Introduction
Ovarian cancer (OvCa) has the highest incidence of
mortality of any gynecological cancer type in the United States,
and is the primary cause of female cancer-associated mortality in
the United States in 2014 (1). Owing
to the mild or absent signs and symptoms during early stage OvCa,
and the lack of a reliable early detection test, OvCa has a
disproportionately poor prognosis, with a 5-year survival rate of
44% between 1995–2007 in the United States (2), therefore further understanding of its
regulatory mechanisms at the molecular level is vital, in order to
identify reliable prognostic biomarkers for the prediction of
survival times of patients with OvCa.
Long non-coding RNAs (lncRNAs) are a group of
non-coding RNAs with >200 nucleotides (3). An increasing number of studies have
demonstrated that lncRNAs are implicated in cancer development, and
their dysregulated expression confers on the cancer cell capability
for tumor initiation and development (4,5). Previous
evidence indicated the functions of the lncRNAs H19 (H19, imprinted
maternally expressed transcript), long stress-induced non-coding
transcript 5 and X-inactive-specific transcript in the
tumorigenesis and progression of OvCa (6). Cheng et al (7), demonstrated that upregulated lncRNA
AB073614 predicts a poor prognosis; however, it has been indicated
that lncRNA homeobox A11 antisense enhances cell proliferation and
invasion in serous OvCa, and is associated with prognosis (8). Chen et al (9) determined the function of lncRNA nuclear
paraspeckle assembly transcript 1 as a clinical prognostic
biomarker for OvCa. Although previous studies have made notable
progress, the prognostic functions of lncRNAs in OvCa and the
underlying mechanisms remain poorly characterized.
In the present study, an in-depth analysis of lncRNA
and mRNA expression profiles, and corresponding clinical
characteristics of patients with OvCa from The Cancer Genome Atlas
(TCGA) data portal was conducted. In contrast with the study by
Zhou et al (10), which
identifies a 10-lncRNA signature for predicting survival using the
competing endogenous RNAs-network driven method, the present study
searched for prognostic lncRNAs based on univariate and
multivariate Cox regression analyses in a training set, and then
validated their prognostic power in a validation set. Furthermore,
Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analysis was performed to
determine the possible biological functions of identified
prognostic lncRNAs. The results of the present study improved the
understanding of the molecular mechanisms of OvCa.
Materials and methods
Data resource
lncRNA and mRNA expression data of OvCa samples,
together with corresponding clinical information were downloaded
from TCGA data portal (gdc-portal.nci.nih.gov). Each sample was annotated
according to its barcode ID. Consequently, a total of 419 samples
with lncRNA and mRNA data, based on the Illumina HiSeq 2000 RNA
Sequencing platform, were collected. Out of these samples, the
samples at early and middle stages according to American Joint
Committee on Cancer standard (11)
with available clinical information (n=353) were selected for the
present study, and randomly and the validation set were combined
and defined as the entire set. Clinical characteristics of these
datasets are listed in Table I.
 | Table I.Clinical characteristics of patients
in the training set, the validation set and the entire set. |
Table I.
Clinical characteristics of patients
in the training set, the validation set and the entire set.
|
Characteristics | Training set
(n=160) | Validation set
(n=177) | Entire set
(n=337) |
|---|
| Age at diagnosis
(mean ± SD) | 59.63±11.18 | 60.11±11.86 | 59.88±11.53 |
| Clinical stage
(I/II/III) | 0/7/153 | 0/15/162 | 0/22/315 |
| Neoplasm
histological grade (G1/G2/G3/G4/-) | 0/12/145/1/2 | 0/32/142/0/3 | 0/44/287/1/5 |
| Lymphatic invasion
(yes/no/-) | 33/28/99 | 54/21/102 | 87/49/201 |
| Tumor recurrence
(yes/no) | 96/64 | 101/76 | 197/140 |
| Status
(deceased/alive) | 86/74 | 107/70 | 193/144 |
| Overall survival
time (mean ± SD, months) | 38.18±27.14 | 34.58±26.99 | 36.29±27.08 |
Screening for differentially expressed
lncRNAs (DELs)
A total of 16 patients with prognosis of <6
months were excluded from the training set (remaining patients,
n=160). No patients were excluded from the validation set. Good
prognosis was defined as patients alive after >24 months, and
poor prognosis was defined as patients who succumbed within 24
months. Patients with good prognosis and poor prognosis were
selected from the training set, and defined as the good prognosis
and poor prognosis groups, respectively. DELs between the groups
were screened using two packages in R3.1.0 (https://www.r-project.org/), Differential Expression
for Sequence Count Data (DESeq) (12)
and edgeR (13). An lncRNA was
considered a significant DEL when false discovery rate (FDR)
<0.05 and |fold change|>1.3. The overlapping DELs between the
two methods were incorporated into the subsequent analyses.
Identification of survival-associated
lncRNAs
Univariate Cox proportional hazards regression model
was employed to evaluate the associations between these significant
DELs and the overall survival (OS) time of the patients with OvCa
in the training set, followed by the log-rank test. The DELs with
log-rank P<0.05 were defined as survival-associated lncRNAs,
which were then subjected to multivariate Cox regression analysis
with OS time as the dependent variable.
Construction of an lncRNA-based risk
scoring system
On the basis of the linear combination of expression
levels of the predictive lncRNAs selected by the multivariate Cox
regression analysis with the regression coefficient, a risk scoring
system was produced to calculate the risk score for each sample
using the following equation (10,14): Risk
score=β1 × Expr1 + β2 × Expr2 + ··· + βn × Exprn, where βn
represents estimated regression coefficient of lncRNAn, and Exprn
represents lncRNAn expression level.
All samples in the training and validation set were
classified into a high-risk (>median risk score) and a low-risk
group (≤median risk score), with the median risk score of the
dataset as the cut-off value.
Prognosis correlation analysis
Univariate Cox regression analysis and Student's
t-test (unpaired) was used to characterize OS time of each risk
group. The log-rank test was then used following univariate
analysis to compare the difference between the two risk groups. The
results were shown by Kaplan-Meier survival curve. With OS time as
the dependent variable, and risk score and clinical variables as
explanatory variables, multivariate Cox regression analysis and
data stratification analysis were conducted to evaluate whether the
risk score was independent of other clinical variables, from which
hazard ratios and 95% confidence intervals (CI) were calculated.
P<0.05 was considered to indicate a statistically significant
difference. To appraise the prognostic power of the lncRNAs-based
risk scoring system, the time-dependent receiver operating
characteristic (ROC) curve analyses were carried out using the pROC
package (15), followed by
calculation of the area under the ROC curves (AUC). All analyses
were conducted by R 3.0.1 software and Bioconductor 1.14.3. The
data are presented as mean ± standard deviation.
GO function and KEGG pathway
enrichment analyses
Spearman correlation coefficients were computed to
assess the association between the prognostic lncRNAs and
corresponding mRNAs, by analyzing expression profiles of the paired
lncRNA and protein-coding genes in all OvCa samples. The
protein-coding genes significantly correlated with at least one
prognostic lncRNA with a Spearman correlation coefficient >0.4
were selected and underwent functional enrichment analyses using
the Database for Annotation, Visualization and Integrated Discovery
(16) tool limited to GO biological
process terms (17) and KEGG pathway
categories (18). GO terms or KEGG
pathways with P<0.05 were considered to indicate a statistically
significant difference.
