Introduction
Allergic rhinitis (AR) is defined as an inflammation
of the lining of the nose and affects up to 40% of the population.
It is characterized by nasal symptoms, including congestion,
sneezing, itching and rhinorrhea. AR is the most common cause of
inflammation of the nasal mucosa (1,2).
Immunoglobulin E (IgE) is a classical antibody that mediates
allergic reactions by sensitizing mast cells and basophils to
allergen activation through binding tightly to a specific receptor
(High-affinity receptor I, FcεRI) (3)
on these cells (4). As the most
common form of non-infectious rhinitis, it is also associated with
an IgE-mediated immune response against allergens (5).
Nucleotide binding and oligomerization domain-like
receptor family pyrin domain-containing 3 (NLRP3) inflammasome
activation is an integral part of the innate immune response in
inflammatory disease (6). The NLRP3
inflammasome is present in variety of cells, including macrophages,
neutrophils, T cells and B cells (7).
The NLRP3 inflammasome is a protein scaffolding complex consisting
of NLRP3, caspase-1 and the adaptor molecule apoptosis-associated
speck-like protein containing a C-terminal caspase recruitment
domain that induces secretion of interleukin-1β (IL-1β) and IL-18
(8,9).
Numerous mechanisms underlying the activation of the NLRP3 have
been identified, including ion channel gating and excessive
reactive oxygen species (ROS) generation (10). The idea that the activation of the
NLRP3 inflammasome was caused by an increase in ROS production was
accepted and it was hypothesized that the NLRP3 inflammasome may be
a general sensor for changes in cellular oxidative stress (11,12).
AR involves systemic inflammation in addition to
nasal inflammation (13). Levels of
acute phase reactants, including high sensitivity C-reactive
protein (14–16), fibrinogen, alpha 1-glycoprotein, alpha
1-antichymotrypsin (16), are not
statistically different between patients with AR and healthy
controls (5,7–9). However,
serum amyloid A (15) and
ceruloplasmin oxidase activity (17)
were higher in AR.
IL-1β is a member of the IL-1 family of ligands.
Although IL-1-targeted drugs are effective against autoinflammatory
disease, they are less effective against autoimmune diseases. Both
AR and asthma are allergen-mediated disorders, so AR shares several
pathogenic similarities with asthma (18). Previous studies have reported on the
pathological roles of IL-1β and the NLRP3 inflammasome in the
development of allergic asthma, though the results were conflicting
(19–23). Nevertheless, the involvement of IL-1β
in AR has not been clearly examined. The present study was designed
to examine the differences in serum IL-1β and PBMCs between
patients with AR and the healthy controls, in order to assess
whether IL-1β participates in the pathological process of AR.
Patients and methods
Patients
A total of 45 patients (Table I), with persistent moderate-severe AR,
and 23 healthy controls were included in the present study. Between
March 2016 and October 2016, these participants were screened at
the Ear, Nose and Throat Department, First Affiliated Hospital of
Jinan University (Guangdong, China). Written, informed consent was
provided according to the procedure approved by the Research Ethics
Committee of the First Affiliated Hospital of Jinan University.
Inclusion criteria for the present study were as follows: Patients
had a detailed clinical history of nasal obstruction, sneezing,
itching, and/or rhinorrhea, sensitization to a minimum of one
perennial allergen and had experienced these symptoms for ≥2 years.
Exclusion criteria were: Nasal polyposis, chronic rhinosinusitis,
excessive septal deviation, bronchial asthma, current smoking of
cigarettes, cardiovascular disease, obesity, diabetes, and other
systemic diseases. Blood samples were gathered from each
participant. Whole blood was centrifuged at 2,451 × g for 15 min at
4°C twice, and sera was collected from the supernatant. Sera
samples were stored at −80°C for subsequent analysis.
 | Table I.Patient clinical characteristics. |
Table I.
Patient clinical characteristics.
| Characteristic | Healthy control
(n) | Allergic rhinitis
(n) |
|---|
| Sex |
|
Male | 13 | 13 |
|
Female | 10 | 32 |
| Age (years) | 28.5±8.2 | 32.3±12.4 |
| AR duration
(years) | Not applicable | 6.31±2.12 |
PBMCs collection and stimulation
PBMCs were isolated by Ficoll gradient
centrifugation and diluted with an equal amount of PBS, overlaid on
Ficoll medium (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA),
and centrifuged at 600 × g for 30 min at 20°C. The PBMCs bands were
aspirated, washed twice with PBS, and re-suspended in cell freezing
medium (90% FBS, Lanzhou Minhai Bio-Engineering, Gansu, China; cat.
no. SA201.02, 10% DMSO; cat. no. D2660, Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) prior to gradient cooling and being stored in
liquid nitrogen for later use. Following the collection of PBMCs
samples, PBMCs were stimulated with 100 ng/ml LPS (cat. no. L4516;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) alone, or with LPS
(100 ng/ml) plus Brefeldin A (dilution, 1:1,000; cat no. 420601;
BioLegend, Inc., San Diego, CA, USA) for 5 h at 37°C. PBMCs then
were collected and stained (30 min, 4°C) with PerCP-A-conjugated
anti-human CD14 (dilution, 1:200; cat no. 325631; BioLegend, Inc.)
and Pacific Blue-conjugated anti-human IL-1β (dilution, 1:200; cat
no. 511710; BioLegend, Inc.) prior to flow cytometric analysis,
which stimulated with LPS added Brefeldin A. For other samples,
which were stimulated with LPS alone, the cell culture supernatants
were gathered and stored at −80°C for later IL-1β concentration
detection.
Flow cytometric analysis
IL-1β levels in lipopolysaccharides (LPS) stimulated
PBMCs were measured through flow cytometry using a BD
Cytofix/Cytoperm™ Fixation/Permeablization kit (cat no.
