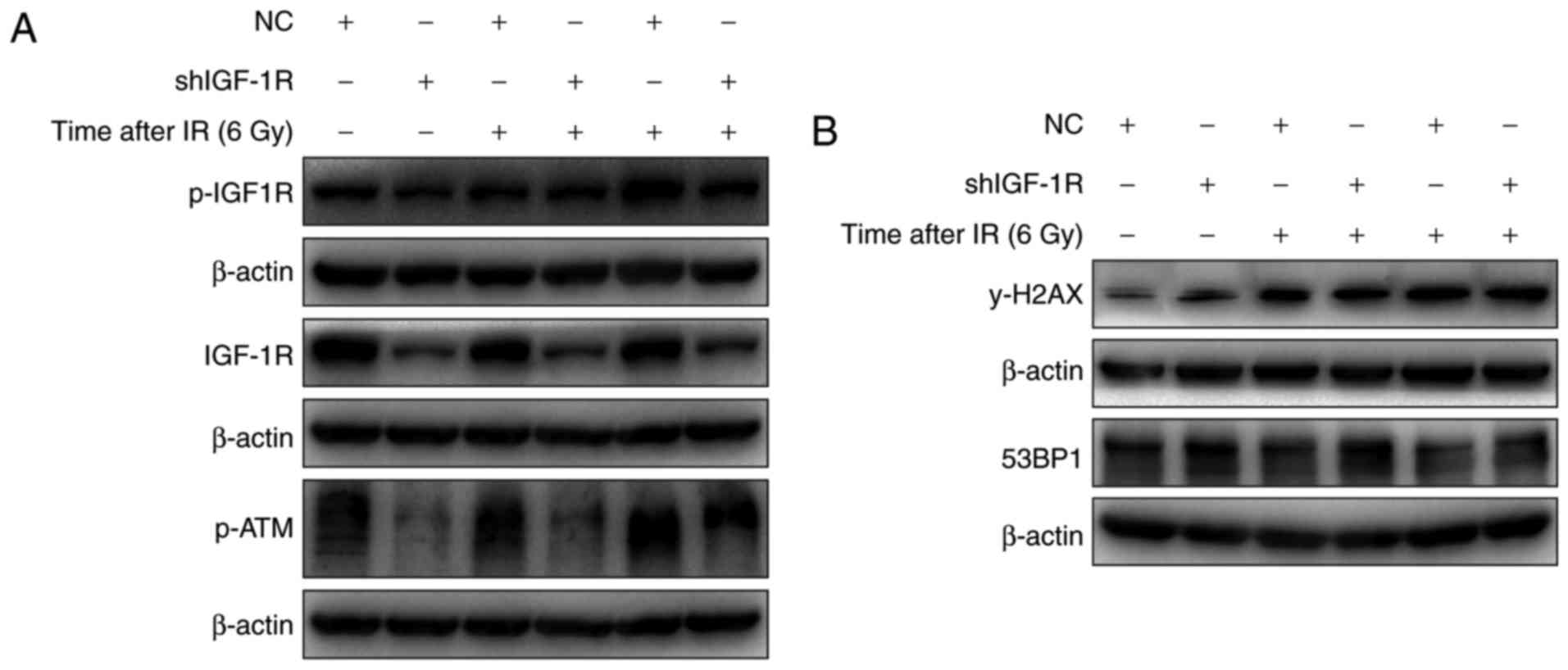
IGF-1R knockdown blocked the
activation of ATM and decreased γ-H2AX levels
The role of ATM activation in the radio-sensitivity
of H520 cells after IGF-1R knockdown was investigated. As shown in
Fig. 5A, irradiation induced the
phosphorylation of IGF-1R in both IGF-1R knockdown and NC cells.
And the levels of phosphorylated IGF-1R were lower in the IGF-1R
knockdown cells compared with the NC cells. Similarly, the
activation of ATM also occurred after irradiation, and the
activation trend was similar to that of IGF-1R phosphorylation.
These results confirm that IGF-IR knockdown regulates the
activation of ATM after irradiation.
