Introduction
Lung cancer is one of the leading causes of
mortality and morbidity globally, and is one of the types of cancer
with the lowest survival rates (1,2). Lung
cancer has a poor prognosis with a 5-year survival rate of <15%
(3). Although novel therapeutic
strategies have emerged (4,5), radiotherapy is still one of the most
effective treatments for lung cancer (6,7).
As an important treatment for cancer, radiation may
inhibit tumor growth, promote tumor cell apoptosis and prolong
patient survival time; however, local or metastatic recurrences
remain impediments to overall survival (8,9).
Additionally, radiation resistance is another obstacle for
radiotherapy (10). Radiation
activates a number of signals, including nuclear factor-κB and
signal transducer and activator of transcription 3 to induce
radiation resistance (11,12). Furthermore, it has been reported that
radiation may promote cancer metastasis; for example, Guerra et
al demonstrated that irradiated patients developed a higher
rate of invasive recurrences compared with non-irradiated patients
(13). Additionally, previous studies
have revealed that radiation promotes the invasion and migration of
numerous tumor cells, including glioma cells (14), hepatocellular cancer cells (15) and breast cancer cells (16). Radiation is reported to induce
invasion by upregulating matrix metalloproteinases (MMPs) in
pancreatic cancer cells (17) and
glioblastoma cells (14). Therefore,
effective strategies are required to increase radiation sensitivity
in cancer cells and intercept radiation-induced cell migration and
invasion. However, the underlying mechanisms of radiation
resistance and radiation-induced cell migration and invasion are
not fully understood.
Specificity protein 1 (Sp1) is a
ubiquitously-expressed transcription factor that is overexpressed
in a number of cancer subtypes (18–20). Sp1
is involved in cell proliferation (21), cell migration and invasion (18) and apoptosis (22). Sp1 is reported to regulate target gene
transcription by binding to the GC-rich sequence in the promoter
region (23). Numerous mechanisms
have been reported to regulate the DNA binding of Sp1 (24,25). For
example, cyclin-dependent kinase (CDK)II increases the DNA-binding
activity of Sp1 by phosphorylating Sp1 at Ser59 (24) and CDKI decreases the DNA-binding
activity of Sp1 by phosphorylating Sp1 at Thr739 (25).
Cyclooxygenase-2 (COX-2) is an enzyme that converts
arachidonic acid into prostaglandins (26). It is overexpressed in numerous tumors
(27–29) and acts on a number of signaling
pathways involved in cell proliferation, apoptosis, migration and
invasion. Therefore, COX-2 is a potential target for cancer
chemotherapy (30,31).
The present study demonstrated that a COX-2
inhibitor, celecoxib, induced apoptosis as well as antagonized cell
migration and invasion by inhibiting the expression level and
DNA-binding ability of Sp1 in radiation-resistant lung cancer
cells.
Materials and methods
Cell culture
NCl-H1650 cells, purchased from American Type
Culture Collection (Manassas, VA, USA), were incubated in RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C in an atmosphere containing
5% CO2.
Antibodies, reagents and small
interfering (si)RNA
The MMP-2 (cat. no. 40994), MMP-9 (cat. no. 13667),
t-JNK (cat. no. 9252), p-JNK (cat. no. 9255), anti-IgG antibodies
(cat. no. 14708) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). The β-actin (sc-58673) and B cell
lymphoma-2 (Bcl-2; sc-509) antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA) and the Sp1 antibody was
purchased from Merck KGaA (Darmstadt, Germany). Secondary rat
antibody conjugated with horseradish peroxidase (cat. no. A0208)
was purchased from Biyuntian Biotechnology (Biyuntian, Beijing,
China). Celecoxib (cat. no. S1261) and anisomycin (cat. no. S7409)
were purchased from Selleck Chemicals (Houston, TX, USA). The small
interfering (si)RNA for Sp1, 5′-UGUAGAGUCUGCCAACUGACCUGUC-3′, was
purchased from Santa Cruz Biotechnology, Inc. The scramble siRNA,
5′-UAGUGCUUACGCAGUUGCUAGACCC-3′, was synthesized by Guangzhou
RiboBio Co., Ltd. (Guangzhou, China).
siRNA transfection
NCL-H1650 or NCL-H1650R cells were plated into
six-well plates at 1×106/well. After the cells reached
95% confluence, they were transfected with or 100 nmol small
interfering RNA (siRNA) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. The cells were cultured in RPMI-1640
medium supplemented with 10% FBS at 37°C in an atmosphere
containing 5% CO2. The cells were harvested 48 h after
transfection.
Identification of radiation-resistant
lung cancer cells
The NCl-H1650 cells were exposed to 4 Gy 60Co
radiation (dose rate was 2.5 Gy/min) and dead cells were removed
every day until the surge of apoptosis had ceased, for one week.
The remaining cells were then exposed to 4 Gy 60Co radiation again
and any dead cells were removed. The aforementioned procedures were
repeated until there was no apoptosis surge observed following
exposure to radiation. The duration of this total process was ~3
months. The radiation-resistant cells were subsequently referred to
as NCl-H1650R cells.
Treatment with radiation
NCL-H1650 cells and NCL-H1650R cells were seeded in
6-well plates at 5×104/well and cultured in RPMI-1640
medium supplemented with 10% FBS at 37°C in an atmosphere
containing 5% CO2. After 24 h, both cell types were
treated with 8 Gy 60Co. The cells were cultured for another 24 h at
37°C, 5% CO2; the cell proliferation apoptosis was
detected.
Treatment with celecoxib, SP600125 or
anisomycin
Celecoxib was diluted in dimethyl sulfoxide (DMSO;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) to produce 0, 20, 40
and 80 µM dilutions of celecoxib. SP600125 (Sigma-Aldrich; Merck
KGaA) was diluted in DMSO (Sigma-Aldrich; Merck KGaA) to make a 20
µM SP600125. Anisomycin (Sigma-Aldrich; Merck KGaA) was diluted in
DMSO (Sigma-Aldrich; Merck KGaA) to make a 20 nM dilution.
NCL-H1650 cells and NCL-H1650R cells were seeded in 6-well plates
at 5×104/well and cultured in RPMI-1640 medium
supplemented with 10% FBS at 37°C in an atmosphere containing 5%
CO2. After the cells were cultured for 24 h, celecoxib,
SP 600125, anisomycin or the vehicle of DMSO were added. The
cells were cultured for another 24 h at 37°C, 5% CO2.