554714; BD Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer's protocol. Once LPS was stimulated, PBMCs were
harvested for immunofluorescent staining. A total of
~106 cells were stained using 50 µl staining buffer
(PBS+1% FBS) with PerCP-A-conjugated anti-human CD14 dilution
(1:200; cat no. 325631; BioLegend, Inc.) for 30 min at 4°C. Cells
were protected from light throughout staining and storage. The
cells were washed twice with PBS and pelleted using centrifugation
for 10 min at 4°C (250 × g). The cells were resuspended and 250 µl
Fixation/Permeabilization solution was added and incubated for 20
min at 4°C. The cells were washed twice with 1× BD
Perm/Wash™ buffer staining in tubes. Then the cells were
resuspended and fixed/permeabilized in 50 µl 1× BD Perm/Wash buffer
with a pre-determined optimal concentration of IL-1β antibody
(dilution, 1:200; cat no. 511710; BioLegend, Inc.) and incubated at
4°C for 30 min in the dark. The cells were washed again twice with
1× BD Perm/Wash™ buffer and resuspended in staining
buffer prior to flow cytometric analyzed using the BD
FACSVerse™ and BD FACSuite™ Software v1.0.5
(BD Biosciences) and FlowJo software v 10.0.6 (FlowJo LLC, Ashland,
OR, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA was extracted from PBMCs using
TRIzol® reagent (cat no. 15596018; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), as described in our
previous study (24), RNA was
analyzed by RT-qPCR using Power SYBR Green PCR Master Mix on a
Real-Time PCR system (cat no. RR820A; Takara Bio, Inc., Otsu,
Japan). The reaction mixture (20 µl) contained 0.5 µl sense primer
(10 µmol/l), 0.5 µl antisense primer (10 µmol/l), 10 µl SYBR-Green,
8 µl H2O and 1 µl cDNA. The thermocycling conditions
were as follows: 95°C for 5 sec and 60°C for 30 sec for 40
circulations. Analysis of the relative gene expression data using
RT-qPCR and the 2−ΔΔCq method (25). Relative expression levels were
normalized to those of GAPDH. The primer sequences are listed in
Table II. Expression was calculated
as relative to GAPDH. All primers were purchased from Sangon
Biotech Co., Ltd., (Shanghai, China).
| Table II.Primer sequences used for RT-qPCR
analysis. |
Table II.
Primer sequences used for RT-qPCR
analysis.
| Primer | Sense (5′-3′) | Antisense
(5′-3′) |
|---|
| NLRP3 |
5′-TAGGTTGAGGTGCTTTGCCA-3′ |
5′-AGACACACTTCCCCAGCATT-3′ |
| IL-1β |
5′-TGAACTGAAAGCTCTCCACC-3′ |
5′-TCTTTCAACACGCAGGACAG-3′ |
| GAPDH |
5′-TCACCAGGGCTGCTTTTAAC-3′ |
5′-TGACGGTGCCATGGAATTTG-3′ |
Mitochondrial ROS levels detected in
PBMCs
PBMCs isolated from AR (n=11) and healthy controls
(n=11) were primed with LPS (100 ng/ml) for 5 h, and then
stimulated with ATP (1 mmol/l; cat no. A6419; Sigma-Aldrich, Merck
KGaA) for 1 h in the absence or presence of Mito-TEMPO (200 µmol/l;
cat no. SML0737; Sigma-Aldrich; Merck KGaA) at 37°C. Following
this, the cells were stained with 5 µmol/l MitoSOX (cat no. M36008,
Invitrogen; Thermo Fisher Scientific, Inc.) for 10 min at 37°C,
protected from light, gated for the CD14+ population,
and analyzed by flow cytometry.
Cytokine analysis
The plasma samples and cell culture supernatants
were analyzed using ELISA kits (IL-1β, cat no. 437004; IL-17A cat
no. 433914; BioLegend, Inc.). The sera were brought to room
temperature, according to the manufacturer's protocol, for the
simultaneous detection of IL-1β and IL-17 in a single sample.
Statistical analysis
The results are expressed as the means ± standard
error, except the age of participants, which were expressed as the
means ± standard deviation. Differences between groups were
evaluated by one-way analysis of variance. Spearman's correlation
analysis was used to assess associations between variables.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed using IBM SPSS
version 20.0 software (IBM Corp., Armonk, NY, USA).
