Introduction
Natural products and their respective metabolites
are valuable resources that have enabled the detection of novel
chemotherapeutic agents (1). Among
these, alkaloids have demonstrated promising anti-cancer effects.
These compounds have also been revealed to elicit effects on a
variety of different targets, including regulators associated with
cell cycle progression, cell apoptosis and drug resistance
inhibition (2). Chaetominine is an
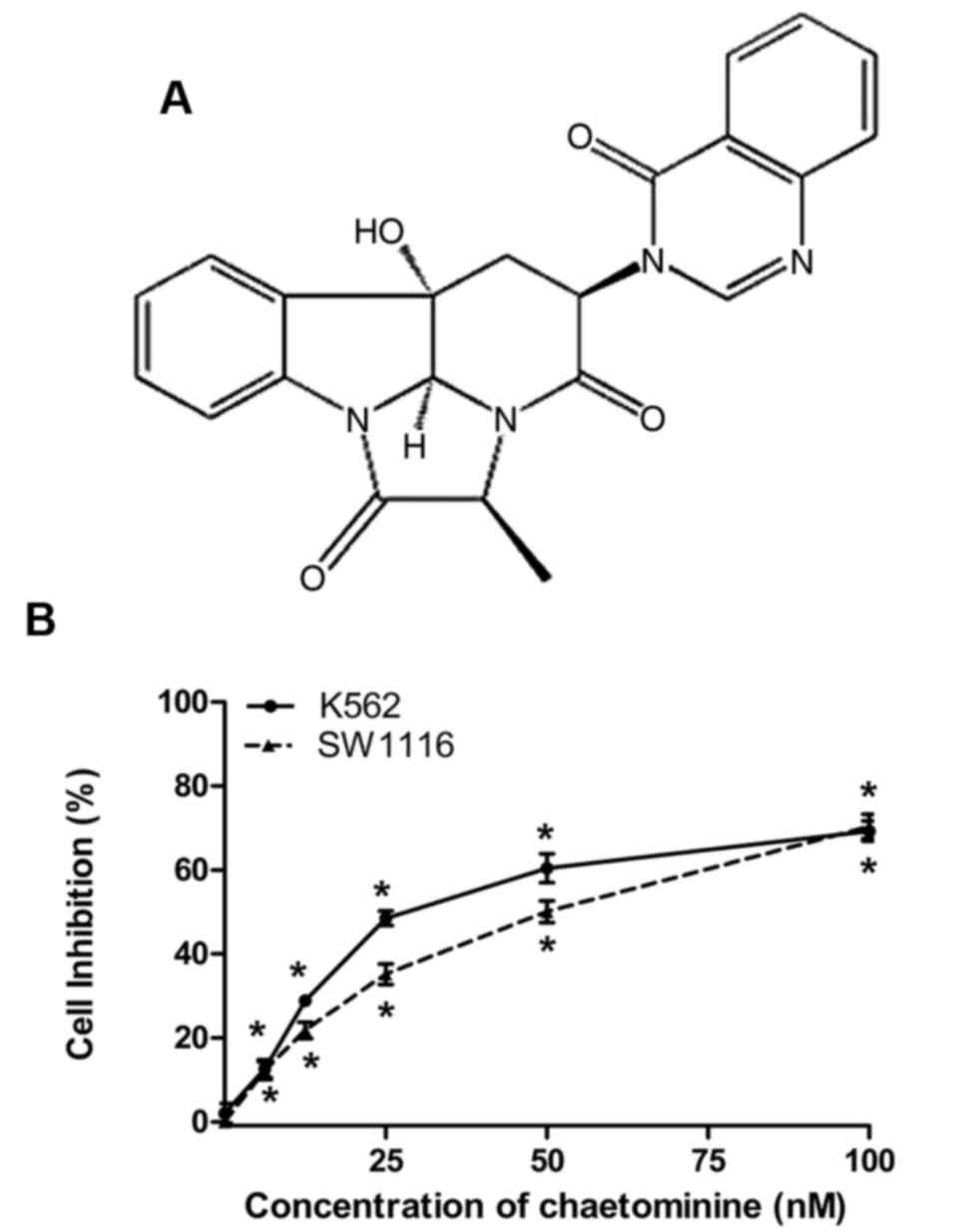
alkaloid (Fig. 1A) isolated from the
metabolites of an endophytic fungus, Aspergillus fumigatus
CY018 (3). A previous study
demonstrated that chaetominine may be lethal to human leukemia K562
cells, with its effects being mediated through the mitochondrial
apoptosis pathway (4). Previous
studies have indicated that similar compounds have the capacity to
inhibit cancer cell growth by inducing cell apoptosis and/or cell
cycle arrest (4–6).