Results
Identification of significant
DELs
Following the deletion of lncRNAs with notably low
expression, 2,035 lncRNAs were acquired from TCGA. According to the
survival time information, 28 patients were included in the poor
prognosis group and 41 patients were included in the good prognosis
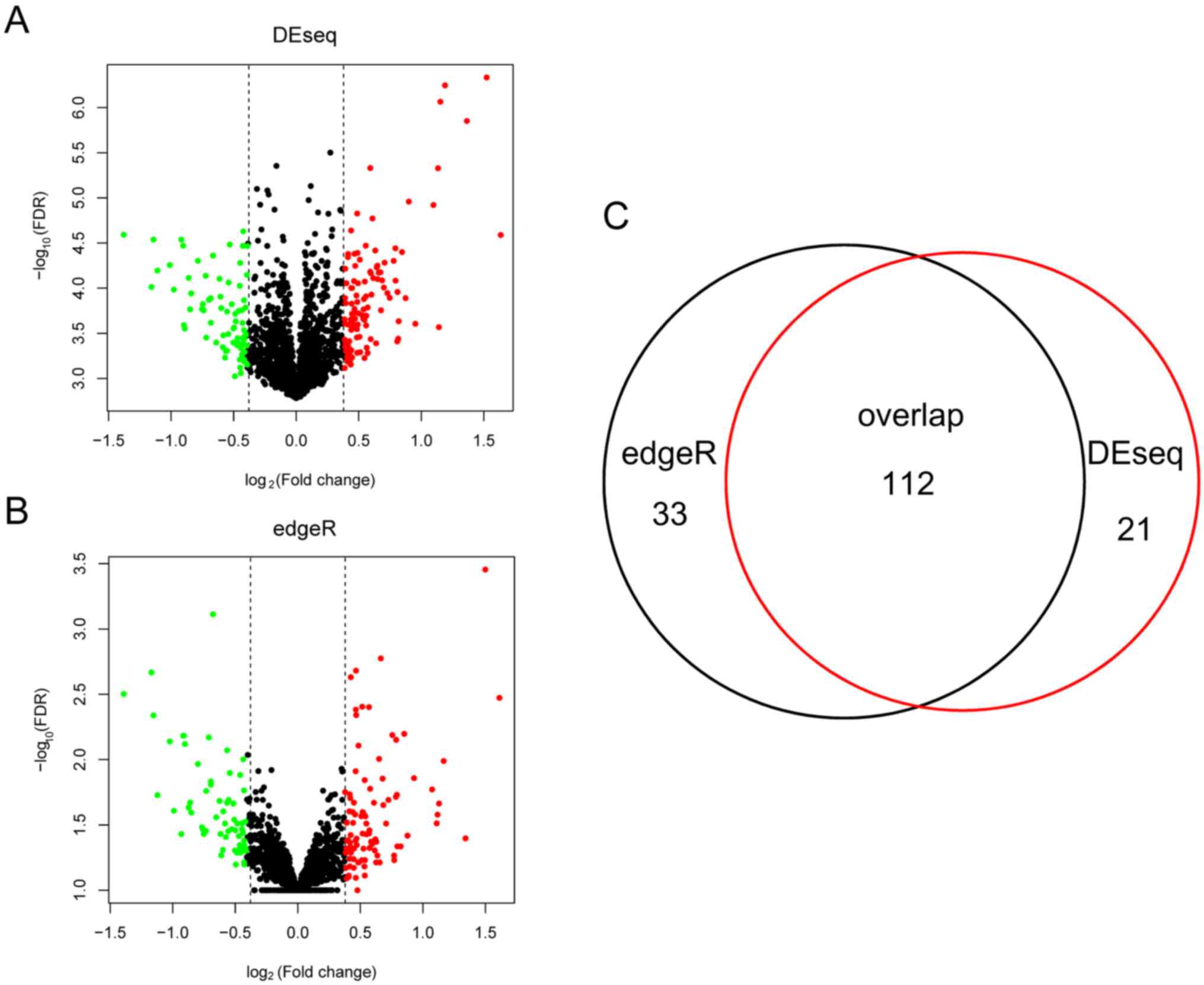
group. As presented in Fig. 1, the
edgeR method detected 145 DELs between the two groups, whereas the
DESeq method identified 133 DELs. A total of 112 DELs were shared
by the two methods and were selected for further analyses.
Three-lncRNAs signature for survival
prediction
Out of the 112 overlapping DELs, 33 lncRNAs were
determined to be significantly associated with survival (P<0.05;
Table II), and were then included in
the multivariate Cox regression analysis. Subsequently, the lncRNAs
KB-1836B5, long intergenic non-protein coding RNA 566 (LINC00566)
and FAM27L were selected following multivariate Cox regression
analysis and used to construct a risk scoring system as follows:
Risk score=[(−1.10205) × Exp KB-1836B5] + [(−0.58589) × Exp
LINC00566] + [(−1.69273) × Exp FAM27L]. Exp was taken as the
expression level of the lncRNA.
| Table II.Three prognostic lncRNAs analyzed by
multivariate Cox regression analysis. |
Table II.
Three prognostic lncRNAs analyzed by
multivariate Cox regression analysis.
| LncRNA | Cox regression
coefficient | 95% CI | P-value |
|---|
| KB-1836B5 | −1.102 | 0.163–0.676 | 0.002a |
| LINC00566 | −0.586 | 0.362–0.857 | 0.008a |
| FAM27L | −1.693 | 0.044–0.761 | 0.020a |
| LINC00706 | 0.71872 | 0.9464–4.4483 | 0.039a |
| LINC01040 | 0.74652 | 0.93732–4.7482 | 0.047a |
| RP11-49K4 | −0.18254 | 0.67114–1.0343 | 0.050 |
| LINC01109 | 0.39972 | 0.80386–2.767 | 0.205 |
| KB-1254G8 | 0.71192 | 0.66577–6.2379 | 0.212 |
| GM140 | 0.92532 |
0.54335–11.7124 | 0.238 |
| LINC01195 | 0.31406 | 0.77485–2.4187 | 0.280 |
| GHET1 | −0.70253 | 0.13395–1.8317 | 0.292 |
| LINC01447 | −0.26729 | 0.46046–1.2724 | 0.303 |
| RP1-84O15 | −0.3337 | 0.37818–1.3566 | 0.306 |
| FOXD1-AS1 | −0.88184 | 0.07468–2.2952 | 0.313 |
| LINC00032 | −0.95171 | 0.05269–2.8292 | 0.349 |
| DIRC3-AS1 | 0.83933 |
0.31433–17.0467 | 0.410 |
| RP11-490B18 | 0.07483 | 0.88091–1.3185 | 0.467 |
| LINC01135 | −0.25925 | 0.37569–1.5848 | 0.480 |
| LINC01162 | 0.14533 | 0.74264–1.8008 | 0.520 |
| LINC00364 | −0.304 | 0.28455–1.9133 | 0.532 |
| RP11-61L19 | −0.11059 | 0.62889–1.2746 | 0.540 |
| LINC00562 | −0.15952 | 0.5056–1.4376 | 0.550 |
| RP11-680F200 | 0.11108 | 0.77155–1.6185 | 0.557 |
| LINC01514 | 0.15865 | 0.63662–2.1573 | 0.610 |
| MIR219A2 | 0.10826 | 0.72297–1.7175 | 0.624 |
| LINC00701 | 0.15113 | 0.58805–2.3007 | 0.664 |
| LINC01349 | −0.14309 | 0.37503–2.0029 | 0.738 |
| TTTY1B | 0.43506 |
0.06779–35.2169 | 0.785 |
| TTTY23 | −0.26192 | 0.0745–7.9493 | 0.826 |
| TTTY3 | −0.19517 | 0.10562–6.4082 | 0.852 |
| LINC00366 | −0.03284 | 0.6815–1.3741 | 0.854 |
| LINC01034 | −0.03165 | 0.48084–1.9521 | 0.929 |
| D21S2088E | 0.0173 | 0.36988–2.7988 | 0.973 |
The 160 patients in the training set were classified
into a high-risk (n=80) and low-risk group (n=80) by the
three-lncRNAs panel-based risk scoring system, with the median risk
score as the cut-off value.