Results
The expression of NLRP3 and
proinflammatory cytokines increased in the PBMCs of patients with
AR
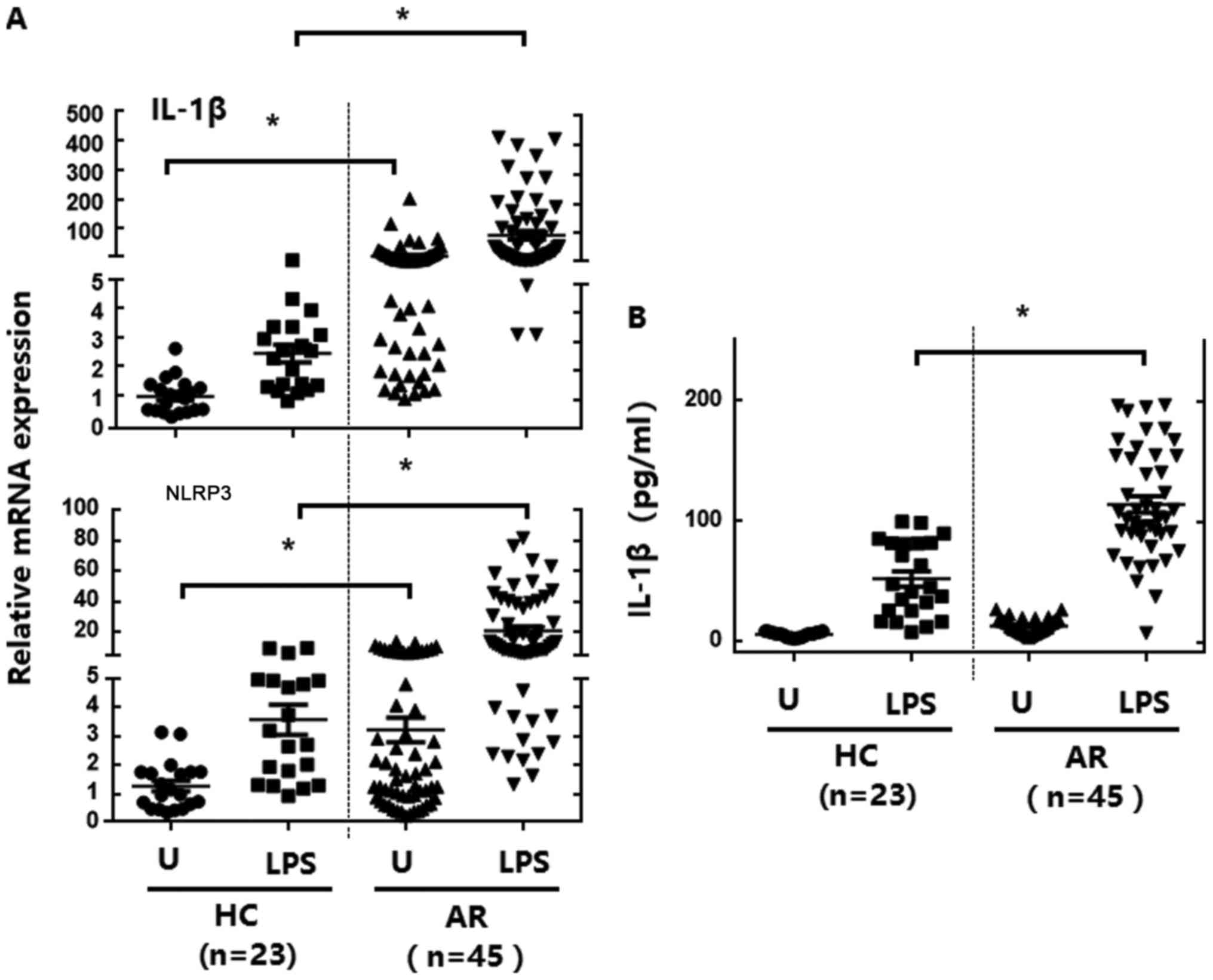
The mRNA expression of IL-1β and NLRP3 in PBMCs from
the 45 patients with AR and 23 healthy controls was detected prior
to and following stimulation with LPS (a TLR4 ligand). Basal levels
of IL-1β and NLRP3 mRNA expression in PBMCs were significantly
upregulated in patients with AR (Fig.
1A). Following LPS stimulation, IL-1β and NLRP3 mRNA expression
levels were significantly increased in PBMCs from patients with AR,
compared with in cells from the healthy controls (Fig. 1A). Basal and LPS-induced production of
IL-1β was significantly elevated in PBMCs (Fig. 1B) from patients with AR. These data
suggest that patients with AR exhibit upregulated inflammatory
cytokine production and NLRP3 expression in their PBMCs, compared
with in those from healthy controls.
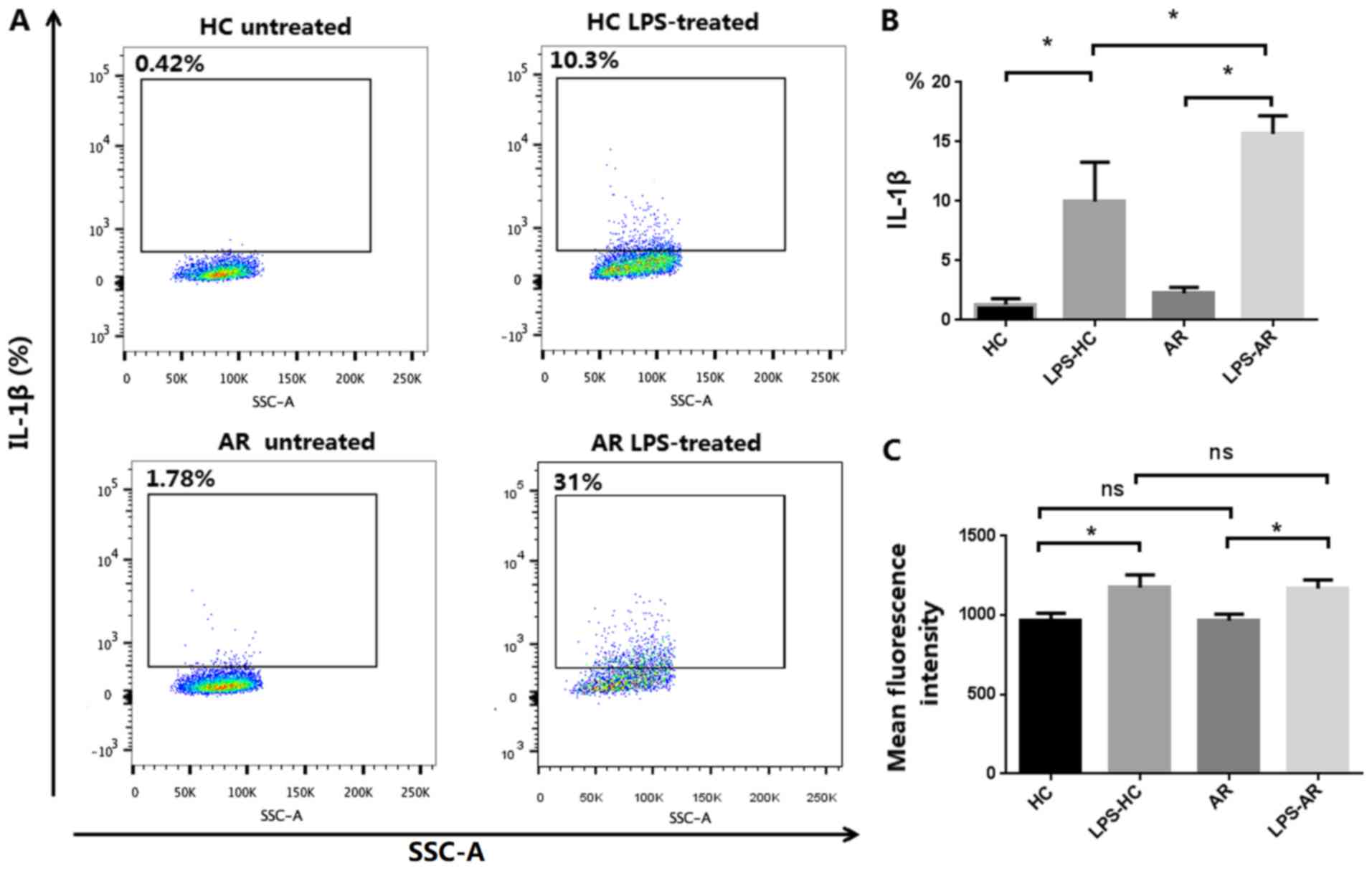
Upregulated IL-1β activation in
monocytes/macrophages and PBMCs in patients with AR
Human monocytes exhibit constitutive inflammasome
activation (26), thus the activation
of IL-1β in monocytes/macrophages was assessed. As shown in
Fig. 2A and B, patients with AR
exhibited a significantly elevated level of IL-1β in LPS-primed
monocytes/macrophages compared with the non-AR healthy controls.
Production of IL-1β was significantly upregulated in PBMCs from
patients with AR, as determined using ELISAs (Fig. 1B). The results indicate that IL-1β
activation is upregulated in the monocytes/macrophages and PBMCs of