The cells were then subject to other treatments, including protein
extraction or 60Co exposure as described below.
Assessment of cell apoptosis
NCL-H1650 and NCL-H1650R cells were washed with PBS
three times and then fixed with 95% ethanol for 5 min at room
temperature and stained with 5 mg/ml DAPI for 3 min at room
temperature. The cells were then washed with PBS and examined under
a fluorescence microscope (magnification, ×20, Nikon Corporation,
Tokyo, Japan) at a wavelength of 385 nm. The cells with nuclear
condensation and fragmentation were identified as apoptotic cells.
The apoptotic cells were counted within five randomly selected
fields. The rate of apoptotic cells was presented as the mean ±
standard deviation (SD).
Transwell assays
For migration assays, NCL-H1650 and NCL-H1650R cells
were seeded in the upper chambers at 105 cells/well in
serum-free RPMI-1640 medium, while the lower chambers contained the
culture medium supplemented with 10% FBS. The cells were incubated
for 12 h at 37°C in an atmosphere containing 5% CO2.
Subsequently, the cells on the top surface of the membrane were
removed and the cells on the bottom surface of the membrane were
fixed with 4% paraformaldehyde for 10 min and stained with 0.01%
crystal violet for 5 min at room temperature. The cells on the
bottom surface of the membrane were then examined under a light
microscope (Nikon Corporation), counted and averaged by the number
within six randomly selected fields, magnification, ×20. For the
Transwell invasion assay, the upper chambers were coated with 20 µg
Matrigel (Sigma-Aldrich; Merck KGaA) to seeding of the NCL-H1650
and NCL-H1650R cells. The medium used and the time and temperature
of incubation were the same as the migration assay.
Western blotting
Cells were lysed with the RIPA lysis buffer (Merck
KGaA) at room temperature for 30 min, and then the lysates were
centrifuged at 12,000 × g 4°C for 30 min. Following centrifugation,
the supernatant was collected. Protein concentrations were
evaluated by BCA protein assay. Subsequently, 20 µg protein per
lane was used for 10% SDS-PAGE electrophoresis and transferred onto
a nitrocellulose membrane (Merck KGaA). The membrane was blocked
with 5% fat-free milk in TBS Tween 20 (TBST) for 1 h at room
temperature and then incubated with primary antibodies (1:1,000
diluted in 5% fat-free milk) for 16 h at 4°C. The membrane was
washed with TBST three times and then incubated with secondary goat
anti-rat IgG antibody conjugated with horseradish peroxidase
(1:10,000 diluted in TBST) for 1 h at room temperature. Following
washing with TBST, the membrane was visualized with Beyo ECL plus
reagents (Biyuntian, Beijing, China). Protein expression levels
were quantified with Image J 1.48 software (National Institutes of
Health, Bethesda, MD, USA). Data were presented as the mean ± SD of
three independent experiments.
Chromatin immunoprecipitation assay
(ChIP)
The ChIP assay was performed using a ChIP assay kit
(Upstate Biotechnology, Inc., Lake Placid, NY, USA). Briefly,
NCl-H1650R cells were cross-linked with 1% formaldehyde at room
temperature for 10 min. The chromatin was sonicated into fragments
ranging between 200–1,000 base pairs using an Ultrasonic Processor
(Toshiba Corporation, Tokyo, Japan) at 4°C for 10 min and was
subsequently pulled down using Sp1 antibodies or the anti-IgG
antibody, which were 1:100 diluted in SDS lysis buffer (Beijing
Pulilai Gene Technology Co., Ltd., Beijing, China) at room
temperature for 1 h. Then the polymerase chain reaction (PCR)
amplification was performed to detect the binding DNA fragment. DNA
polymerase was purchased form Promega Corporation (Madison, WI,
USA) The primers used for amplifying the fragments of the MMP-2
promoter containing the Sp1-binding site were adopted from a
previous study (32) and were as
follows: Sense, 5′-GTCCTGGCAATCCCTTTG-3′ and antisense,
5′-GGGGAAAAGAGGTGGAGAAA-3′. PCR reaction step was as follows:
Initial denaturation (95°C, 5 min); and Reaction [denaturation
(95°C, 1 min), annealing (55°C, 1 min), extension (72°C, 30 sec)]
for 30 cycles). PCR reaction was performed in Eppendorf
Mastercycler nexus (Eppendorf, Hamburg, Germany). The PCR products
were analyzed on 1.5% agarose gel. The densitometry of the bands
was evaluated with Image J 1.48 software. Data were presented as
the mean ± SD of three independent experiments.
Statistical analysis
Statistical analysis was performed using SPSS v11.5
for Windows (SPSS, Inc., Chicago, IL, USA). All data were presented
as the mean ± SD. The intergroup comparison was performed by using
single-factor analysis of variance. P<0.05 was considered to
indicate a statistically significant difference.