The expression of γ-H2AX, which is associated with
apoptosis, was increased in H520 cells after irradiation. To
evaluate the effect of IGF-1R on apoptosis induced by irradiation,
the effect of irradiation on the levels of γ-H2AX after IGF-1R
knockdown was determined using western blotting. As shown in
Fig. 5B, the levels of γ-H2AX were
increased after IGF-1R knockdown in H520 cells, and irradiation
also up-regulated the levels of γ-H2AX after irradiation for 48 and
72 h. However, compared with the NC cells, the levels of γ-H2AX
demonstrated a smaller increase in the IGF-1R knockdown cells after
irradiation. These results illustrate that IGF-1R knockdown
attenuated irradiation-induced apoptosis by up-regulating levels of
γ-H2AX.
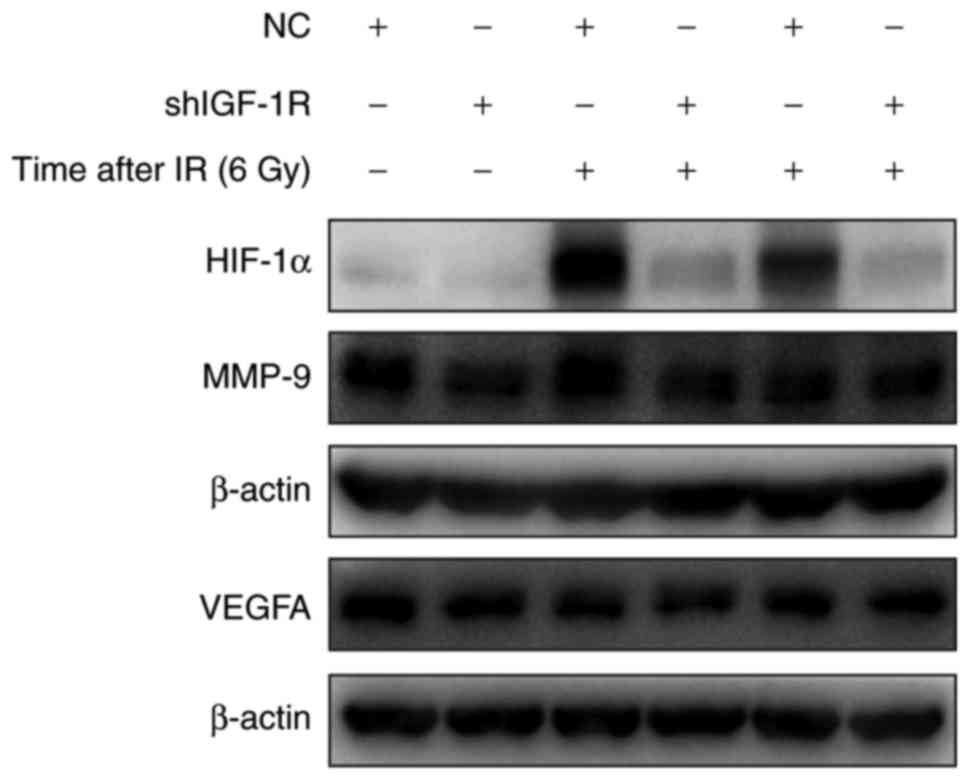
IGF-1R knockdown attenuated
irradiation-induced HIF-1α signaling in H520 cells
HIF-1 is a critical gene in tumor resistance and is
the downstream of IGF-1R. To explore the mechanism of IGF-1R in
radio-resistance of H520 cells, the effect of irradiation on HIF-1α
expression was assessed. As shown in Fig.
6, after IGF-1R knockdown, the expression of HIF-1α was
decreased in H520 cells. In addition, irradiation caused a
significant increase in HIF-1α expression. Compared with the NC
cells, the expression of HIF-1α was significantly down-regulated in
IGF-1R knockdown cells after irradiation. Furthermore, the proteins
derived from downstream genes of HIF-1α, such as MMP-9 and VEGFA,
also showed a similar decrease in IGF-1R knockdown cells. After
irradiation for 48 h, the decrease was more obvious in the IGF-1R
knockdown cells compared with the NC cells.
Discussion
Radiotherapy is frequently applied for the treatment
of lung cancer. However, for many patients, the results are still
unsatisfactory because of radio-resistance. Therefore, exploring
the mechanism underlying treatment resistance is important. Novel
therapeutic approaches, such as up-regulating tumor suppressive
genes or down-regulating oncogenes, which can enhance the
radio-sensitivity or chemo-sensitivity of tumor cells, are urgently
required.
IGF-1R is a transmembrane receptor tyrosine kinase,
which is frequently over-expressed in tumors, and involved in cell
proliferation, protection against apoptosis, cell invasion and
metastasis. IGF-1R is over-expressed in lung squamous carcinoma and
patients with such tumors demonstrate resistance to conventional
anti-cancer treatments including radiotherapy and chemotherapy.