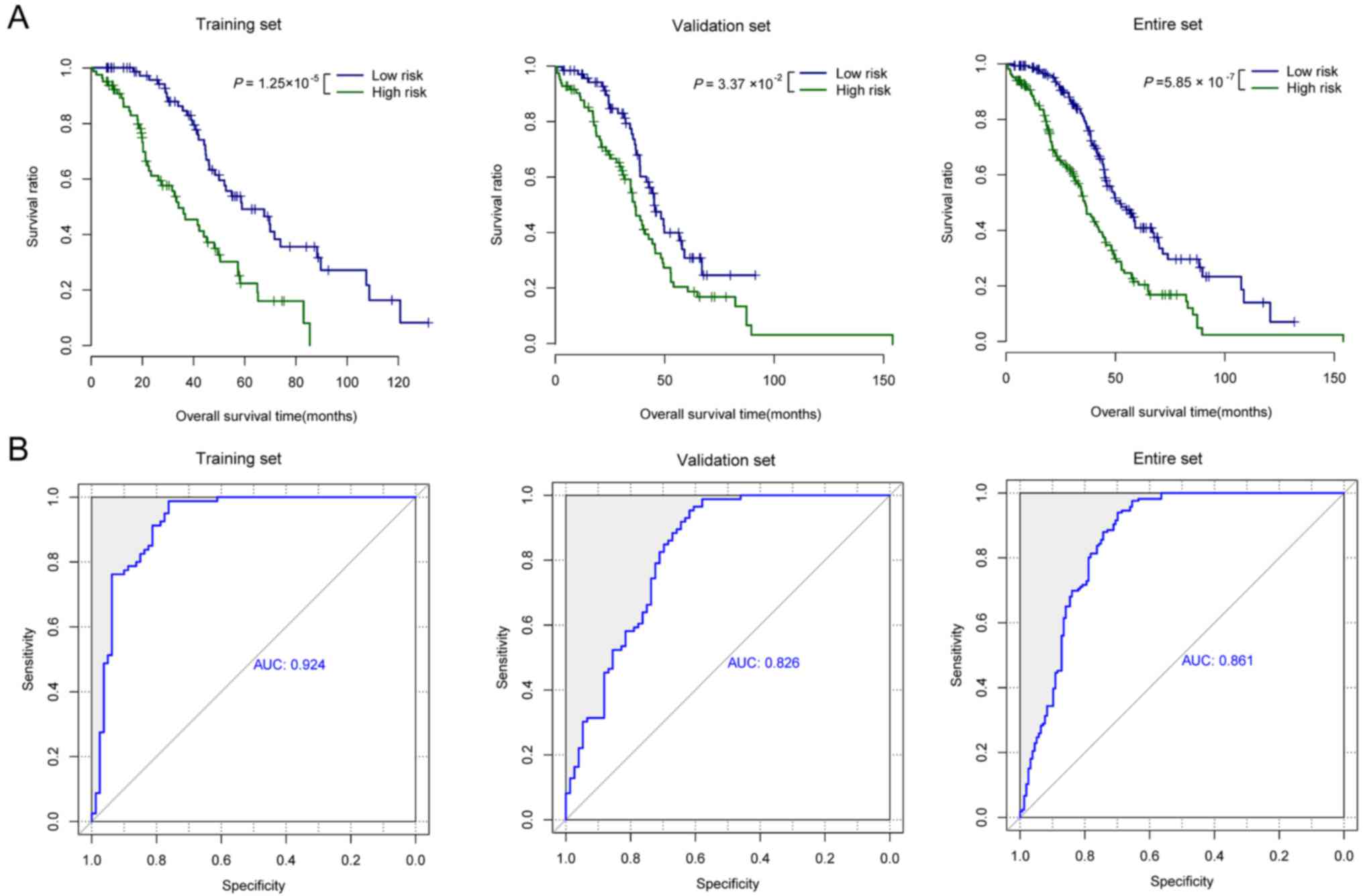
The Kaplan-Meier survival analysis demonstrated
notable differences in OS time between the two risk groups
(28.29±21.25 months for the high-risk group vs. 48.07±28.85 months
for the low-risk group; log-rank P<0.001; Fig. 2A). The AUC for the three-lncRNAs panel
in the training set was 0.924 (Fig.
2B). In order to validate the prognostic capability of the
three-lncRNAs panel, 177 patients of the validation set were
grouped into high-risk and low-risk groups with the risk scoring
system based on the three-lncRNAs derived from the training set.
The results of the validation set (32.47±21.02 months for the
high-risk group vs 36.66±31.79 months for the low-risk group;
log-rank P<0.001; Fig. 2A) were
similar to the observations for the training set. The AUC of the
validation set was 0.826 (Fig. 2B).
The entire set was also classified into a high-risk and low-risk
group with the same three-lncRNAs panel and the β value derived
from the training set. Similarly, the OS time was significantly
different between the two risk groups (32.47±21.02 months for the
high-risk group vs. 36.66±31.79 months for the low-risk group;
log-rank P<0.001; Fig. 2A), with
an AUC of 0.861 (Fig. 2B).