Results
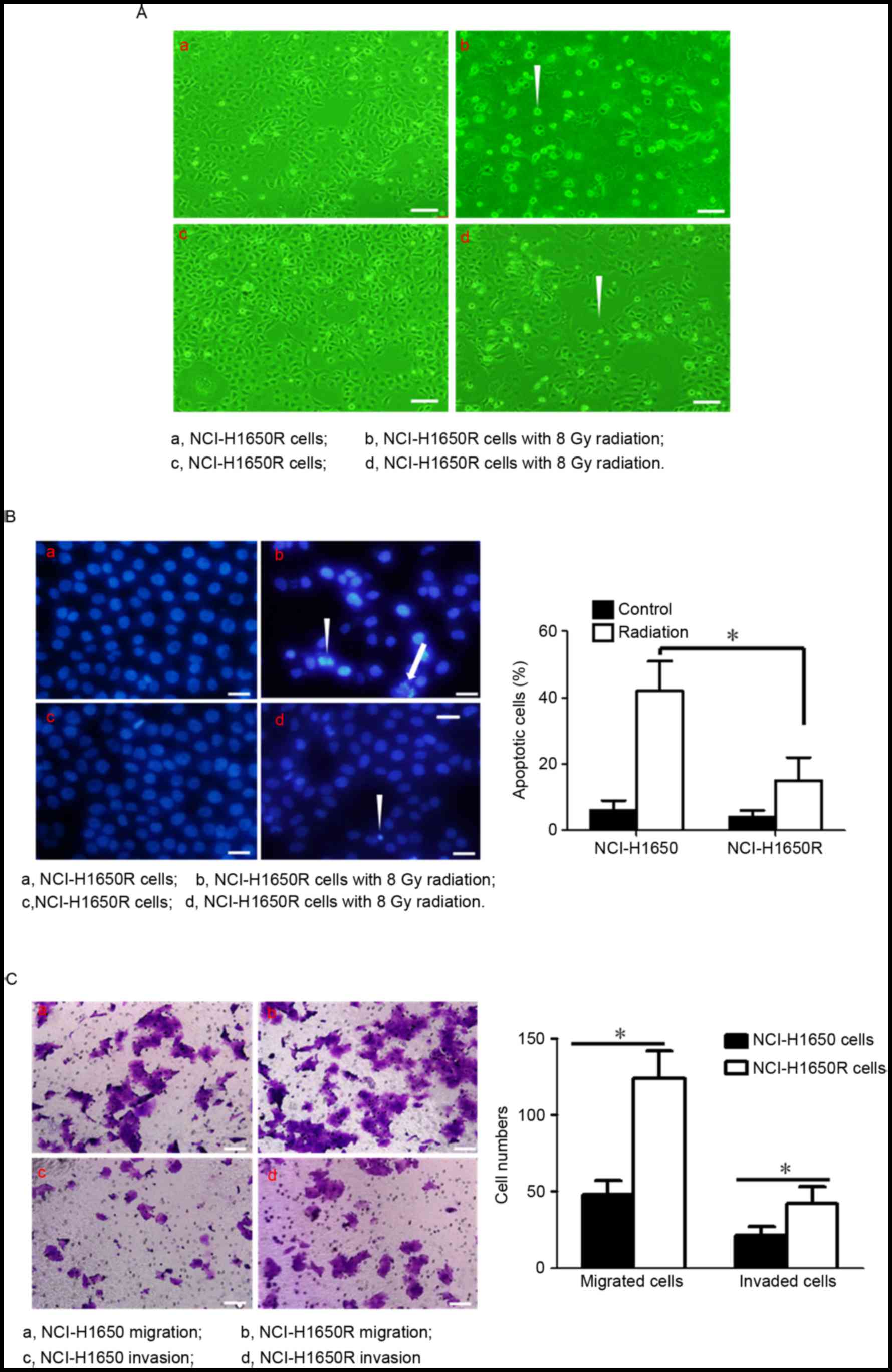
Radiation-resistant lung cancer cells
revealed high potential for radiation resistance, cell migration
and cell invasion
The present study we selected radiation-resistant
cells from the NCl-H1650 cell lines using discontinuous 4 Gy 60Co,
referred to as NCl-H1650R. To investigate whether NCL-H1650R cells
were more resistant than NCL-H1650 cells, NCl-H1650 cells and
NCL-H1650R cells were exposed to radiation (8 Gy60Co) and
subsequently cultured for 24 h. Radiation significantly induced
apoptosis and inhibited cell proliferation in NCl-H1650 cells;
however, NCl-H1650R cells demonstrated only slight induction of
apoptosis and inhibition of cell proliferation (Fig. 1A and B). The present study further
investigated whether migration and invasion differed in NCl-H1650R
cells compared with their parental cells. As presented in Fig. 1C, NCl-H1650R cells had higher
migration and invasion abilities compared with NCl-H1650 cells.
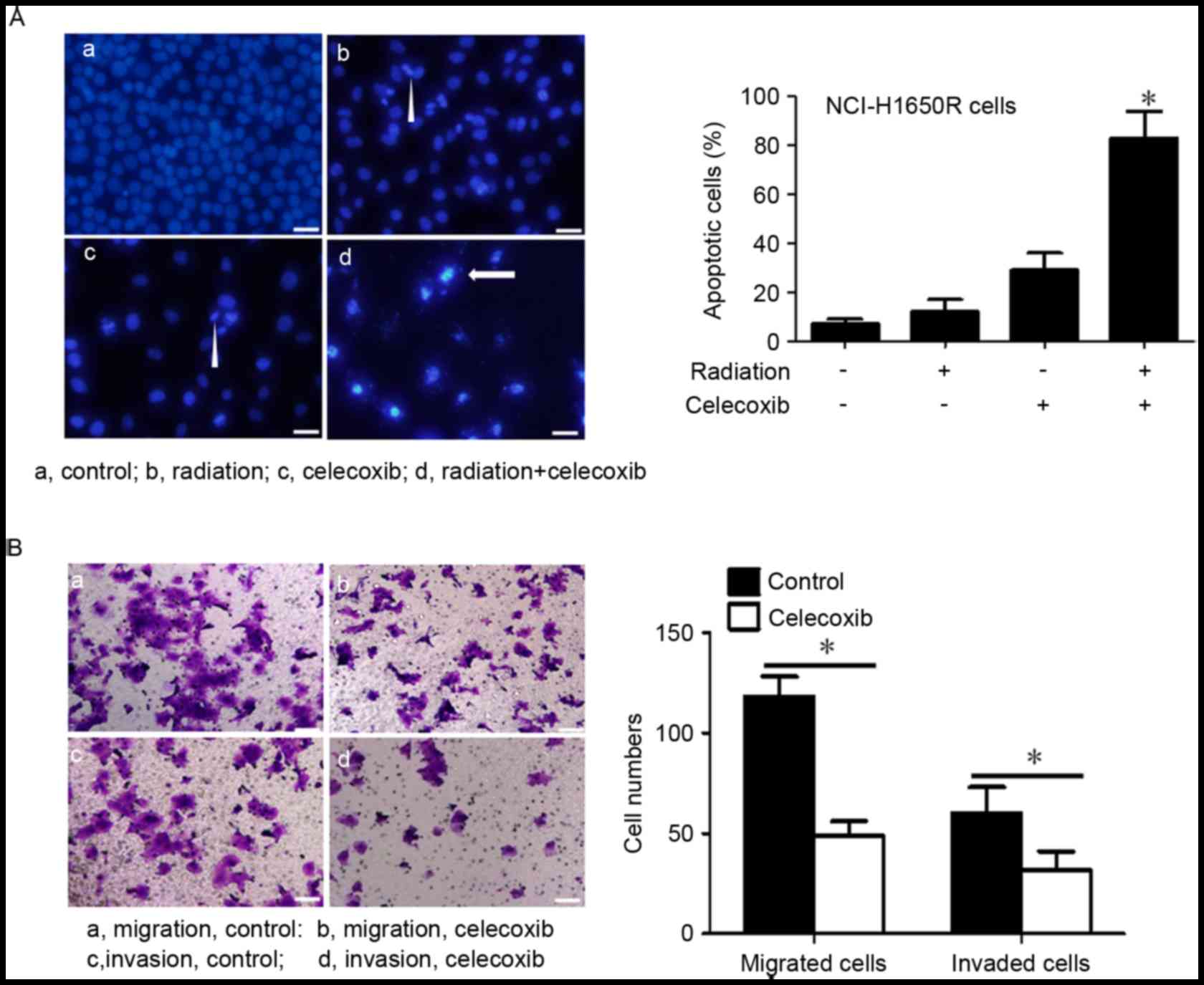
COX-2 selective inhibitor celecoxib
induced apoptosis and inhibition of cell migration and invasion in
radiation-resistant lung cancer cells
COX-2 inhibitors have previously been reported to
induce radiation sensitivity (33);
therefore, the present study investigated whether the COX-2
inhibitor, celecoxib, induced radiation sensitivity and inhibited
cell migration and invasion in radiation-resistant cells.