Therefore, IGF-1R has attracted increased attention as an
anti-cancer treatment target (14).
Various strategies have been employed to inhibit IGF-1R expression
and function, including anti-sense oligonucleotides, specific
IGF-1R inhibitors, antibodies to IGF-IR, or dominant negative
IGF-1R mutants, which have been shown to inhibit in vitro or
in vivo growth of these tumors, enable reversal of the
transformed phenotype and induce apoptosis (15–17). Some
researchers have shown that the activation of IGF-1R signaling
pathways induces chemo-resistance in a variety of malignancies
(18). It has been reported that
siRNA, antisense or monoclonal antibody-mediated inhibition of
IGF-1R can enhance the chemo-sensitivity of human esophageal
squamous carcinoma and breast cancer cells (19,20).
However, there have been no reports concerning the correlation
between IGF-1R expression and the radio-resistance of lung squamous
carcinoma cells. In the present study, the therapeutic potential of
a lentivirus-mediated shRNA targeting IGF-1R, combined with
radiotherapy, for the treatment of human lung squamous carcinoma
was investigated. RNA interference with shRNA was used to stably
target IGF-1R, and can effectively down-regulate the expression of
IGF-1R in human H520 lung squamous carcinoma cells.
Exposure to irradiation immediately triggers DSBs,
which results in recruitment of phosphorylated H2AX to the DSB
sites. This is the early response to irradiation-induced DSBs.
Subsequently, DNA damage repair pathways are activated to prevent
cell division and repair the damage to promote cell survival. The
pathways are initiated within a few h after irradiation, and the
phosphorylation of H2AX is crucial for the repair process. However,
cell apoptosis will be induced if the DNA damage is unrepaired.
DSBs are the most important lesion in the induction of apoptosis.
In the current study, it was found that H2AX is phosphorylated at
30 min after irradiation, and returns to basal levels at 4 h after
irradiation. The results indicate that DNA damage repair occurs at
30 min after irradiation in H520 cells, and the phosphorylation of
H2AX at Ser 139 is an early response to DSBs induced by
irradiation. Meanwhile, we found that the levels of phosphorylation
ATM were obviously increase at 30 min after irradiation. In
addition, the levels of 53BP1, another important protein associated
with the DNA damage response were also determined, and the
expression of 53BP1 increased at 30 min and returned to basal
levels at 6 h after irradiation in H520 cells, which is similar to
the trend of H2AX activation after irradiation. Furthermore, it was
demonstrated that the levels of phosphorylated H2AX reached a
second peak at 48 h after irradiation for 6 Gy in H520 cells, and
this process was sustained to 72 h. Phosphorylation of ATM was
activation simultaneously at 48 h after exposuring to irradiation.
These results indicated that the DNA damage repair may evoked at 30
min, and once repair process are failure, the cell will occur cell
cycle arrest and/or apoptosis. Therefor, it will make sense that
H520 cells appear first peak of gamma H2AX at 30 min after
irradiation. When DNA damage exceeds repair, cell will die, but
this process will not be actively accepted by dying cell, we think
residual cell may activate some survival signals to resist death,
such as p-AKT pathway (21). Our
results have also shown that the levels of p-AKT highly increased
at 48 h after irrdiation and may explain the levels of
phosphorylated H2AX reaching a second peak at 48 h after
irradiation. And this hypothesis needs us to further investigated.
In IGF-1R knockdown cells, it was found that the levels of
phosphorylated H2AX were up-regulated compared with NC cells. After
irradiation, the levels of phosphorylated H2AX increased both in
the IGF-1R knockdown cells and NC cells, and the increase was
sustained to 72 h. The apoptosis analyses confirmed that greater
levels of apoptosis were induced in IGF-1R knockdown cells compared
with NC cells after irradiation. These results suggest that the
levels of phosphorylated H2AX induced by irradiation can be
attributed to cell apoptosis after IGF-1R knockdown in H520
cells.