Association of the expression levels
of the three prognostic lncRNAs with OS time
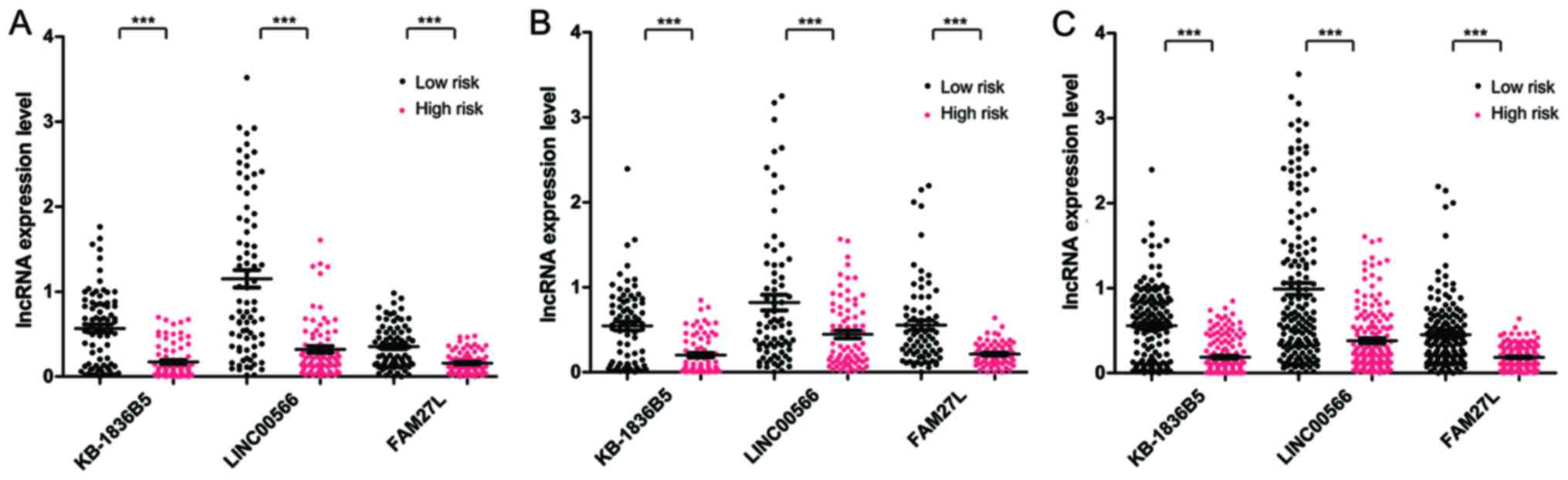
Expression levels of KB-1836B5, LINC00566 and FAM27L
were significantly different between the high-risk and low-risk
group in the training set, the validation set and the entire set
(P<0.005, Fig. 3). All samples in
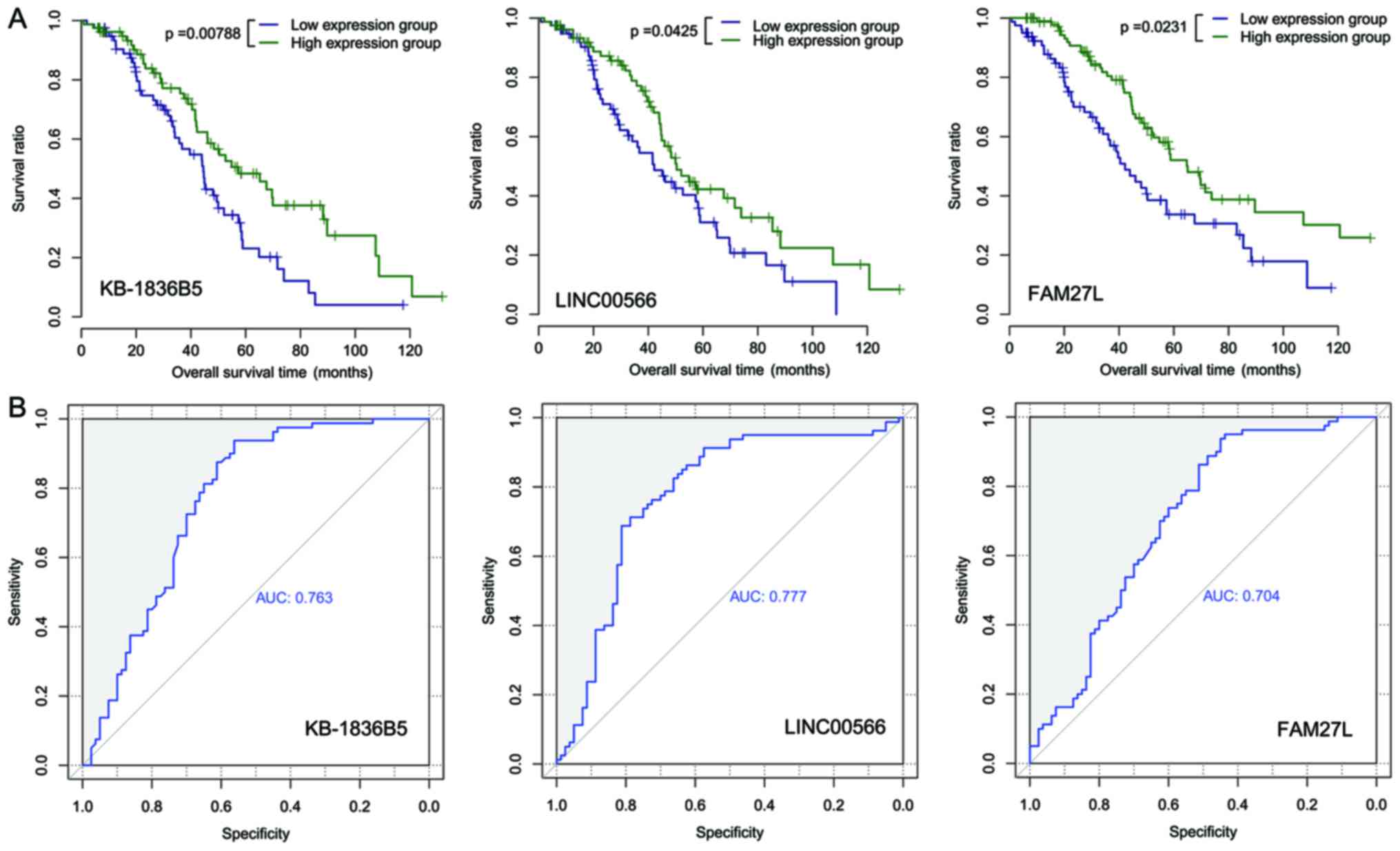
the training set (n=160) were assorted into a high-expression and a
low-expression group. There were notable differences in the OS time
between the two expression groups for each prognostic lncRNA
(P=0.0079 and AUC=0.763 for KB-1836B5; P=0.043 and AUC=0.777 for
LINC00566; and P=0.023 and AUC=0.704 for FAM27L; Fig. 4).
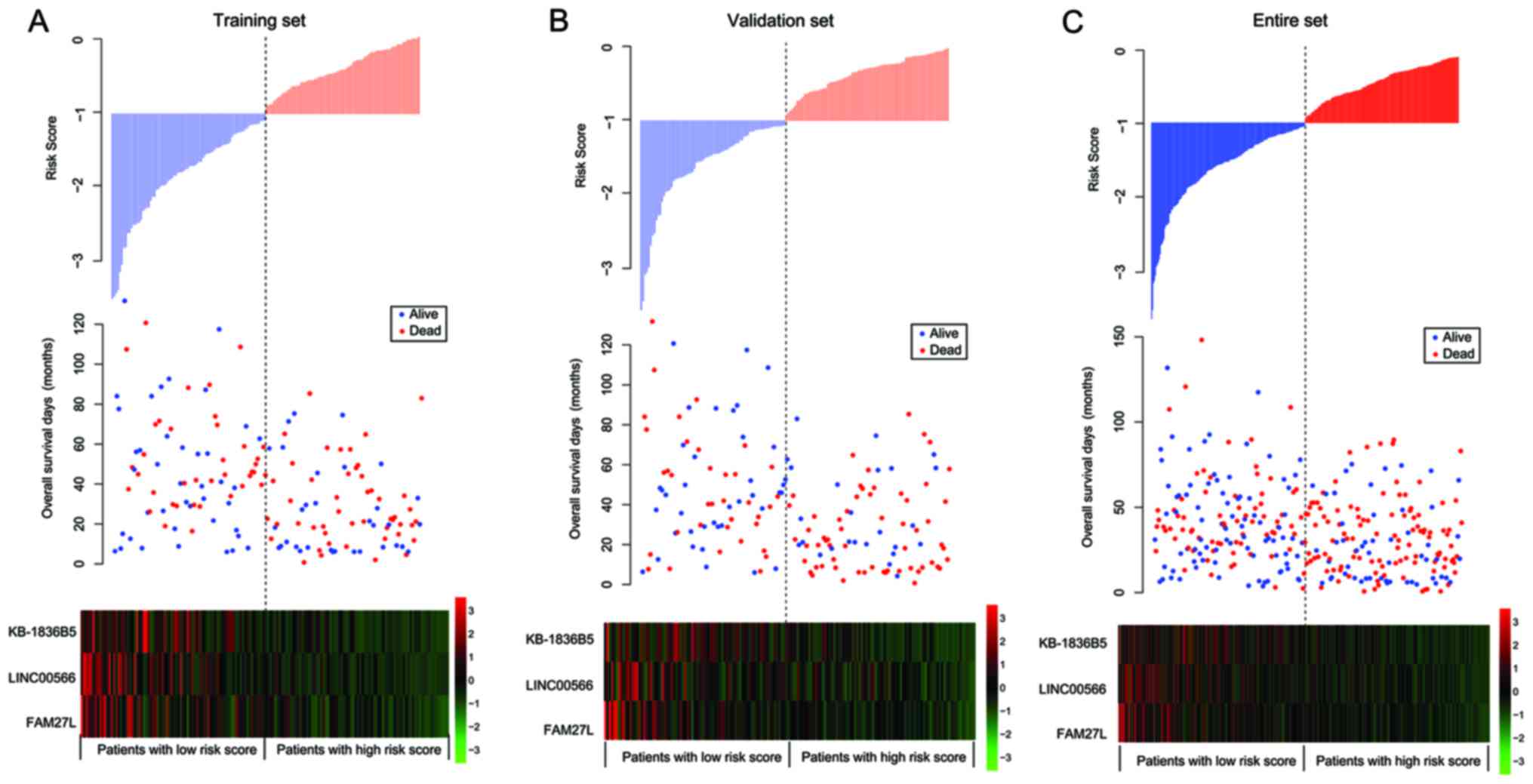
The distribution of the three-lncRNAs risk score, OS
time of patients and the expression profiles of the three
prognostic lncRNAs in the training, validation and entire sets is
presented in Fig. 5. The expression
levels of KB-1836B5, LINC00566 and FAM27L were upregulated in the
low-risk patients, compared with the high-risk patients.