NCl-H1650R cells were treated with celecoxib, which were
subsequently exposed to 8 Gy60Co radiation and then cultured for 24
h. Celecoxib inhibited cell proliferation and induced apoptosis in
NCl-H1650R cells (Fig. 2A).
Furthermore, when treated with celecoxib and radiation, these cells
revealed significantly higher rates of apoptosis compared with the
other groups. The present study also observed that celecoxib
treatment significantly inhibited NCl-H1650R cell migration and
invasion (Fig. 2B).
Sp1 overexpression was responsible for
radiation resistance and the high potential of cell migration and
invasion in radiation-resistant lung cancer cells
It has previously been reported that Sp1 was
involved in numerous cell processes, including cell proliferation
(21), migration and invasion
(18) and apoptosis (22), thus the present study investigated
whether Sp1was responsible for radiation resistance and the
increased cell migration and invasion in radiation-resistant lung
cancer cells. As presented in Fig.
3A, Sp1 was overexpressed in NC1-H1650R cells compared with
NC1-H1650 cells, and this was associated with an upregulation of
MMP-2, MMP-9 and Bcl-2 compared with NCl-H1650 cells. Subsequently,
the present study knocked-down Sp1 in NCl-H1650R cells with Sp1
siRNA. Knockdown of Sp1 resulted in the downregulation of MMP-2,
MMP-9 and Bcl-2 (Fig. 3B).
Furthermore, a combination of the knockdown of Sp1 and radiation
exposure significantly induced cell apoptosis in NCl-H1650R cells
(Fig. 3C). Additionally, the present
study revealed that the knockdown of Sp1 inhibited cell migration
and invasion in NCl-H1650R cells (Fig.
3D).
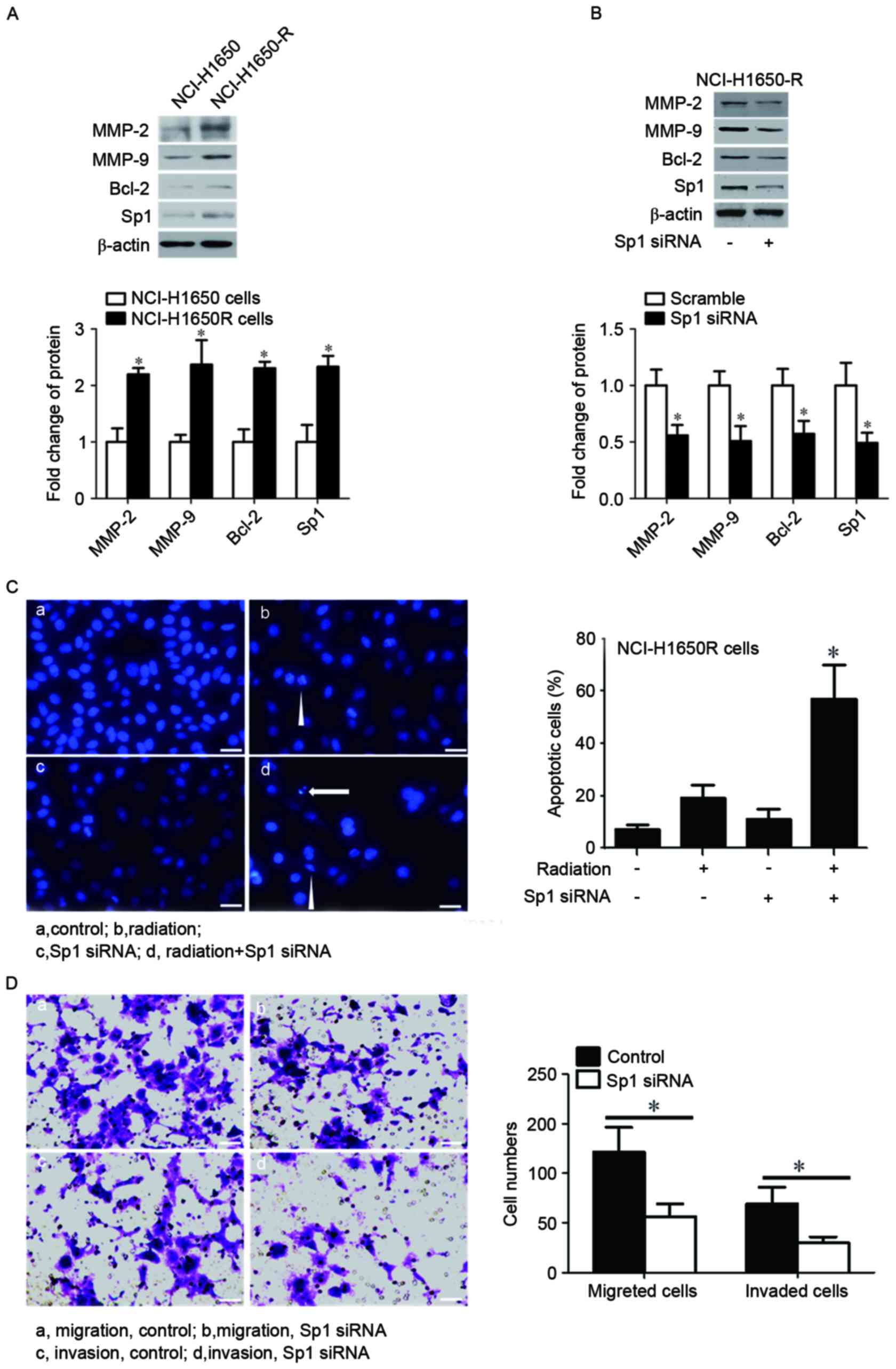 | Figure 3.Sp1 was overexpressed in and
responsible for radiation resistance and increased cell migration
and invasion in radiation-resistant cancer cells. Protein
expression levels of Sp1, MMP2, MMP9 and Bcl-2 were determined by
western blotting in (A) NCL-H1650 and NCL-H1650R cells, and (B)
NCL-H1650R cells transfected with control or Sp1 siRNA. β-actin
served as an internal control. (C) The left panel presents
fluorescent photomicrographs of NCL-H1650R cells. The cells were
stained with DAPI following the indicated treatments and those
cells with nuclear condensation (triangle) or fragmentation
(arrows) were identified as apoptotic cells. Scale bars: 20 µm. The
right panel presents the apoptotic rate of cells following various
treatments. (D) Photomicrographs of migrated or invaded NCL-H1650R
cells following the indicated treatments, as assessed by Transwell
assay. The cell counts of migrated or invaded cells from 6–10
separate fields are presented on the right. The data are presented
as the mean ± standard deviation. Scale bars: 20 µm. *P<0.05 vs.