Efficient regulation of the cell cycle is crucial to
the process of cell survival and involves the prevention of
uncontrolled cell division alongside the detection and repair of
genetic damage associated with tumorigenesis (7). Checkpoints are pivotal components of the
cell cycle regulative machinery and are governed by effector
kinases, including ataxia telangiectasia mutated (ATM) and ATM and
Rad3-related (ATR) proteins. The predominant downstream transducers
of checkpoints include checkpoint kinase 1 and checkpoint kinase 2
(Chk1 and Chk2) as well as p53 (8).
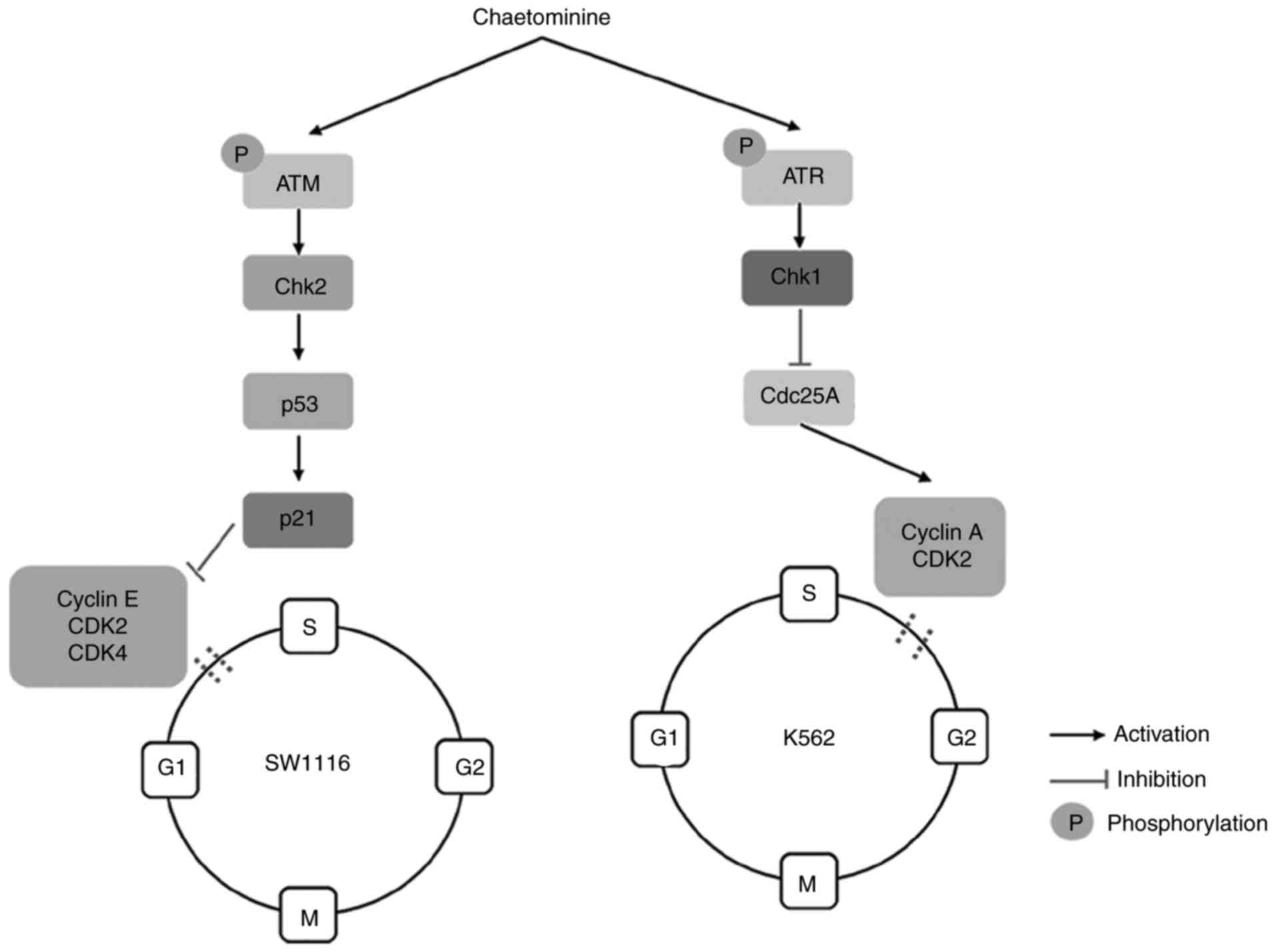
The activation of the p53-p21 cascade in the ATM/Chk2/p53 signaling
pathway facilitates the induction of G1-phase arrest
(7). Conversely, S-phase arrest is
primarily triggered by multiple pathways that involve the
inhibition of cell division cycle 25A (cdc25A). These pathways
transmit checkpoint signals to cyclin-dependent kinases (CDKs),
which form complexes with cyclins, resulting in cell cycle arrest
(9). CDK2 and CDK4 are responsible
for G1/S transitions during the cell cycle. These events
occur following the interaction of kinases with their respective
cyclin complex subunits. The binding of cyclin E with
G1-phase CDK2 promotes the transition of
G1-to S-phase, while cyclin A is required to activate
CDK2 for progression through the S-phase (9,10). Once
cell cycle arrest occurs, related signaling pathways are activated,
leading to the initiation of the cell death program. This results
in the inhibition of cancer cell growth. Accordingly, biomedical
studies are focused on the identification and evaluation of novel
inhibitors of protein kinases that are restricted to the cell cycle
(2).