patients with AR.
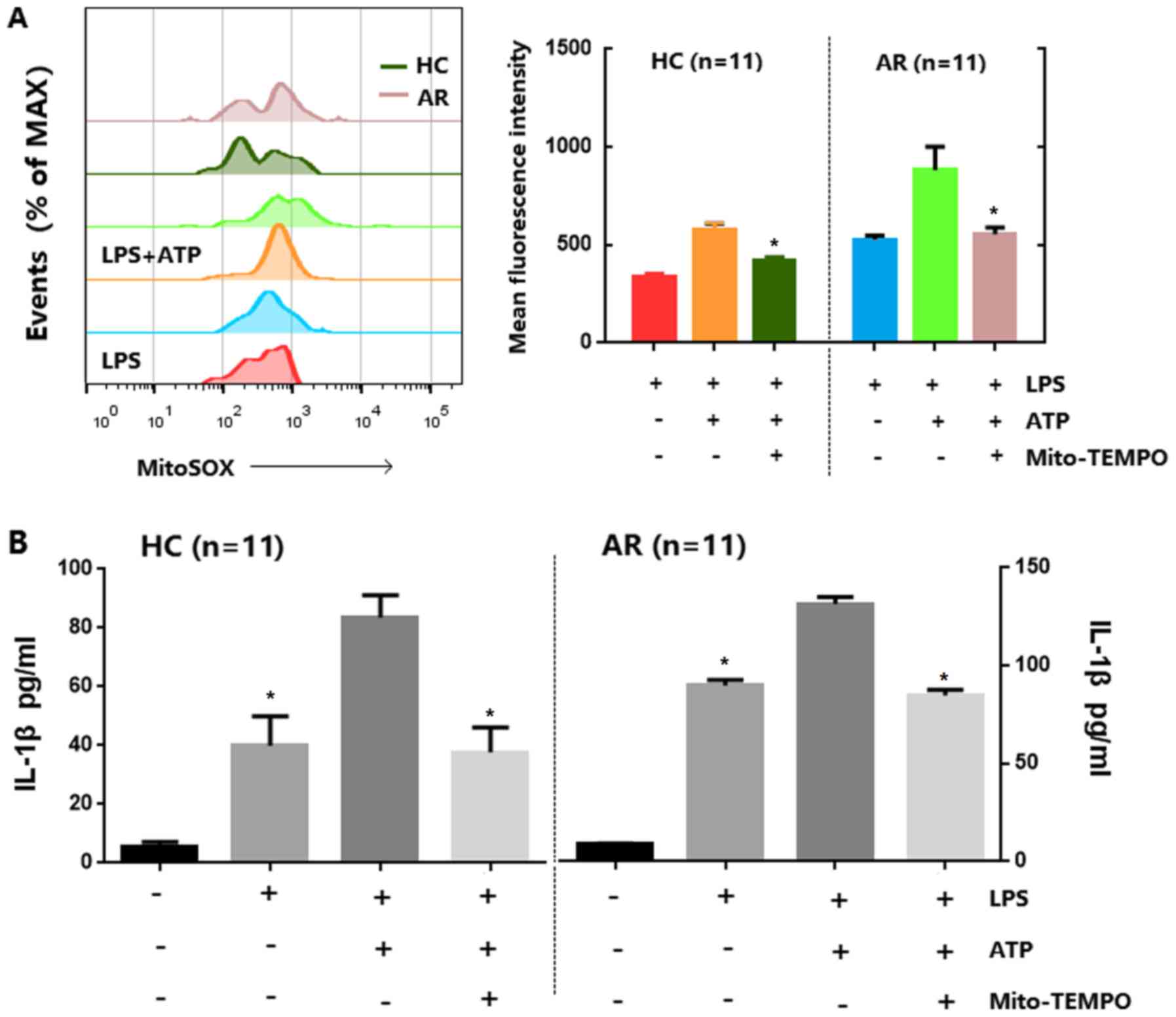
Mitochondrial ROS and NLRP3 are
required for IL-1β synthesis in monocytes/macrophages and PBMCs in
patients with AR
Studies have demonstrated that ROS derived from
mitochondria have been involved in NLRP3 inflammasome activation
(27,28), which could be indirectly implicated in
IL-1β production. Therefore, the present study investigated whether
inflammasome stimuli enhance mitochondrial ROS generation in
CD14+ cell fractions of PBMCs, and whether this was
greater in patients with AR, compared with in healthy controls. As
presented in Fig. 3A, LPS-primed
PBMCs were pretreated with ATP alone or ATP plus Mito-TEMPO, which
inhibits mitochondrial ROS production, and the results revealed
that the generation of mitochondrial ROS in LPS-primed PBMCs
stimulated with ATP was higher in cells from patients with AR than
in cells from healthy controls and ATP-induced IL-1β secretion in
LPS-primed PBMCs were inhibited in Mito-TEMPO groups. Subsequently,
it was investigated whether mitochondrial ROS are required for
IL-1β secretion in PBMCs. As shown in Fig. 3B, the mitochondria-targeting
antioxidant Mito-TEMPO was used, which could inhibit mitochondrial
ROS production to pre-treat PBMCs. Mito-TEMPO was shown to
significantly inhibit ATP-induced IL-1β secretion in LPS-primed
PBMCs in the examined groups (n=11). These results suggest that the
monocyte/macrophage fractions of PBMCs from patients with AR
exhibit raised mitochondrial ROS levels.