Previous studies have shown that IGF-1R may enhance
DNA repair through activation of IGF-1R/p38 signaling after
irradiation in mouse embryo fibroblasts and breast cancer cells
(22,23). Irradiation can induce DSBs and
activate related kinases, such as ATM, ATR and DNA protein kinase,
which can phosphorylate histone H2AX (24). Furthermore, phosphorylated-H2AX is an
important marker of radiation-induced DSBs and recruits other DNA
repair proteins including 53BP1, MDC1 (mediator of DNA damage check
point protein1) and BRCA1 (breast cancer type 1 susceptibility
protein) to DSB sites (25). 53BP1 is
regulated by ATM after DNA damage. ATM-deficient cells show no
53BP1 hyperphosphorylation and reduced 53BP1 foci formation in
response to radiation compared with cells expressing wild-type ATM.
53BP1 is an ATM substrate that is involved early in the DNA
damage-signaling pathways and is regulated by ATM after irradiation
(26). ATM has a critical role in the
response to DNA damage (27).
However, little research has focused on the association between
IGF-1R and ATM activation. This study is the first to show that
IGF-IR knockdown results in significantly decreased levels of
phosphorylated ATM in lung squamous carcinoma cells. The levels of
phosphorylated ATM in lung squamous carcinoma cells were increased
following irradiation. Furthermore, though the increase was more
obvious in IGF-1R knockdown cells after irradiation, the levels of
phosphorylated ATM were lower in IGF-1R knockdown cells compared
with NC cells. These results suggest that IGF-1R signaling has a
distinct effect on ATM activation, and IGF-1R knockdown enhances
radio-sensitivity by attenuating the activation of ATM/H2AX/53BP1
signaling.
Hypoxia, a tumor-specific microenvironment, is
associated with radiation resistance, because the depletion of
oxygen affects the radiolysis of H2O and reduces the production of
reactive and cytotoxic species, and radiation-induced DNA damage is
fixed under normoxia. HIF-1 has been reported to play a key role in
hypoxia-related radio-resistance (28). HIF-1 is a heterodimeric transcription
factor, including an α-subunit (HIF-1α) and a β-subunit (HIF-1β),
and its activity is mainly dependent on the HIF-1α subunit. HIF-1
binds to its cognate DNA sequence, the hypoxia-responsive element,
and induces the expression of several factors, such as MMP-9 and
VEGF. MMP-9 is involved in the breakdown of extracellular matrix in
physiological processes, including angiogenesis, and tumor cell
migration. Previous studies have reported that EGF/EGFR signaling
activates downstream PI3K/Akt to induce FoxO1 nuclear exclusion,
which activates MMP-9 to promote the invasiveness and metastasis of
NSCLC (29). VEGF has been reported
to not only induce angiogenesis but also protect endothelial cells
from the cytotoxic effects of irradiation and consequently increase
tumor radio-resistance (30). The
hypoxic microenvironment of the tumor is one of the main reasons
underlying radio-resistance (31,32).
Hypoxia can induce the expression of several genes
including HIF-1, VEGF and MMP-9, which regulate the survival and
invasiveness of tumor cells (33–35).
However, the association between IGF-1R and HIF-1α in
radio-resistance has not been fully elucidated. In the current
study, to assess the mechanism of IGF-1R in the radio-sensitivity
of H520 cells, the effect of IGF-1R down-regulation and hypoxia on
the radio-sensitivity of H520 cells was investigated. The results
show that irradiation has an effect on the microenvironment of
tumor cells, in terms of increasing the expression of the
hypoxia-related gene, HIF-1α, after irradiation in H520 cells.
However, the expression of HIF-1α was decreased after IGF-1R
knockdown. Irradiation induced HIF-1α expression, but the increase
was inhibited in IGF-1R knockdown cells compared with NC cells.
These results illustrate that HIF-1α plays an important role in the
response to irradiation, and IGF-1R knockdown increases
radio-sensitivity through decreasing the expression of HIF-1α.
The levels of MMP-9 and VEGF were analyzed using
western blotting: The expression of MMP-9 in H520 cells after
treatment with radiation and IGF-1R knockdown is shown in Fig. 6. The cells showed a decrease in the
expression of MMP-9 upon inhibition of HIF-1α expression. However,
there was no obvious change in the expression of VEGFA because of
IGF-1R silencing. The results reveal that the expression of MMP-9
is influenced by HIF-1α levels, whereby MMP-9 levels appear to
decrease upon a decrease in HIF-1α levels. Previous studies have
reported that both MMP-9 and VEGFA can be influenced by different
levels of HIF-1α. It is possible that the radio-sensitivity of H520
cells is influenced by the hypoxic pathway.