NCI-H1650 cells, scramble siRNA or control. Sp1, specificity
protein 1; MMP, matrix metalloproteinase; Bcl-2, B cell lymphoma-2;
NCL-H1650R, radiation resistant NCI-H1650 cells; siRNA, short
interfering RNA. |
Celecoxib inhibited expression and
DNA-binding of Sp1
In order to confirm whether Sp1 was responsible for
celecoxib-induced cell apoptosis and inhibition of cell migration
and invasion in radiation-resistant cancer cells, the present study
treated NCl-H1650R cells with celecoxib. Sp1 was downregulated
following celecoxib treatment, and this was associated with a
corresponding downregulation of MMP-2, MMP-9 and Bcl-2 (Fig. 4A). As c-Jun N-terminal kinase (JNK)
has been demonstrated to increase Sp1 protein stability, the
inhibition of JNK may decrease E-Ras-induced Sp1 phosphorylation,
and therefore decrease the protein stability of Sp1 (34). The present study hypothesized that the
JNK signaling pathway mediated the celecoxib-induced downregulation
of Sp1. The present study detected the activation of JNK in
radiation resistant NC1-H1650R cells and their parental NC1-H1650
cells. NCL-H1650 cells and NCL-H1650R cells were exposed to 8
Gy60Co radiation and then cultured for 24 h. As presented in
Fig. 4B, there was no change in the
level of JNK phosphorylation, although Sp1 was overexpressed in
NCl-H1650R cells compared with their parental cells. However,
inhibition of the phosphorylation of JNK by the JNK inhibitor,
SP600125, downregulated Sp1 in NCl-H1650R cells (Fig. 4C). Celecoxib was revealed to inhibit
JNK phosphorylation, whereas the activation of JNK by anisomycin
(that induced JNK phosphorylation by activating mitogen-activated
protein kinase kinase) was able to reverse celecoxib-induced
inhibition of JNK phosphorylation, Sp1 downregulation and the
corresponding changes of MMP-2, MMP-9 and Bcl-2 (Fig. 4D). These results indicated that JNK
was involved in celecoxib-induced Sp1 downregulation, radiation
sensitivity, inhibition of cell migration and invasion.
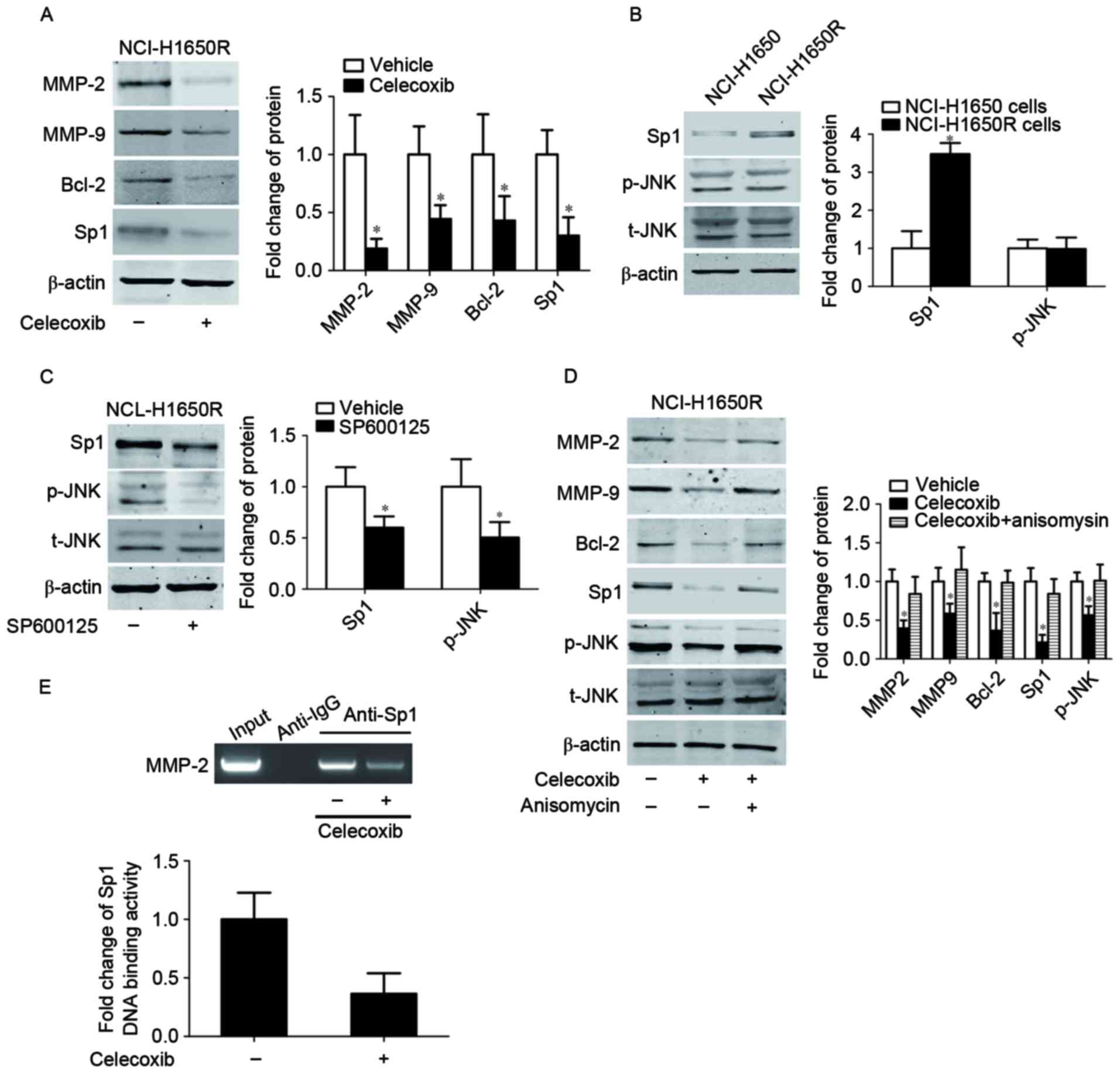 | Figure 4.Celecoxib inhibited the expression
and DNA-binding of Sp1. (A) Protein expression levels of Sp1, MMP2,
MMP9 and Bcl-2 were determined by western blotting following
celecoxib treatment. β-actin served as an internal control. Protein
expression levels of t-JNK, p-JNK and Sp1 in (B) NCL-H1650 and
NCL-H1650R cells and (C) NCL-H1650R cells treated with SP600125 or
vehicle. *P<0.05 vs. vehicle group. (D) Protein expression
levels of t-JNK, p-JNK, Sp1, MMP2, MMP9 and Bcl-2 following
treatment with celecoxib and/or anisomycin. *P<0.05 vs. vehicle
group. (E) Chromatin immunoprecipitation assays were performed in
NCL-H1650R cells with anti-Sp1 or anti-IgG antibodies, using
primers to amplify the region of the MMP-2 promoter containing the
Sp1-binding site. Sp1, specificity protein 1; MMP, matrix
metalloproteinase; Bcl-2, B cell lymphoma-2; JNK, c-Jun N-terminal
kinase; t, total; p, phosphorylated; NCL-H1650R, radiation
resistant NCI-H1650 cells. |
As a transcription factor, the transcription
activity of Sp1 depends on its DNA binding ability, thus the
present study performed a ChIP assay on NCl-H1650R cells using
primers to amplify the region in the MMP-2 promoter containing the
Sp1 binding site, in order to determine whether celecoxib treatment
altered Sp1 binding to the MMP-2 promoter. As presented in Fig. 4E, specific DNA fragment was amplified
from the chromatin pulled down by the anti-Sp1 antibody (but not by
the anti-IgG antibody) and this pulled-down fragment was decreased
following celecoxib treatment. These results indicated that the
COX-2 selective inhibitor inhibited Sp1 DNA-binding activity.
Discussion
The present study demonstrated that the COX-2
selective inhibitor, celecoxib, induced cell apoptosis and the