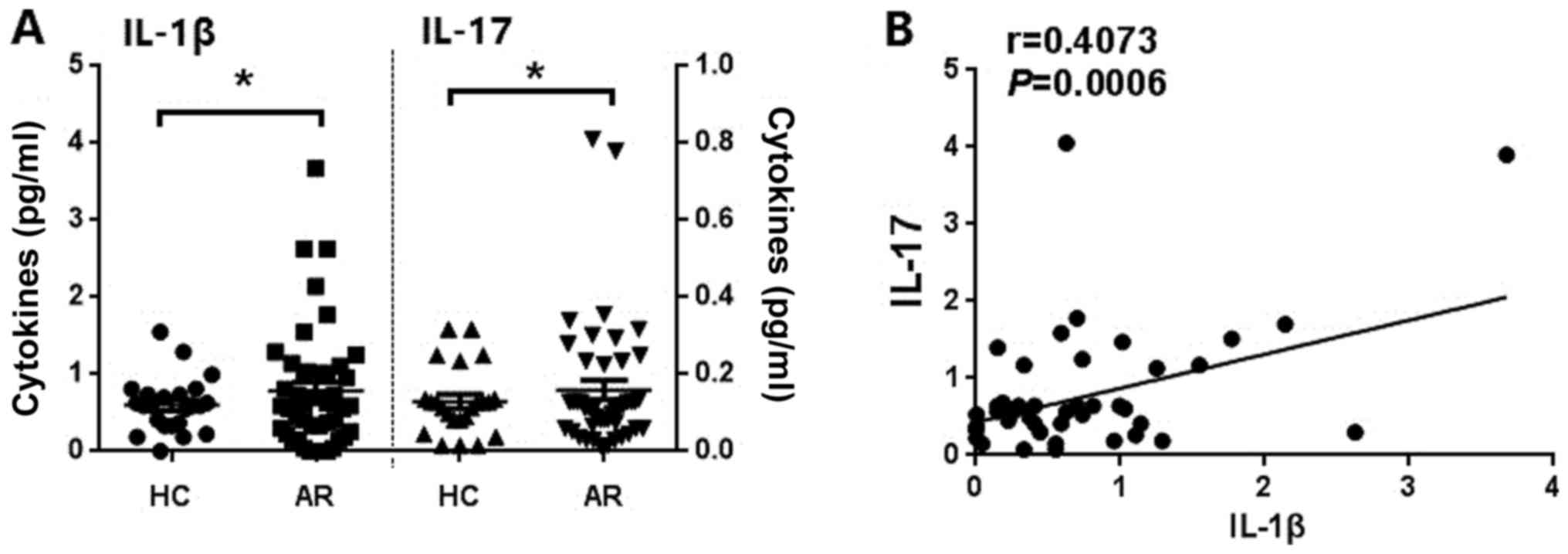
Levels of IL-1β and IL-17 are
increased in patients with AR and are positively correlated with
each other
Previous studies in patients with AR indicated that
IL-17 is also likely to be involved in the pathogenesis of AR
(29–33). Potential differences in the basal
level of IL-1β in patients with AR and healthy controls, whilst
simultaneously detecting the serum IL-17 levels in patients with AR
were investigated. As shown in Fig.
4, patients with AR had increased serum levels of IL-1β and
IL-17 (Fig. 4A) when compared with
the healthy controls. Serum IL-1β and IL-17 were also identified to
be positively correlated with one other (Fig. 4B). These data suggest that IL-1β may
be involved in the development of AR by inducing the production of
IL-17.
Discussion
AR affects up to 40% of the global population, and
its incidence is increasing (34–36). The
symptoms of AR are inconvenient for the sufferer, and AR is a
strong risk factor for other chronic respiratory diseases,
including chronic rhinosinusitis and asthma. Characteristics of AR
include an inflammatory reaction sustained by T-helper 2 cell (Th2)
polarization (37). The Th bias is
evidenced by the PBMCs of patients with AR. Th2 cells
preferentially produce the cytokine IL-4 that promotes IgE
production and inhibits Th1 (38,39). The
dichotomy in function between Th1 and Th2 cells has been modified
by the discovery of another T-lymphocyte subset, namely Th17 cells
(40). IL-17 is one of the cytokines
produced by Th17 cells. IL-17 could recruit macrophages and had
also been identified as a survival factor for airway macrophages
(41). Serum levels of IL-17 are
elevated in a number of disorders, including rheumatoid arthritis
(42), acute hepatic injury (43) and AR (31). Conversely, specific studies regarding
serum IL-17 levels in patients with AR have presented conflicting
results, which may be explained by varying clinical characteristics
of patients and the type of sensitization used between studies
(32,44,45).