Radioresistance is considered to be a majoy
obstacles of ineffective treatment. Resistance to apoptosis shown
in the presented study maybe one of the reasons. Meanwhile, the
other mechanisms, such as autophagic cell death, may also
considered as a new way resulting to radioresistance. As shown in
various studies, autophagic cell death was considered as the second
type of programmed cell death (36–38). The
occurrence of apoptosis in the presented study does not exclude the
chance of autophagy incidence. Moreover, these two processes may
exist simultaneously (39,40). Some of our results (Data not shown)
have indicated that irradiation induces the occurrence of
autophapy, which was confirmed by an increase expression of
autophagic marker LC3-II. However, more investigations are needed
to further clarify the mechanisms.
In conclusion, the results of the present study
indicated that IGF-1R may play an important role in the
radioresistance of SCC cells. And the underlying mechanism is
probably related to the decreased expression of proteins involved
in ATM/H2AX/53BP1 DNA damage repair and the HIF-1α/MMP-9 hypoxic
pathway, which results in the induction of apoptosis and increased
radio-sensitivity. These findings suggest that targeting of IGF-1R
may represent a new approach for lung SCC radiation treatment.
Acknowledgements
Not applicable.
Funding
This work was funded by a seed fund of Ren Ji
Hospital, School of Medicine, Shanghai Jiao Tong University (grant
no. RJZZ13-026) and the clinical study with combined traditional
Chinese medicine and western medicine of Shanghai Municipal
Committee for health and family planning (grant no.
ZHYY-ZXYJHZX-201610).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XXL performed the experiments, drafted the
manuscript and analyzed the data. HYC assisted with the data
analysis. XX and MY assisted with the experiments. HBC participated
in design of irradiation experiments. LX participated in the cell
irradiation experiments. YLH, JMT and DZ assisted with the
experiments. YRB participated in the study design and coordination
and helped to revised the manuscript. XMM was responsible for the
study design and final approval of the manuscript. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IGF-1R
|
insulin-like growth factor-1
receptor
|
|
SCC
|
lung squamous cell carcinoma
|
|
ATM
|
ataxia-telangiectasia mutated
|
|
H2AX
|
H2A histone family member X
|
|
53BP1
|
p53 binding protein 1
|
|
HIF-1α
|
hypoxia inducible factor 1 alpha
|
|
MMP-9
|
matrix metallopeptidase 9
|
|
NSCLC
|
non-small-cell lung cancer
|
|
DSBs
|
double strand breaks
|
|
MDC1
|
mediator of DNA damage check point
protein1
|
|
BRCA1
|
breast cancer type 1 susceptibility
protein
|
|
shRNA
|
Short hairpin RNA
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dong JC, Gao H, Zuo SY, Zhang HQ, Zhao G,
Sun SL, Han HL, Jin LL, Shao LH, Wei W and Jin SZ: Neuropilin 1
expression correlates with the Radio-resistance of human
non-small-cell lung cancer cells. J Cell Mol Med. 19:2286–2295.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Juchum M, Gunther M and Laufer SA:
Fighting cancer drug resistance: Opportunities and challenges for
mutation-specific EGFR inhibitors. Drug Resist Updat. 20:12–28.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumarakulasinghe NB, van Zanwijk N and Soo
RA: Molecular targeted therapy in the treatment of advanced stage
non-small cell lung cancer (NSCLC). Respirology. 20:370–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valenciano A, Henríquez-Hernández LA,
Moreno M, Lloret M and Lara PC: Role of IGF-1 receptor in radiation
response. Transl Oncol. 5:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao H, Dong W, Shen H, Xu J, Zhu L, Liu Q
and Du J: Combinational therapy enhances the effects of anti-IGF-1R
mAb figitumumab to target small cell lung cancer. PLoS One.