inhibition of cell migration and invasion via the JNK/Sp1 signaling
pathway. The results of the present study provide novel evidence
that radiation-resistant cells may contribute to local or
metastatic recurrence, and that celecoxib maybe used to enhance the
radiation sensitivity of cancer cells and inhibit cell migration
and invasion.
Radiation-resistant cancer cells are more capable of
surviving radiation and have a high potential for proliferation,
migration and invasion. Radiation is an effective treatment for
cancer; however, local or metastatic recurrences remain common
outcomes (13). Therefore, radiation
resistance and radiation-induced migration and invasion have
attracted increasing attention (14–16,35). The
present study established one radiation-resistant lung cancer cell
line from NCl-H1650 cells. Notably, a subpopulation of
radiation-resistant cells already exists in a number of cell lines,
including the MCF-7/C6 breast cancer cell line (36) and the nasopharyngeal carcinoma CNE-2
cell line (37), and these
radiation-resistant cancer cells are responsible for cancer
recurrence following radiotherapy, including local or metastatic
recurrence. The present study revealed that these
radiation-resistant cancer cells were not only resistant to
radiation, but also exhibited increased cell migration and invasion
compared with non-resistant cells, which may be the reason local or
metastasis recurrences occur. As a result, these
radiation-resistant cancer cells have become a target for
decreasing local or metastatic cancer recurrence.
The overexpression of Sp1 was responsible for
increased levels of radiation resistance, cell migration and
invasion irradiation-resistant lung cancer cells. The results of
the present study revealed that Sp1 was overexpressed in
radiation-resistant cancer cells, with a corresponding upregulation
of MMPs and Bcl-2. Knockdown of Sp1 inhibited cell proliferation,
migration and invasion. These results were consistent with numerous
previous studies that demonstrated that Sp1 promoted cell
proliferation, migration and invasion by regulating MMPs and Bcl-2
(38–40). To the best of our knowledge, this is
the first study to reveal that Sp1 is responsible for radiation
resistance and increased cell migration and invasion in
radiation-resistant lung cancer cells. Drugs that downregulate or
inactivate Sp1 may be used to antagonize cancer radiation
resistance and metastasis following radiation treatment.
Celecoxib was demonstrated to enhance radiation
sensitivity and decrease cell migration and invasion, at least
partially, by antagonizing the Sp1-induced upregulation of MMPs and
Bcl-2. The present study observed that celecoxib may significantly
induce radiation sensitivity and inhibit cell migration and
invasion of the radiation-resistant lung cancer cells. The present
study also observed that celecoxib significantly reduced Sp1
expression and correspondingly downregulated MMP-2, MMP-9 and
Bcl-2. Considering that Sp1 serves a crucial function in cancer
cell progression, the results of the present study suggested that
decreased Sp1 expression levels induced by the COX-2 inhibitor may
have contributed to the antagonism of radiation-resistance and cell
migration and invasion in radiation-resistant lung cancer cells. In
addition to other previously reported mechanisms (41,42), the
induction of radiation sensitivity maybe another important
underlying mechanism of COX-2 inhibitors. A phase II clinical study
has previously revealed that celecoxib is able to improve the
radiotherapy outcome of patients with rectal cancer (43). Therefore, the results of the present
study further supported the suggestion that COX-2 inhibitors may be
promising enhancers of tumor radiotherapy.