In the present study, patients with AR were
identified as having higher serum IL-17 levels compared with the
healthy controls. In obesity-associated asthma, IL-17A was
hypothesized to cause airway hyper-reactivity (46). Furthermore, IL-17 serves a critical
role in, and is associated with, the clinical severity of AR
(31,32). IL-17 producing T cells are associated
with polysensitization in patients with AR, though not with
bronchial hyper-responsiveness (47).
The results of the present study revealed that NLRP3
and IL-1β expression in PBMCs from patients with AR were elevated
compared with the healthy controls. There was also an upregulated
maturation of IL-1β activation in monocytes/macrophages and PMBCs
in patients with AR. A previous study demonstrated that nitric
oxide could sustain IL-1β expression in human dendritic cells by
enhancing their capacity to induce IL-17-production (48). T-regulatory cells highly expressed
IL-1R1, and IL-1β could induce prominent activation of p38 and
c-Jun N-terminal kinases, which are involved in IL-17 production
(49). IL-1β directly causes airway
hyper-reactivity by inducing IL-17A production in
obesity-associated asthma (46). This
was corroborated by the results of the present study, which
identified a positive correlation between serum IL-1β and IL-17
levels in patients with AR. Therefore, it is hypothesized that the
high levels of serum IL-1β may have a role in the pathogenesis of
AR through inducing IL-17-production.
There is evidence to suggest that an excess of ROS
serve an important role in the pathogenesis of airway inflammation
(50–53). Furthermore, previous studies have
suggested that abnormalities in mitochondria are associated with
the development of asthma (54,55) and
that mitochondrial ROS serve critical roles in the pathogenesis of
allergic airway inflammation through modulation of NLRP3
inflammasome activation (56).
However, the association between mitochondrial ROS generation and
AR is not well understood. In the present study,
monocyte/macrophage fractions of PBMCs from patients with AR
demonstrated increased production of mitochondrial ROS prior to and
following treatment with NLRP3 inflammasome stimuli, suggesting
that the elevated production of mitochondrial ROS affects NLRP3
inflammasome activation in patients with AR. Following inhibition
of mitochondrial ROS by Mito-TEMPO, significantly decreased IL-1β
secretion from LPS-primed PBMCs from patients with AR was observed,
suggesting that mitochondrial ROS are responsible for NLRP3
inflammasome activation in AR. Mitochondrial ROS production
increases regularly due to defective mitochondrial homeostasis in
macrophages, which render mitochondria more susceptible to damage
by inflammasome stimuli (28).
Furthermore, mitochondrial membrane permeability transition and
mitochondrial ROS generation are required for the activation of
caspase-1 and IL-β secretion (28).
Therefore, mitochondria ROS serve an important role in the
upregulation of inflammatory responses via acting as
signal-transduction molecules (57).
Although the data of the present study suggest that
activation of the NLRP3 inflammasome is associated with AR
pathogenesis, it has its limitations. At present, the roles of
inflammatory cytokines in AR primarily focus on nasal local
inflammation in patients with AR, which is highly significant
compared with systemic inflammation. The level of systemic
inflammation and local inflammation is not consistent between a
number of diseases. Therefore, further experiments are required to
verify the exact role of IL-1β in AR. However, despite these
limitations, the findings of the present study may provide novel
avenues for anti-AR therapies, as well as for a variety of
inflammatory diseases.
Acknowledgements
Not applicable.
Funding
National Natural Science Foundation of China (grant
no. 3150050081), China Postdoctoral Science Foundation (grant no.
2015M572411), Natural Science Foundation of Guangdong Province,
China (grant no. 2015A030313312), and Foundation Sciences Jinan
University (grant no. 11615479).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HJ and HY initially proposed the study. Wrote the
protocol: QS, ZL, DL and GC. Wrote the manuscripts: QS. Collected
the samples and data: QS, ZL, DL, GC, QW. Analyzed the data: QS,
SL, HJ. Reviewed drafts of the paper: QS, GC, SL, HY.
Ethics approval and consent to
participate
The present study was approved by Research Ethics
Committee of the First Affiliated Hospital of Jinan University
[approval no. 2016 (022)]. All patients provided written informed
consent for participation in the present study.
Consent for publication
Informed consent was obtained from all patients
included in the present study for the publication of the associated
data and the accompanying images.
Competing interests
The authors declare that they have no compeing
interests.
References
|
1
|
Varshney J and Varshney H: Allergic
rhinitis: An overview. Indian J Otolaryngol Head Neck Surg.
67:143–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greiner AN, Hellings PW, Rotiroti G and
Scadding GK: Allergic rhinitis. Lancet. 378:2112–2122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hong JY, Bae JH, Lee KE, Kim M, Kim MH,
Kang HJ, Park EH, Yoo KS, Jeong SK, Kim KW, et al: Antibody to
FcεRIα suppresses immunoglobulin E binding to high-affinity
receptor i in allergic inflammation. Yonsei Med J. 57:1412–1419.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gould HJ, Sutton BJ, Beavil AJ, Beavil RL,
McCloskey N, Coker HA, Fear D and Smurthwaite L: The biology of IGE
and the basis of allergic disease. Annu Rev Immunol. 21:579–628.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ciprandi G, Marseglia GL, Castagnoli R,
Valsecchi C, Tagliacarne C, Caimmi S and Licari A: From IgE to
clinical trials of allergic rhinitis. Expert Rev Clin Immunol.