Chaetominine has been demonstrated to exhibit toxic
effects against the human leukemia cell line K562 and the human
colon cancer cell line SW1116 (11).
However, the molecular mechanisms that underpin the cytotoxic
effects of chaetominine are yet to be elucidated fully. Following a
previous study that observed the cytotoxic and apoptotic effects of
K562 cells (4), the present study
hypothesized that chaetominine may alter cell cycle progression in
these two cancer cell lines. The apoptotic effects induced by
chaetominine on SW1116 cells and cell cycle regulation in SW1116
and K562 cells following treatment with chaetominine were also
assessed. The inhibitory effects on cell growth promoted by
chaetominine are likely to vary depending on the cell type that is
exposed to the compound. Additionally, the molecular mechanisms
involved in chaetominine-induced cell cycle arrest in K562 and
SW1116 cells were elucidated in the present study.
Materials and methods
Reagents
Chaetominine was extracted from a liquid culture of
A. fumigatus CY018. The purity of the preparation was determined to
be 99.8% (4). MTT was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Cell culture
Human leukemia and colon cancer cell lines, K562 and
SW1116, were obtained from the Shanghai Institute for Biological
Sciences (Shanghai, China). K562 cells were cultured in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) and SW1116 cells were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS. Cells were maintained for 1-2 days at 37°C in a
humidified incubator containing 5% CO2.
Cell viability assay
The effect of chaetominine on cancer cell viability
was evaluated using a MTT assay. Chaetominine was dissolved in 1 ml
dimethyl sulfoxide at a concentration of 1 mM (DMSO; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) for the following assays. Cells
were seeded in 96-well plates at a concentration of 105
cells/ml. All cell lines were incubated with 100, 50, 25, 12.5,
6.25 or 0 nM chaetominine for 48 h, and 1/10,000 (v/v, 0.01 µl DMSO
in 100 µl reaction system) DMSO was used instead of chaetominine
for the control groups. Each well was supplemented with 20 µl MTT
(5 mg/ml, diluted in RPMI-1640 medium to dissolve the purple
formazan) and incubated for a further 4 h prior to testing. The
absorbance was subsequently measured using a microplate reader
(SpectraMax® i3; Molecular Devices LLC, Sunnyvale, CA,
USA) at an experimental wavelength of 570 nm and a reference
wavelength of 630 nm. The half maximal inhibitory concentration
(IC50) values were calculated using GraphPad Prism 5
(GraphPad Software, Inc., La Jolla, CA, USA).
Annexin V-Fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining
Apoptotic rate was measured using flow cytometry.
Cells were treated with 100, 50, 25 and 0 nM chaetominine for 24 h
in 6-well plates. Cells were then harvested with PBS and incubated
with Annexin V-FITC at 20-25°C for 10 min in the dark using an
Annexin V-FITC/PI kit (Nanjing Keygen Biotech Co., Ltd., Nanjing,
China). Cells were subsequently suspended in a mixture of Annexin V
and PI buffer following centrifugation at room temperature, at a
speed of 200 × g for 5 min. Apoptotic cells were analyzed using a
flow cytometer (FACSAria; BD Biosciences, San Jose, CA, USA).