Celecoxib downregulated Sp1 by inactivating the JNK
signaling pathway. It has been reported that the activation of JNK
may increase Sp1 stability (44). The
present study observed that the inactivation of JNK may
downregulate Sp1 and that the activation of JNK by anisomycin may
antagonize celecoxib-induced Sp1 downregulation and the
corresponding changes to MMP-9, MMP-2 and Bcl-2 expression levels.
Of note, the present study did not observe higher activation of JNK
in radiation resistant lung cancer cells compared with their
parental cells. This may be because JNK was not responsible for the
overexpression of Sp1 in radiation-resistant lung cancer cells, but
the inactivation of JNK following treatment with the COX-2
inhibitor did contribute to the downregulation of Sp1, increased
radiation sensitivity and the inhibition of cell migration and
invasion.
Celecoxib treatment decreased Sp1 DNA-binding
activity. The transcription activity of Sp1 depends on its
DNA-binding ability to the promoters of target genes (32,38). The
present study demonstrated that celecoxib decreased Sp1 DNA-binding
activity to the MMP-2 promoters, indicating that celecoxib
downregulated MMP-2 by decreasing the binding activity of Sp1 to
the MMP-2 promoters, and may act via the same mechanism for MMP-9
and Bcl-2. A number of factors may affect Sp1 DNA-binding activity,
including CDKII (24) and CDKI
(25); however, the determination of
which factor is involved in the celecoxib-induced decrease of Sp1
DNA-binding activity requires further investigation.
In conclusion, the present study revealed that Sp1
was overexpressed in radiation-resistant lung cancer cells and that
COX-2 inhibitors induced radiation sensitivity and decreased cell
migration and invasion in radiation-resistant lung cancer cells via
the inactivation of the JNK/Sp1 signaling pathway and a decrease in
Sp1 DNA-binding activity. Based on these findings, COX-2 inhibitors
may be used in combination with radiation therapy to enhance the
radiation sensitivity of cancer cells and reduce cancer
metastasis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RL cultured the cells, established the radio
resistance cell line and treated the cells with celecoxib and
radiation. QT conducted the transfection experiments and western
blot assay. QL performed the CCK-8 cell proliferation assay and
detected the cell apoptosis.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Parkin DM and Steliarova-Foucher
E: Estimates of cancer incidence and mortality in Europe in 2008.
Eur J Cancer. 46:765–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Siegel R, Ward E, Murray T, Xu J,
Smigal C and Thun MJ: Cancer statistics, 2006. CA Cancer J Clin.
56:106–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guan Z, Yu X, Wang H, Wang H, Zhang J, Li
G, Cao J and Teng L: Advances in the targeted therapy of
liposarcoma. Onco Targets Ther. 8:125–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Duong CP, Yong CS, Kershaw MH, Slaney CY
and Darcy PK: Cancer immunotherapy utilizing gene-modified T cells:
From the bench to the clinic. Mol Immunol. 67:46–57. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kong FM, Zhao J, Wang J and Faivre-Finn C:
Radiation dose effect in locally advanced non-small cell lung
cancer. J Thorac Dis. 6:336–347. 2014.PubMed/NCBI
|
|
7
|
Brady LW, Kramer S, Levitt SH, Parker RG
and Powers WE: Radiation oncology: Contributions of the United
States in the last years of the 20th century. Radiology. 219:1–5.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alamanda VK, Song Y, Shinohara E, Schwartz
HS and Holt GE: Postoperative radiation boost does not improve
local recurrence rates in extremity soft tissue sarcomas. J Med
Imaging Radiat Oncol. 58:633–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao CN, Luo JW, Gao L, Xu GZ, Li SY and
Xiao JP: Recurrence of nasopharyngeal carcinoma in the parotid
region after definitive intensity-modulated radiotherapy. J Oral
Maxillofac Surg. 71:1993–1997. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Milas L, Raju U, Liao Z and Ajani J:
Targeting molecular determinants of tumor chemo-radioresistance.
Semin Oncol. 32 Suppl 9:S78–S81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brach MA, Gruss HJ, Kaisho T, Asano Y,
Hirano T and Herrmann F: Ionizing radiation induces expression of
interleukin 6 by human fibroblasts involving activation of nuclear
factor-kappa B. J Biol Chem. 268:8466–8472. 1993.PubMed/NCBI
|
|
12
|
Yin ZJ, Jin FG, Liu TG, Fu EQ, Xie YH and
Sun RL: Overexpression of STAT3 potentiates growth, survival, and
radioresistance of non-small-cell lung cancer (NSCLC) cells. J Surg
Res. 171:675–683. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guerra LE, Smith RM, Kaminski A, Lagios MD
and Silverstein MJ: Invasive local recurrence increased after
radiation therapy for ductal carcinoma in situ. Am J Surg.
196:552–555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wild-Bode C, Weller M, Rimner A, Dichgans
J and Wick W: Sublethal irradiation promotes migration and
invasiveness of glioma cells: Implications for radiotherapy of
human glioblastoma. Cancer Res. 61:2744–2750. 2001.PubMed/NCBI
|
|
15
|
Cheng JC, Chou CH, Kuo ML and Hsieh CY:
Radiation-enhanced hepatocellular carcinoma cell invasion with
MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction
pathway. Oncogene. 25:7009–7018. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paquette B, Baptiste C, Therriault H,
Arguin G, Plouffe B and Lemay R: In vitro irradiation of basement
membrane enhances the invasiveness of breast cancer cells. Br J
Cancer. 97:1505–1512. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qian LW, Mizumoto K, Urashima T, Nagai E,
Maehara N, Sato N, Nakajima M and Tanaka M: Radiation-induced
increase in invasive potential of human pancreatic cancer cells and
its blockade by a matrix metalloproteinase inhibitor, CGS27023.
Clin Cancer Res. 8:1223–1227. 2002.PubMed/NCBI
|
|
18
|
Hsu TI, Wang MC, Chen SY, Yeh YM, Su WC,
Chang WC and Hung JJ: Sp1 expression regulates lung tumor
progression. Oncogene. 31:3973–3988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zannetti A, Del Vecchio S, Carriero MV,
Fonti R, Franco P, Botti G, D'Aiuto G, Stoppelli MP and Salvatore
M: Coordinate up-regulation of Sp1 DNA-binding activity and
urokinase receptor expression in breast carcinoma. Cancer Res.