Independence of the prognostic
performance of the three-lncRNAs panel from other clinical
variables
Univariate and multivariate Cox regression, and data
stratification analyses were performed in order to determine
whether the prognostic power of the three-lncRNAs signature was
independent of other clinical variables, including neoplasm
histological grade, lymphatic invasion and tumor recurrence.
Univariate Cox regression analysis determined that the
three-lncRNAs risk score was significantly associated with OS time
in the training set (P<0.001; 95% CI=1.736–4.258), the
validation set (P=0.034; 95% CI=0.969–2.135) and the entire set
(P<0.001; 95% CI=1.476–2.636; Table
III). Furthermore, via multivariate Cox regression analyses,
the three-lncRNAs risk score was determined to be an independent
predictor of survival in the training set (P<0.001; 95%
CI=2.787–15.389), the validation set (P=0.013; 95% CI=1.241–6.200)
and the entire set (P≤0.001; 95% CI=2.487–7.549; Table III).
| Table III.Univariate and multivariate Cox
regression analyses with three-lncRNAs risk score and other
clinical variables. |
Table III.
Univariate and multivariate Cox
regression analyses with three-lncRNAs risk score and other
clinical variables.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Training set
(n=160) |
|
|
|
|
|
|
|
Three-lncRNAs risk score
(low/high risk) | 2.718 | 1.736–4.258 |
<0.001a | 6.548 | 2.787–15.389 |
<0.001a |
| Age
(≤60/>60) | 1.171 | 0.763–1.798 | 0.471 | 1.879 | 0.938–3.764 | 0.075 |
|
Neoplasm histological grade
(G1+G2/G3+G4) | 0.978 | 0.370–2.584 | 0.964 | 1.207 | 0.476–3.063 | 0.692 |
|
Lymphatic invasion
(yes/no) | 1.281 | 0.659–2.488 | 0.466 | 1.115 | 0.556–2.235 | 0.759 |
| Tumor
recurrence (yes/no) | 1.139 | 0.686–1.888 | 0.615 | 0.355 | 0.149–0.849 | 0.020 |
| Validation set
(n=177) |
|
|
|
|
|
|
|
Three-lncRNAs risk score
(low/high risk) | 1.439 | 0.969–2.135 | 0.033a | 2.773 | 1.241–6.200 | 0.013a |
| Age,
years (≤60/>60) | 1.350 | 0.913–1.997 | 0.133 | 1.195 | 0.489–2.920 | 0.696 |
|
Neoplasm histological grade
(G1+G2/G3+G4) | 1.887 | 1.115–3.192 | 0.016a | 4.229 | 0.828–5.613 | 0.083 |
|
Lymphatic invasion
(yes/no) | 1.129 | 0.399–1.965 | 0.765 | 0.998 | 0.388–2.570 | 0.997 |
| Tumor
recurrence (yes/no) | 1.193 | 0.552–1.273 | 0.408 | 0.809 | 0.353–1.856 | 0.617 |
| Entire set
(n=337) |
|
|
|
|
|
|
|
Three-lncRNAs risk score
(low/high risk) | 1.972 | 1.476–2.636 |
<0.001a | 4.333 | 2.487–7.549 |
<0.001a |
| Age,
years (≤60/>60) | 1.260 | 0.947–1.686 | 0.112 | 1.454 | 0.871–2.428 | 0.152 |
|
Neoplasm histological grade
(G1+G2/G3+G4) | 1.584 | 1.063–2.362 | 0.024a | 1.931 | 0.928–4.020 | 0.079 |
|
Lymphatic invasion
(yes/no) | 1.133 | 0.681–1.884 | 0.632 | 1.148 | 0.670–1.966 | 0.615 |
| Tumor recurrence
(yes/no) | 1.059 | 0.686–1.300 | 0.726 | 0.620 | 0.357–1.079 | 0.091 |
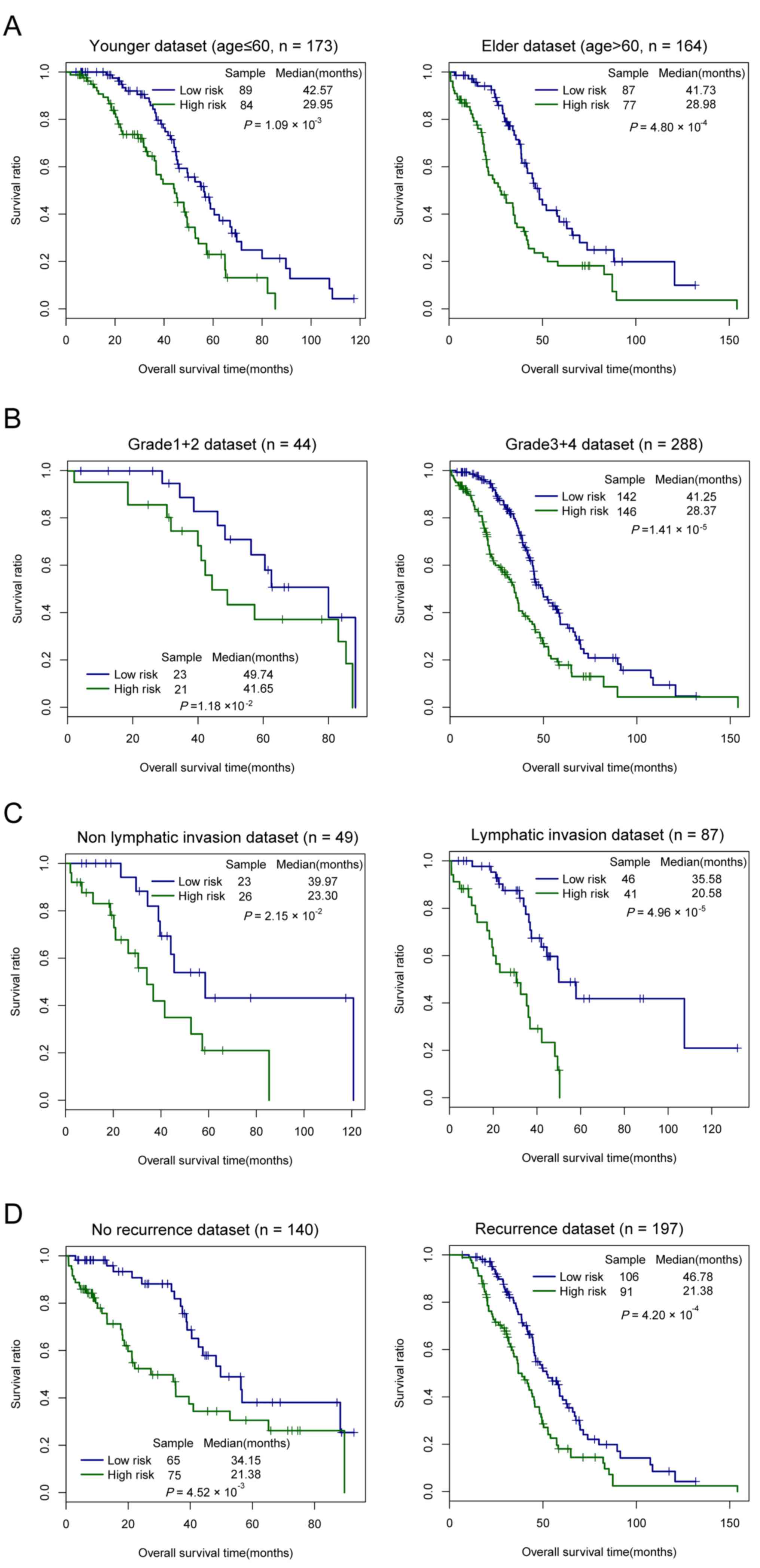
Data stratification analyses were carried out
according to age, neoplasm histological grade, lymphatic invasion
and tumor recurrence, individually (Table IV; Fig.
6). All patients in the entire set were stratified by age into
a younger (≤60 years) and elder dataset (>60 years). The younger
dataset was further divided according to the three-lncRNAs
signature into a low-risk and a high-risk group, which had
significantly different OS time (median, 42.57 vs. 29.29 months,
respectively; log-rank P=0.001; Fig.
6A). Similarly, the elder dataset was further classified into a
low-risk and high-risk group, and the difference in OS time between
the two risk groups was also significant (median, 41.73 vs. 28.98
months, respectively; log-rank P<0.001; Fig. 6A). Subsequently, all patients of the
entire set were stratified by neoplasm histological grade into a
grade 1+2 and grade 3+4 dataset. These datasets was classified by
the three-lncRNAs signature into a low-risk group with longer OS
time and a high-risk group with shorter OS time (median, 49.74 vs.
41.65 months, respectively; log-rank P=0.012, for the grade 1+2