11:1321–1333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Segovia JA, Chang TH, Winter VT, Coalson
JJ, Cagle MP, Pandranki L, Bose S, Baseman JB and Kannan TR: NLRP3
is a critical regulator of inflammation and innate immune cell
response during mycoplasma pneumoniae infection. Infect Immun.
86:pii: e00548. –17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pétrilli V, Dostert C, Muruve DA and
Tschopp J: The inflammasome: A danger sensing complex triggering
innate immunity. Curr Opin Immunol. 19:615–622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Agostini L, Martinon F, Burns K, McDermott
MF, Hawkins PN and Tschopp J: NALP3 forms an IL-1beta-processing
inflammasome with increased activity in Muckle-Wells
autoinflammatory disorder. Immunity. 20:319–325. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dubyak GR: P2×7 receptor regulation of
non-classical secretion from immune effector cells. Cell Microbiol.
14:1697–1706. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding W, Guo H, Xu C, Wang B, Zhang M and
Ding F: Mitochondrial reactive oxygen species-mediated NLRP3
inflammasome activation contributes to aldosterone-induced renal
tubular cells injury. Oncotarget. 7:17479–17491. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abais JM, Zhang C, Xia M, Liu Q, Gehr TW,
Boini KM and Li PL: NADPH oxidase-mediated triggering of
inflammasome activation in mouse podocytes and glomeruli during
hyperhomocysteinemia. Antioxid Redox Signal. 18:1537–1548. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bousquet J, Khaltaev N, Cruz AA, Denburg
J, Fokkens WJ, Togias A, Zuberbier T, Baena-Cagnani CE, Canonica
GW, van Weel C, et al: Allergic rhinitis and its impact on asthma
(ARIA) 2008 update (in collaboration with the World Health
Organization, GA(2)LEN and AllerGen). Allergy. 63 Suppl 86:S8–S160.
2008. View Article : Google Scholar
|
|
14
|
Yildirim YS, Apuhan T, Koçoğlu E, Simşek T
and Kazaz H: High sensitivity C-reactive protein levels in chronic
rhinosinusitis and allergic rhinitis. Kulak Burun Bogaz Ihtis Derg.
21:266–269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Büyüköztürk S, Gelincik AA, Genç S, Koçak
H, Oneriyidogan Y, Erden S, Dal M and Colakoglu B: Acute phase
reactants in allergic airway disease. Tohoku J Exp Med.
204:209–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Steiner I, Sobieska M, Pucher B,
Grzegorowski M and Samborski W: Examination of acute phase proteins
concentrations in children with allergic rhinitis. Ann Acad Med
Stetin. 52:33–37. 2006.(In Polish). PubMed/NCBI
|
|
17
|
Yalcin AD, Gumuslu S, Parlak GE, Bisgin A,
Yildiz M, Kargi A and Gorczynski RM: Systemic levels of
ceruloplasmin oxidase activity in allergic asthma and allergic
rhinitis. Immunopharmacol Immunotoxicol. 34:1047–1053. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khan DA: Allergic rhinitis and asthma:
Epidemiology and common pathophysiology. Allergy Asthma Proc.
35:357–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eisenbarth SC, Colegio OR, O'Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Besnard AG, Guillou N, Tschopp J, Erard F,
Couillin I, Iwakura Y, Quesniaux V, Ryffel B and Togbe D: NLRP3
inflammasome is required in murine asthma in the absence of
aluminum adjuvant. Allergy. 66:1047–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Allen IC, Jania CM, Wilson JE, Tekeppe EM,
Hua X, Brickey WJ, Kwan M, Koller BH, Tilley SL and Ting JP:
Analysis of NLRP3 in the development of allergic airway disease in
mice. J Immunol. 188:2884–2893. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kool M, Willart MA, van Nimwegen M, Bergen
I, Pouliot P, Virchow JC, Rogers N, Osorio F, Sousa Reis e C,
Hammad H and Lambrecht BN: An unexpected role for uric acid as an
inducer of T helper 2 cell immunity to inhaled antigens and
inflammatory mediator of allergic asthma. Immunity. 34:527–540.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marichal T, Ohata K, Bedoret D, Mesnil C,
Sabatel C, Kobiyama K, Lekeux P, Coban C, Akira S, Ishii KJ, et al:
DNA released from dying host cells mediates aluminum adjuvant
activity. Nat Med. 17:996–1002. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi Q, Luo S, Jin H, Cai J, Jia H, Feng L
and Lu X: Insulin-producing cells from human adipose tissue-derived
mesenchymal stem cells detected by atomic force microscope. Appl
Microbiol Biotechnol. 94:479–486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Netea MG, Nold-Petry CA, Nold MF, Joosten
LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G,
Heinhuis B, Devesa I, et al: Differential requirement for the
activation of the inflammasome for processing and release of
IL-1beta in monocytes and macrophages. Blood. 113:2324–2335. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu G, Zhang L, Wang DY, Xu R, Liu Z, Han
DM, Wang XD, Zuo KJ and Li HB: Opposing roles of IL-17A and IL-25
in the regulation of TSLP production in human nasal epithelial
cells. Allergy. 65:581–589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Semik-Orzech A, Barczyk A, Wiaderkiewicz R
and Pierzchala W: Interleukin 17 and RANTES levels in induced
sputum of patients with allergic rhinitis after a single nasal
allergen challenge. Ann Allergy Asthma Immunol. 103:418–424. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ciprandi G, Fenoglio D, De Amici M,
Quaglini S, Negrini S and Filaci G: Serum IL-17 levels in patients
with allergic rhinitis. J Allergy Clin Immunol. 122:650–651.e2.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ciprandi G, De Amici M, Murdaca G,
Fenoglio D, Ricciardolo F, Marseglia G and Tosca M: Serum
interleukin-17 levels are related to clinical severity in allergic
rhinitis. Allergy. 64:1375–1378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ciprandi G, Filaci G, Battaglia F and
Fenoglio D: Peripheral Th-17 cells in allergic rhinitis: New
evidence. Int Immunopharmacol. 10:226–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maio S, Baldacci S, Carrozzi L, Pistelli
F, Angino A, Simoni M, Sarno G, Cerrai S, Martini F, Fresta M, et
al: Respiratory symptoms/diseases prevalence is still increasing: A
25-yr population study. Respir Med. 110:58–65. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Long A, McFadden C, DeVine D, Chew P,
Kupelnick B and Lau J: Management of allergic and nonallergic
rhinitis. Evid Rep Technol Assess (Summ). 1–6. 2002.PubMed/NCBI
|
|
36
|
Meltzer EO: The prevalence and medical and
economic impact of allergic rhinitis in the United States. J
Allergy Clin Immunol. 99:S805–S828. 1997.PubMed/NCBI
|
|
37
|
Qiu S, Du Y, Duan X, Geng X, Xie J, Gao H
and Yang PC: B cell immunity in allergic nasal mucosa induces T
helper 2 cell differentiation. J Clin Immunol. 32:886–895. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jayasekera NP, Toma TP, Williams A and
Rajakulasingam K: Mechanisms of immunotherapy in allergic rhinitis.