60:1546–1551. 2000.PubMed/NCBI
|
|
20
|
Chiefari E, Brunetti A, Arturi F, Bidart
JM, Russo D, Schlumberger M and Filetti S: Increased expression of
AP2 and Sp1 transcription factors in human thyroid tumors: A role
in NIS expression regulation? BMC Cancer. 2:352002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang JP, Zhang H, Wang HB, Li YX, Liu GH,
Xing S, Li MZ and Zeng MS: Down-regulation of Sp1 suppresses cell
proliferation, clonogenicity and the expressions of stem cell
markers in nasopharyngeal carcinoma. J Transl Med. 12:2222014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cho JJ, Chae JI, Yoon G, Kim KH, Cho JH,
Cho SS, Cho YS and Shim JH: Licochalcone A, a natural chalconoid
isolated from Glycyrrhiza inflata root, induces apoptosis via Sp1
and Sp1 regulatory proteins in oral squamous cell carcinoma. Int J
Oncol. 45:667–674. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dynan WS and Tjian R: The
promoter-specific transcription factor Sp1 binds to upstream
sequences in the SV40 early promoter. Cell. 35:79–87. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Borja Fojas P, Collins NK, Du P,
Azizkhan-Clifford J and Mudryj M: Cyclin A-CDK phosphorylates Sp1
and enhances Sp1-mediated transcription. EMBO J. 20:5737–5747.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chuang JY, Wang SA, Yang WB, Yang HC, Hung
CY, Su TP, Chang WC and Hung JJ: Sp1 phosphorylation by
cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding
activity during mitosis in cancer cells. Oncogene. 31:4946–4959.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang HW: COX-2 regulation of
prostaglandins in synaptic signaling. Sheng Li Ke Xue Jin Zhan.
40:317–320. 2009.(In Chinese). PubMed/NCBI
|
|
27
|
Lim HY, Joo HJ, Choi JH, Yi JW, Yang MS,
Cho DY, Kim HS, Nam DK, Lee KB and Kim HC: Increased expression of
cyclooxygenase-2 protein in human gastric carcinoma. Clin Cancer
Res. 6:519–525. 2000.PubMed/NCBI
|
|
28
|
Tucker ON, Dannenberg AJ, Yang EK, Zhang
F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT
and Fahey TJ III: Cyclooxygenase-2 expression is up-regulated in
human pancreatic cancer. Cancer Res. 59:987–990. 1999.PubMed/NCBI
|
|
29
|
Molina MA, Sitja-Arnau M, Lemoine MG,
Frazier ML and Sinicrope FA: Increased cyclooxygenase-2 expression
in human pancreatic carcinomas and cell lines: Growth inhibition by
nonsteroidal anti-inflammatory drugs. Cancer Res. 59:4356–4362.
1999.PubMed/NCBI
|
|
30
|
Brown JR and DuBois RN: Cyclooxygenase as
a target in lung cancer. Clin Cancer Res. 10:4266s–4269s. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pruthi RS, Derksen E and Gaston K:
Cyclooxygenase-2 as a potential target in the prevention and
treatment of genitourinary tumors: A review. J Urol. 169:2352–2359.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang CH, Chang HC and Hung WC: p16
inhibits matrix metalloproteinase-2 expression via suppression of
Sp1-mediated gene transcription. J Cell Physiol. 208:246–252. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Meng Z and Gan YH: Activating PTEN by
COX-2 inhibitors antagonizes radiation-induced AKT activation
contributing to radiosensitization. Biochem Biophys Res Commun.
460:198–204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kwon YW, Jang S, Paek JS, Lee JW, Cho HJ,
Yang HM and Kim HS: E-Ras improves the efficiency of reprogramming
by facilitating cell cycle progression through JNK-Sp1 pathway.
Stem Cell Res. 15:481–494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohuchida K, Mizumoto K, Murakami M, Qian
LW, Sato N, Nagai E, Matsumoto K, Nakamura T and Tanaka M:
Radiation to stromal fibroblasts increases invasiveness of
pancreatic cancer cells through tumor-stromal interactions. Cancer
Res. 64:3215–3222. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo L, Xiao Y, Fan M, Li JJ and Wang Y:
Profiling global kinome signatures of the radioresistant MCF-7/C6
breast cancer cells using MRM-based targeted proteomics. J Proteome
Res. 14:193–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peng G, Cao RB, Li YH, Zou ZW, Huang J and
Ding Q: Alterations of cell cycle control proteins SHP1/2, p16,
CDK4 and cyclin D1 in radioresistant nasopharyngeal carcinoma
cells. Mol Med Rep. 10:1709–1716. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kou XX, Hao T, Meng Z, Zhou YH and Gan YH:
Acetylated Sp1 inhibits PTEN expression through binding to PTEN
core promoter and recruitment of HDAC1 and promotes cancer cell
migration and invasion. Carcinogenesis. 34:58–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duan H, Heckman CA and Boxer LM: Histone
deacetylase inhibitors down-regulate bcl-2 expression and induce
apoptosis in t(14;18) lymphomas. Mol Cell Biol. 25:1608–1619. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qiu T, Zhou X, Wang J, Du Y, Xu J, Huang
Z, Zhu W, Shu Y and Liu P: MiR-145, miR-133a and miR-133b inhibit
proliferation, migration, invasion and cell cycle progression via
targeting transcription factor Sp1 in gastric cancer. FEBS Lett.
588:1168–1177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang MY, Lee HT, Chen CM, Shen CC and Ma
HI: Celecoxib suppresses the phosphorylation of STAT3 protein and
can enhance the radiosensitivity of medulloblastoma-derived cancer
stem-like cells. Int J Mol Sci. 15:11013–11029. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim YM, Jeong IH and Pyo H: Celecoxib
enhances the radiosensitizing effect of 7-hydroxystaurosporine
(UCN-01) in human lung cancer cell lines. Int J Radiat Oncol Biol
Phys. 83:e399–e407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang LW, Hsiao CF, Chen WT, Lee HH, Lin
TC, Chen HC, Chen HH, Chien CR, Lin TY and Liu TW: Celecoxib plus
chemoradiotherapy for locally advanced rectal cancer: A phase II
TCOG study. J Surg Oncol. 109:580–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chuang JY, Wang YT, Yeh SH, Liu YW, Chang
WC and Hung JJ: Phosphorylation by c-Jun NH2-terminal kinase 1
regulates the stability of transcription factor Sp1 during mitosis.
Mol Biol Cell. 19:1139–1151. 2008. View Article : Google Scholar : PubMed/NCBI
|