dataset; median, 41.25 vs. 28.37 months, respectively; log-rank
P<0.001, for the grade 3+4 dataset; Fig. 6B). Similarly, all patients were
stratified by lymphatic invasion into a non-lymphatic invasion and
lymphatic invasion dataset. In the two datasets, differences in OS
time between the low-risk and high-risk groups were significant
(median, 39.97 vs. 23.30 months, respectively; log-rank P=0.022,
for the non-lymphatic invasion dataset; median, 35.58 vs. 20.58
months, respectively; log-rank P≤0.001, for the lymphatic invasion
dataset; Fig. 6C). Subsequently, all
patients were stratified by tumor recurrence into a no recurrence
and a recurrence dataset. Significant differences between the
low-risk and high-risk groups were observed in the two datasets
(median 34.15 vs. 21.38 months, respectively; log-rank P=0.005, for
the no recurrence dataset; median, 46.78 vs. 21.38 months,
respectively; log-rank P<0.001, for the recurrence dataset;
Fig. 6D). These data indicated that
the prognostic value of the three-lncRNAs panel is independent from
age, neoplasm histological grade, lymphatic invasion and tumor
recurrence.
| Table IV.Results of data stratification
analysis. |
Table IV.
Results of data stratification
analysis.
|
| Univariate
analysis |
|---|
|
|
|
|---|
| Variables | HR | 95% CI | P-value |
|---|
| Age, years |
|
|
|
| ≤60
(n=173) | 2.012 | 1.323–3.061 | 0.001 |
| >60
(n=164) | 2.085 | 1.380–3.148 | <0.001 |
| Neoplasm
histological grade |
|
|
|
| G1+G2
(n=44) | 1.949 | 0.844–4.500 | 0.012 |
| G3+G4
(n=288) | 1.993 | 1.460–2.721 | <0.001 |
| Lymphatic
invasion |
|
|
|
| Yes
(n=49) | 3.940 | 2.032–7.642 | <0.001 |
| No
(n=87) | 2.761 | 1.161–6.563 | 0.022 |
| Tumor
recurrence |
|
|
|
| Yes
(n=197) | 1.845 | 1.313–2.594 | <0.001 |
| No
(n=140) | 2.274 | 1.290–4.009 | 0.005 |
Potential functions of the
three-lncRNAs signature in OvCa tumorigenesis
It has been demonstrated that lncRNAs affect various
cellular processes such as organ or tissue development, cellular
transport or metabolic processes via regulating protein-coding
genes (19). For the purpose of
determining the potential functions of the three prognostic lncRNAs
in OvCa, the protein-coding genes associated with ≥1 of the three
prognostic lncRNAs with a Spearman correlation coefficient >0.4
were selected. Subsequently, GO function and KEGG pathway
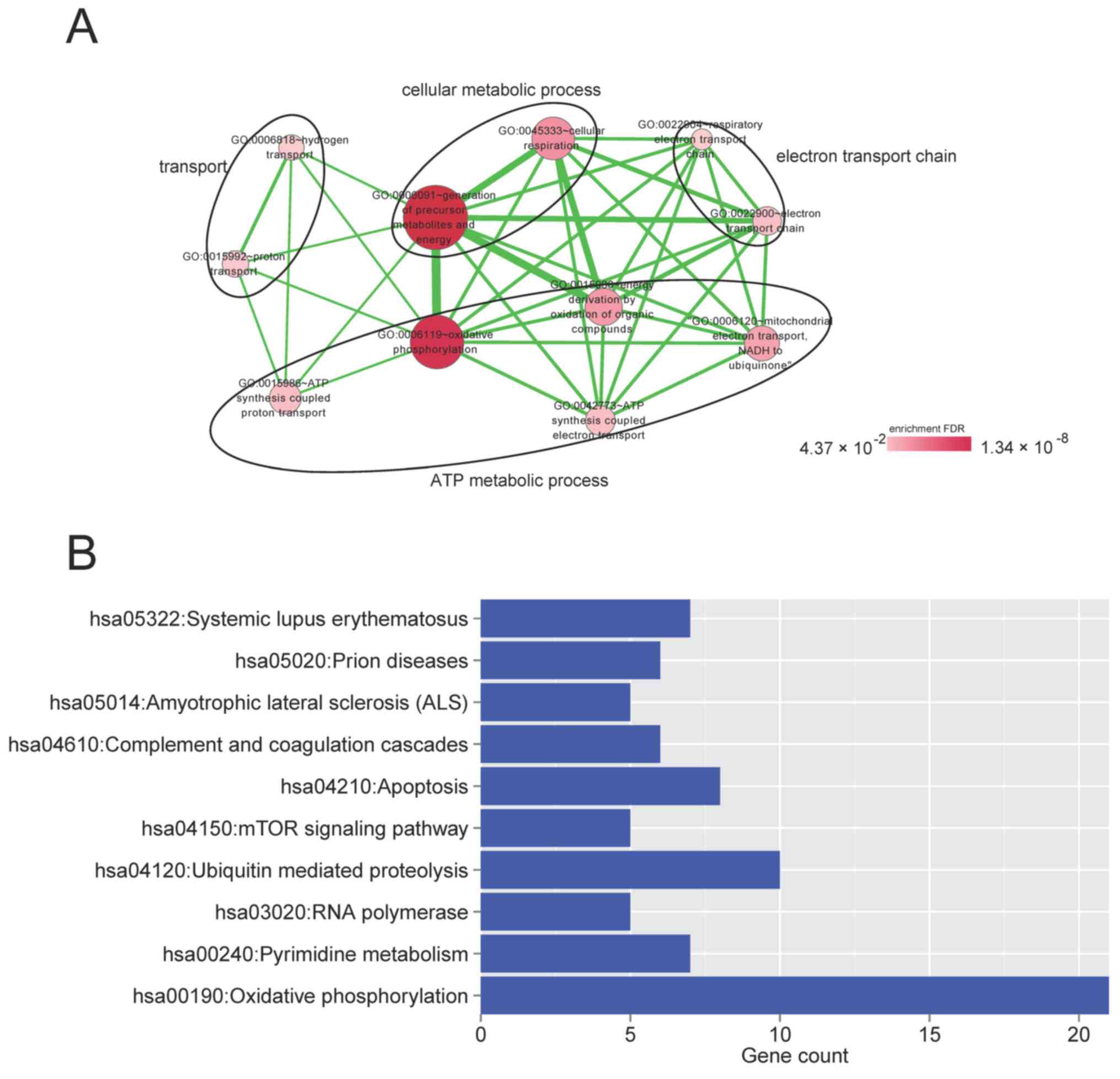
enrichment analyses were performed for these genes. As presented in
Fig. 7A, these genes were
significantly associated with 13 GO terms (P<0.05), which were
categorized into four functional clusters, ATP metabolic process,
transport, electron transport chain and cellular metabolic process.
Furthermore, 10 pathways were significantly associated with these
genes (P<0.05), consisting of oxidative phosphorylation, prion
diseases, RNA polymerase, apoptosis, ubiquitin-mediated
proteolysis, complement and coagulation cascades, pyrimidine
metabolism, systemic lupus erythematosus, mechanistic target of
rapamycin (mTOR) signaling pathway and amyotrophic lateral
sclerosis pathways (Fig. 7B).
Discussion
OvCa has the highest malignancy incidence of all
gynecological malignancy types globally in 2013 (20). LncRNAs may serve as independent
biomarkers for survival prediction, due to lncRNAs not coding
proteins (21). To investigate an
lncRNA-based signature for predicting the prognosis of patients
with OvCa, the present study initially selected 112 DELs between
patients with good prognosis and poor prognosis using DESeq and
edgeR methods. On the basis of the results of univariate and
multivariate Cox regression analysis, a three-lncRNAs signature
(lncRNA KB-1836B5, LINC00566 and FAM27L) for survival prediction
was determined and used to construct a risk scoring system. The
three-lncRNAs signature-based risk scoring system classified the
patients in the training set into a high-risk and low-risk group,
indicating significantly different OS time. The risk stratification
capability of the three-lncRNAs signature was confirmed in the