10:e01358442015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takahashi T, Uehara H, Ogawa H, Umemoto H,
Bando Y and Izumi K: Inhibition of EP2/EP4 signaling abrogates
IGF-1R-mediated cancer cell growth: Involvement of protein kinase
C-θ activation. Oncotarget. 6:4829–4844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim JS, Kim ES, Liu D, Lee JJ, Behrens C,
Lippman SM, Hong WK, Wistuba II, Lee E and Lee HY: Activation of
insulin-like growth factor 1 receptor in patients with non-small
cell lung cancer. Oncotarget. 6:16746–16756. 2015.PubMed/NCBI
|
|
9
|
Iwasa T, Okamoto I, Suzuki M, Hatashita E,
Yamada Y, Fukuoka M, Ono K and Nakagawa K: Inhibition of
insulin-like growth factor 1 receptor by CP-751,871 radiosensitizes
non-small cell lung cancer cells. Clin Cancer Res. 15:5117–5125.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morgan MA and Lawrence TS: Molecular
pathways: Overcoming radiation resistance by targeting DNA damage
response pathways. Clin Cancer Res. 21:2898–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wakasugi M, Sasaki T, Matsumoto M, Nagaoka
M, Inoue K, Inobe M, Horibata K, Tanaka K and Matsunaga T:
Nucleotide excision repair-dependent DNA double-strand break
formation and ATM signaling activation in mammalian quiescent
cells. J Biol Chem. 289:28730–28737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sears CR, Cooney SA, Chin-Sinex H,
Mendonca MS and Turchi JJ: DNA damage response (DDR) pathway
engagement in cisplatin radiosensitization of non-small cell lung
cancer. DNA Repair (Amst). 40:35–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Lu P, Liang Z, Zhang Z, Shi W, Cai
X and Chen C: Increased insulin-like growth factor 1 receptor
(IGF1R) expression in small cell lung cancer and the effect of
inhibition of IGF1R expression by RNAi on growth of human small
cell lung cancer NCI-H446 cell. Growth Factors. 33:337–346. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao H, Dong W, Qu X, Shen H, Xu J, Zhu L,
Liu Q and Du J: Metformin enhances the therapy effects of
anti-IGF-1R mAb figitumumab to NSCLC. Sci Rep. 6:310722016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furukawa J, Wraight CJ, Freier SM, Peralta
E, Atley LM, Monia BP, Gleave ME and Cox ME: Antisense
oligonucleotide targeting of insulin-like growth factor-1 receptor
(IGF-1R) in prostate cancer. Prostate. 70:206–218. 2010.PubMed/NCBI
|
|
16
|
Wang J, Huang F, Bai Z, Chi B, Wu J and
Chen X: Curcumol inhibits growth and induces apoptosis of
colorectal cancer LoVo cell line via IGF-1R and p38 MAPK pathway.
Int J Mol Sci. 16:19851–19867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou X, Shen F, Ma P, Hui H, Pei S, Chen
M, Wang Z, Zhou W and Jin B: GSK1838705A, an IGF-1R inhibitor,
inhibits glioma cell proliferation and suppresses tumor growth in
vivo. Mol Med Rep. 12:5641–5646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ramcharan R, Aleksic T, Kamdoum WP, Gao S,
Pfister SX, Tanner J, Bridges E, Asher R, Watson AJ, Margison GP,
et al: IGF-1R inhibition induces schedule-dependent sensitization
of human melanoma to temozolomide. Oncotarget. 6:39877–39890. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y, Zhu C, Peng Z, Dai Y and Gu Y:
Lentivirus-mediated short-hairpin RNA targeting IGF-1R inhibits
growth and lymphangiogenesis in breast cancer. Oncol Rep.
28:1778–1784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao H and Gu X: Silencing of insulin-like
growth factor-1 receptor enhances the radiation sensitivity of
human esophageal squamous cell carcinoma in vitro and in vivo.
World J Surg Oncol. 12:3252014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahlberg SH, Gustafsson AS, Pendekanti PN,
Glimelius B and Stenerlöw B: The influence of AKT isoforms on
radiation sensitivity and DNA repair in colon cancer cell lines.