Biomed Pharmacother. 61:29–33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ngoc PL, Gold DR, Tzianabos AO, Weiss ST
and Celedón JC: Cytokines, allergy, and asthma. Curr Opin Allergy
Clin Immunol. 5:161–166. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pawankar R, Hayashi M, Yamanishi S and
Igarashi T: The paradigm of cytokine networks in allergic airway
inflammation. Curr Opin Allergy Clin Immunol. 15:41–48. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sergejeva S, Ivanov S, Lötvall J and
Lindén A: Interleukin-17 as a recruitment and survival factor for
airway macrophages in allergic airway inflammation. Am J Respir
Cell Mol Biol. 33:248–253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hussein MR, Fathi NA, El-Din AM, Hassan
HI, Abdullah F, Al-Hakeem E and Backer EA: Alterations of the
CD4(+), CD8 (+) T cell subsets, interleukins-1beta, IL-10, IL-17,
tumor necrosis factor-alpha and soluble intercellular adhesion
molecule-1 in rheumatoid arthritis and osteoarthritis: Preliminary
observations. Pathol Oncol Res. 14:321–328. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yasumi Y, Takikawa Y, Endo R and Suzuki K:
Interleukin-17 as a new marker of severity of acute hepatic injury.
Hepatol Res. 37:248–254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guo C, Chen G and Ge R: IL-23, rather than
IL-17, is crucial for the development of ovalbumin-induced allergic
rhinitis. Mol Immunol. 67:436–443. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lv H, Lu B, Qian XJ, Huang JA and Qiu TF:
Serum IL-17 & eotaxin levels in asthmatic patients with
allergic rhinitis. Pak J Med Sci. 32:700–704. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim HY, Lee HJ, Chang YJ, Pichavant M,
Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, et
al: Interleukin-17-producing innate lymphoid cells and the NLRP3
inflammasome facilitate obesity-associated airway hyperreactivity.
Nat Med. 20:54–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tsvetkova-Vicheva VM, Gecheva SP,
Komsa-Penkova R, Velkova AS and Lukanov TH: IL-17 producing T cells
correlate with polysensitization but not with bronchial
hyperresponsiveness in patients with allergic rhinitis. Clin Transl
Allergy. 4:32014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Obregon C, Graf L, Chung KF, Cesson V and
Nicod LP: Nitric oxide sustains IL-1β expression in human dendritic
cells enhancing their capacity to induce IL-17-producing T-cells.
PLoS One. 10:e01201342015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li L, Kim J and Boussiotis VA:
IL-1β-mediated signals preferentially drive conversion of
regulatory T cells but not conventional T cells into
IL-17-producing cells. J Immunol. 185:4148–4153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim SR, Lee KS, Park SJ, Min KH, Lee MH,
Lee KA, Bartov O, Atlas D and Lee YC: A novel dithiol amide CB3
attenuates allergic airway disease through negative regulation of
p38 mitogen-activated protein kinase. Am J Respir Crit Care Med.
183:1015–1024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Riedl MA and Nel AE: Importance of
oxidative stress in the pathogenesis and treatment of asthma. Curr
Opin Allergy Clin Immunol. 8:49–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ciencewicki J, Trivedi S and Kleeberger
SR: Oxidants and the pathogenesis of lung diseases. J Allergy Clin
Immunol. 122:456–468; quiz 469–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Aguilera-Aguirre L, Bacsi A,
Saavedra-Molina A, Kurosky A, Sur S and Boldogh I: Mitochondrial
dysfunction increases allergic airway inflammation. J Immunol.
183:5379–5387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Heinzmann A, Thoma C, Dietrich H and
Deichmann KA: Identification of common polymorphisms in the
mitochondrial genome. Allergy. 58:830–831. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Raby BA, Klanderman B, Murphy A, Mazza S,
Camargo CA Jr, Silverman EK and Weiss ST: A common mitochondrial
haplogroup is associated with elevated total serum IgE levels. J
Allergy Clin Immunol. 120:351–358. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho
SH and Lee YC: NLRP3 inflammasome activation by mitochondrial ROS
in bronchial epithelial cells is required for allergic
inflammation. Cell Death Dis. 5:e14982014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Naik E and Dixit VM: Mitochondrial
reactive oxygen species drive proinflammatory cytokine production.
J Exp Med. 208:417–420. 2011. View Article : Google Scholar : PubMed/NCBI
|