validation and entire set. Furthermore, the results of the
multivariate Cox regression analysis and data stratification
analysis demonstrated that the prognostic value of the
three-lncRNAs signature was independent of age, neoplasm
histological grade, lymphatic invasion and tumor recurrence. It is
notable that histology was not a significant predictor of survival,
and established risk factors for the patient's outcome, including
tumor stage and residual tumor, were not included in the
multivariate model. Although tumor stage is an important prognostic
factor in OvCa (20), only
early-stage samples were selected for analysis in the present
study, with the majority of samples being stage II and III;
therefore, the samples were considered to be at a ‘fixed’ stage,
and were not used in the analysis. Furthermore, there are numerous
clinical features that are associated with the prognosis of
patients with OvCa (22,23), but clinical information for these
features were not available for all patients in the datasets
downloaded from TCGA. Furthermore, the aim of the multivariate Cox
regression analyses was to investigate whether the three-lncRNAs
signature was a significant variable, and whether it was
independent of other clinical variables; thus, only available
clinical features were analyzed in the present study. The results
of the present study demonstrated that the three-lncRNAs panel may
be a promising independent biomarker to predict the OS time of
patients with OvCa.
It has been demonstrated that lncRNAs serve a
function in a number of biological processes by functioning as
important regulators of gene regulation at transcriptional,
posttranscriptional and epigenetic levels (24,25);
therefore, the present study investigated the protein-coding genes
regulated by the three prognostic lncRNAs, in order to determine
their possible biological function in the molecular mechanisms of
OvCa. In the present study, the protein-coding genes associated
with ≥1 of the three prognostic lncRNAs (Spearman correlation
coefficient >0.4) were selected. Results of GO function
enrichment analysis demonstrated that these genes were
significantly associated with ATP metabolic process, transport,
electron transport chain and cellular metabolic process.
Furthermore, these genes were significantly enriched in a number of
KEGG signaling pathways, including the mTOR signaling pathway,
ubiquitin-mediated proteolysis, and complement and coagulation
cascade pathways. mTOR, a member of phosphoinositide
3-kinase-associated kinase family of protein kinases, is a
serine/threonine protein kinase (26). The mTOR signaling pathway serves a
central function in a number of major cellular processes, including
cell growth, cell proliferation and cell survival, and is being
identified to be involved in an increasing number of diseases,
including cancer, obesity and type 2 diabetes (27,28).
Previous studies have demonstrated that mTOR pathway activation is
frequently observed in OvCa and is involved in tumorigenesis and
progression, indicating this pathway as a potential therapeutic
target for OvCa (29–31). Ubiquitin-mediated proteolysis serves a
pivotal function in protein turnover, thereby exerting a regulatory
effect on carcinogenesis-associated cellular processes, including
cell cycle, apoptosis and gene transcription (32). Furthermore, proteasome inhibitors have
attracted considerable interest as a treatment option for solid
tumor types (33). Complement is a
central part of innate immunity, and is also involved in the
adaptive immune response, inflammation and other biological
processes (34). Emerging studies
have demonstrated that complement activation exerts a
tumor-promoting effect by strengthening tumor growth and metastasis
(35,36). Results of the present study indicated
that the lncRNAs KB-1836B5, LINC00566 and FAM27L may exert an
effect on mTOR signaling pathway, ubiquitin-mediated proteolysis,
and complement and coagulation cascade pathways via gene
regulation, thus influencing OvCa cancerogenesis and
progression.