Tumour Biol. 35:3525–3534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Héron-Milhavet L, Karas M, Goldsmith CM,
Baum BJ and LeRoith D: Insulin-like growth factor-I (IGF-I)
receptor activation rescues UV-damaged cells through a p38
signaling pathway. Potential role of the IGF-I receptor in DNA
repair. J Biol Chem. 276:18185–18192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Héron-Milhavet L and LeRoith D:
Insulin-like growth factor I induces MDM2-dependent degradation of
p53 via the p38 MAPK pathway in response to DNA damage. J Biol
Chem. 277:15600–15606. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Osipov AN, Pustovalova M, Grekhova A,
Eremin P, Vorobyova N, Pulin A, Zhavoronkov A, Roumiantsev S,
Klokov DY and Eremin I: Low doses of X-rays induce prolonged and
ATM-independent persistence of γH2AX foci in human gingival
mesenchymal stem cells. Oncotarget. 6:27275–27287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurashige T, Shimamura M and Nagayama Y:
Differences in quantification of DNA double-strand breaks assessed
by 53BP1/γH2AX focus formation assays and the comet assay in
mammalian cells treated with irradiation and N-acetyl-L-cysteine. J
Radiat Res. 57:312–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baldock RA, Day M, Wilkinson OJ, Cloney R,
Jeggo PA, Oliver AW, Watts FZ and Pearl LH: ATM localization and
heterochromatin repair depend on direct interaction of the
53BP1-BRCT2 domain with γH2AX. Cell Rep. 13:2081–2089. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lima M, Bouzid H, Soares DG, Selle F,
Morel C, Galmarini CM, Henriques JA, Larsen AK and Escargueil AE:
Dual inhibition of ATR and ATM potentiates the activity of
trabectedin and lurbinectedin by perturbing the DNA damage response
and homologous recombination repair. Oncotarget. 7:25885–25901.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ghattass K, Assah R, El-Sabban M and
Gali-Muhtasib H: Targeting hypoxia for sensitization of tumors to
radio- and chemotherapy. Curr Cancer Drug Targets. 13:670–685.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pei J, Lou Y, Zhong R and Han B: MMP9
activation triggered by epidermal growth factor induced FoxO1
nuclear exclusion in non-small cell lung cancer. Tumour Biol.
35:6673–6678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyasaka A, Oda K, Ikeda Y, Sone K, Fukuda
T, Inaba K, Makii C, Enomoto A, Hosoya N, Tanikawa M, et al:
PPI3K/mTOR pathway inhibition overcomes radioresistance via
suppression of the HIF1-α/VEGF pathway in endometrial cancer.
Gynecol Oncol. 138:174–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harada H: Hypoxia-inducible factor
1-mediated characteristic features of cancer cells for tumor
radioresistance. J Radiat Res. 57(Suppl 1): i99–i105. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Helbig L, Koi L, Brüchner K, Gurtner K,
Hess-Stumpp H, Unterschemmann K, Pruschy M, Baumann M, Yaromina A
and Zips D: Hypoxia-inducible factor pathway inhibition resolves
tumor hypoxia and improves local tumor control after single-dose
irradiation. Int J Radiat Oncol Biol Phys. 88:159–166. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fotia C, Massa A, Boriani F, Baldini N and
Granchi D: Hypoxia enhances proliferation and stemness of human
adipose-derived mesenchymal stem cells. Cytotechnology.
67:1073–1084. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fu Z, Chen D, Cheng H and Wang F:
Hypoxia-inducible factor-1α protects cervical carcinoma cells from
apoptosis induced by radiation via modulation of vascular
endothelial growth factor and p53 under hypoxia. Med Sci Monit.
21:318–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao Q, Guo M, Zeng W, Wang Y, Yang L, Pang
X, Li H, Suo Y, Jiang X and Yu C: Matrix metalloproteinase 9
secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell
migration in vitro. Stem Cells Int. 2015:8363902015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ondrej M, Cechakova L, Durisova K, Pejchal
J and Tichy A: To live or let die: Unclear task of autophagy in the
radiosensitization battle. Radiother Oncol. 119:265–275. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Milczarek M, Wiktorska K, Mielczarek L,
Koronkiewicz M, Dąbrowska A, Lubelska K, Matosiuk D and Chilmonczyk
Z: Autophagic cell death and premature senescence: New mechanism of
5-fluorouracil and sulforaphane synergistic anticancer effect in
MDA-MB-231 triple negative breast cancer cell line. Food Chem
Toxicol. 111:1–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jo GH, Bögler O, Chwae YJ, Yoo H, Lee SH,
Park JB, Kim YJ, Kim JH and Gwak HS: Radiation-induced autophagy
contributes to cell death and induces apoptosis partly in malignant
glioma cells. Cancer Res Treat. 47:221–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ryan F, Khodagholi F, Dargahi L,
Minai-Tehrani D and Ahmadiani A: Temporal pattern and crosstalk of
necroptosis markers with autophagy and apoptosis associated
proteins in ischemic hippocampus. Neurotox Res. Jan 8. 2018, (Epub
ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suzuki R, Kang Y, Li X, Roife D, Zhang R
and Fleming JB: Genistein potentiates the antitumor effect of
5-Fluorouracil by inducing apoptosis and autophagy in human
pancreatic cancer cells. Anticancer Res. 34:4685–4692.
2014.PubMed/NCBI
|