Currently, the investigation of lncRNAs is in the
early stages. According to the literature surveyed, there are few
studies involving lncRNAs KB-1836B5, LINC00566 and FAM27L. To the
best of our knowledge, their involvement in OvCa has not been
reported previously. The present study determined and validated a
three-lncRNAs predictive signature via comprehensive analysis,
based on lncRNA expression files downloaded from TCGA.
However, two limitations of the present study should
be mentioned. First, the number of patients in the training set
(n=160) and validation set (n=177) is limited, which may affect the
prediction accuracy of this three-lncRNAs prognostic signature;
therefore, further work is required to verify the results of the
present study in a larger cohort of patients prior to applications
of these data in the clinic. Secondly, the present results were all
derived from bioinformatics analysis of clinical data, and no
direct in vitro experimental validations were performed.
Although bioinformatics has been a reliable method to select the
genetic factors implicated in the cellular progress, molecular
function, and even tumorigenesis and its prognosis, and provides a
possible method to screen the majority of potential factors from a
huge information pool, it is considered that the present study will
be more reliable following validation with cell or animal
experiments; therefore, determining the potential factors is the
first step to accelerate the study of tumor mechanisms, and further
in vitro analyses required to validate the present
results.
In conclusion, the present study identified and
validated a three-lncRNAs signature for survival prediction in
OvCa. The prognostic capability of this signature was independent
of other clinical variables and may be recommended as a promising
prognostic biomarker for OvCa. The three prognostic lncRNAs are
associated with several cellular processes and signaling pathways,
including the mTOR signaling pathway, ubiquitin-mediated
proteolysis, and complement and coagulation cascades pathways. The
present study provides an insight into the involvement of lncRNAs
into the molecular mechanisms of OvCa.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Fund for Outstanding Young Scholars (grant no. 81322039);
the National Natural Science Foundation (grant no. 81602884); and
the National Nature Science Foundation of China (grant nos.
81472442 and 81272871).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HJL performed data analyses and wrote the
manuscript. MG, MZ and XRW contributed significantly to the data
analyses and provided important suggestions. WJC and YKX conceived
and designed the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. Ca A Cancer J Clin. 65:5–29. 2014. View Article : Google Scholar
|
|
2
|
Baldwin LA, Huang B, Miller RW, Tucker T,
Goodrich ST, Podzielinski I, DeSimone CP, Ueland FR, van Nagell JR
and Seamon LG: Ten-year relative survival for epithelial ovarian
cancer. Obstet Gynecol. 120:612–618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dey BK, Mueller AC and Dutta A: Long
non-coding RNAs as emerging regulators of differentiation,
development, and disease. Transcription. 5:e9440142014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta (BBA)-Gene Regul Mech.
1839:1097–1109. 2014. View Article : Google Scholar
|
|
5
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:12532015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren C, Li X, Wang T, Zhao C, Liang T, Zhu
Y, Li M, Yang C, Zhao Y and Zhang GM: Functions and mechanisms of
long noncoding RNAs in ovarian cancer. Int J Gynecol Cancer.
25:566–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng Z, Guo J, Chen L, Luo N, Yang W and
Qu X: A long noncoding RNA AB073614 promotes tumorigenesis and
predicts poor prognosis in ovarian cancer. Oncotarget.
6:25381–25389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yim GW, Kim HJ, Kim LK, Kim SW, Kim S, Nam
EJ and Kim YT: Long non-coding RNA HOXA11 antisense promotes cell
proliferation and invasion and predicts patient prognosis in serous
ovarian cancer. Cancer Res Treat. 49:656–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen ZJ, Zhang Z, Xie BB and Zhang HY:
Clinical significance of up-regulated lncRNA NEAT1 in prognosis of
ovarian cancer. Eur Rev Med Pharmacol Sci. 20:3373–3377.
2016.PubMed/NCBI
|
|
10
|
Zhou M, Wang X, Shi H, Cheng L, Wang Z,
Zhao H, Yang L and Sun J: Characterization of long non-coding
RNA-associated ceRNA network to reveal potential prognostic lncRNA
biomarkers in human ovarian cancer. Oncotarget. 7:12598–12611.
2016.PubMed/NCBI
|
|
11
|
Bristow RE, Palis BE, Chi DS and Cliby WA:
The national cancer database report on advanced-stage epithelial
ovarian cancer: Impact of hospital surgical case volume on overall
survival and surgical treatment paradigm. Gynecol Oncol.
118:262–267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anders S and Huber W: Differential
expression of RNA-Seq data at the gene level-the DESeq package.
Embl. 2012.
|
|
13
|
Robinson MD, Mccarthy DJ and Smyth GK:
edgeR: A bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang P, Wang Y, Bo H, Zou X and Mao JH: A
novel gene expression-based prognostic scoring system to predict
survival in gastric cancer. Oncotarget. 7:55343–55351.
2016.PubMed/NCBI
|
|
15
|
Hanley JA: The robustness of the
‘binormal’ assumptions used in fitting ROC curves. Med Decis
Making. 8:197–203. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID bioinformatics resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35:169–175. 2007. View Article : Google Scholar
|
|
17
|
Consortium GO: Gene ontology consortium:
Going forward. Nucleic Acids Res. 43:1049–1056. 2015. View Article : Google Scholar
|
|
18
|
Minoru K, Yoko S, Masayuki K, Miho F and
Mao T: KEGG as a reference resource for gene and protein
annotation. Nucleic Acids Res. 44:457–462. 2016. View Article : Google Scholar
|
|
19
|
Qi L, Liu C, Yuan X, Kang S, Miao R, Xiao
H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction of
long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jayson GC, Kohn EC, Kitchener HC and
Ledermann JA: Ovarian cancer. Lancet. 384:1376–1388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nina H and Damjan G: Long non-coding RNA
in cancer. Int J Mol Sci. 14:4655–4669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rooth C: Ovarian cancer: Risk factors,
treatment and management. Br J Nurs. 22:S23–S30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hunn J and Rodriguez GC: Ovarian cancer:
Etiology, risk factors, and epidemiology. Clin Obstet Gynecol.
55:3–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nature
Rev Genet. 15:7–21. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11:592013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dello Russo C, Lisi L, Feinstein DL and
Navarra P: mTOR kinase, a key player in the regulation of glial
functions: Relevance for the therapy of multiple sclerosis. Glia.
61:301–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mabuchi S, Kuroda H, Takahashi R and
Sasano T: The PI3K/AKT/mTOR pathway as a therapeutic target in
ovarian cancer. Gynecol Oncol. 137:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Zeng J and Shen K: PI3K/AKT/mTOR
signaling pathway as a therapeutic target for ovarian cancer. Arch
Gynecol Obstet. 290:1067–1078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dobbin ZC and Landen CN: The importance of
the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int
J Mol Sci. 14:8213–8227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bassermann F, Eichner R and Pagano M: The
ubiquitin proteasome system-implications for cell cycle control and
the targeted treatment of cancer. Biochim Biophys Acta.
1843:150–162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Johnson DE: The ubiquitin-proteasome
system: Opportunities for therapeutic intervention in solid tumors.
Endocr Relat Cancer. 22:1–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Varela JC and Tomlinson S: Complement: An
overview for the clinician. Hematol Oncol Clin North Am.
29:409–427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Afshar-Kharghan V: The role of the
complement system in cancer. J Clin Invest. 127:780–789. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pio R, Corrales L and Lambris JD: The role
of complement in tumor growth. Tumor Microenviron Cell Stress.
229–262. 2013.
|