Annexin-V FITC positive and PI negative results indicated early
apoptosis, while Annexin-V FITC positive and PI positive results
indicated late apoptosis. The FACSAria was equipped with BD
FACSDiva software v6.0 (BD Biosciences).
Cell cycle assay
The effects of chaetominine on cell cycle
progression were analyzed using a Cell Cycle Detection kit (Nanjing
Keygen Biotech Co., Ltd.). Cells were treated with 40 nM
chaetominine for 0, 12, 24 of 48 h, or 40, 20, 10 or 0 nM
chaetominine for 24 h. Following treatment, cells were washed with
PBS and fixed overnight at 4°C in 70% ethanol. Cells were then
incubated with 100 µl RNase at 37°C for 30 min and stained for 30
min in the dark with 400 µl PI solution at 4°C. The percentage of
cells in different phases was monitored using a flow cytometer
(FACSAria; BD Biosciences) at 488 nm. The FACSAria was equipped
with BD FACSDiva software v6.0 (BD Biosciences).
Western blot assay
Whole lysates were prepared with cell lysis buffer
(Biotech Well, Shanghai, China) following incubation with 40, 20,
10 or 0 nM chaetominine for 24 h. Lysates were subsequently washed
with PBS and protein concentrations were determined using a
bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Protein samples (20 µg/lane) containing 0.01%
bromophenol blue were separated using 10% SDS-PAGE and were
subsequently transferred onto polyvinylidene difluoride membranes
(EMD Millipore, Billerica, MA, USA). The PVDF membranes were
blocked at room temperature for 2 h with 5% bovine serum albumin
(diluted in TBS-Tween-20; TBS-T) and incubated overnight with the
appropriate primary antibody (diluted with blocking buffer) at 4°C.
Primary antibodies against ATM (dilution, 1:500; cat. no.
SC-377239), phosphorylated (p)-ATM (Ser1981; dilution, 1:500; cat.
no. SC-47739), Chk1 (dilution, 1:200; cat. no. SC-8408), Chk2
(dilution, 1:200; cat. no. SC-5278), p53 (dilution, 1:200; cat. no.
SC-126), p21 (dilution, 1:200; cat. no. SC-469), GAPDH (dilution,
1:1,000; cat. no. SC-69778) and β-actin (dilution, 1:1,000; cat.
no. SC-47778) were obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). ATR (dilution, 1:1,000; cat. no. 2790), p-ATR
(Ser428; dilution, 1:1,000; cat. no. 2853), Cdc25A (dilution,
1:1,000; cat. no. 3652), cyclin A (dilution, 1:1,000, cat. no.
4656) and CDK2 (dilution, 1:1,000; cat. no. 2546) primary
antibodies were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Horseradish peroxidase-conjugated goat
anti-mouse (dilution, 1:1,000; cat. no. A0216) and goat anti rabbit
(dilution, 1:1,000; cat. no. A0208) secondary IgG (H+L) antibodies
were purchased from Beyotime Institute of Biotechnology (Haimen,
China). Following three sequential TBS-T washes, the membranes were
incubated overnight with horseradish peroxidase-labeled secondary
antibodies at 4°C. An enhanced chemiluminescence kit (Biotech Well)
was used to visualize the immunoreactions. Protein levels were
determined following the detection of chemiluminescent signals. The
signals were quantified using Quantity One v4.62 software (Bio-Rad
Laboratories Inc., Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using a Total RNA Extraction
kit (Nanjing Keygen Biotech Co., Ltd.). RNA samples were quantified
using UV spectrophotometry at a wavelength of 260 nm. Reverse
transcription into cDNA was performed using a PrimeScript™ RT
reagent kit (Takara Biotechnology Co., Ltd, Dalian, China) with 500
ng total RNA. cDNA was subsequently utilized for PCR analysis. The
qPCR reaction was conducted using SYBR® Premix Ex Taq™
II (Takara Biotechnology Co., Ltd.) on a CFX96 Touch™ PCR Detection
system (Bio-Rad Laboratories, Inc.). The conditions for PCR
amplification were as follows: 40 cycles of initial duration at
94°C for 30 sec, annealing at 60°C for 30 sec and extension at 72°C
for 1 min. The primer sequences used were as follows: Cyclin A,
5′-TCCATGTCAGTGCTGAGAGGA-3′ (forward), 5′-GAAGGTCCATGAGACAAGGC-3′
(reverse); CDK2, 5′-GCTTTCTGCCATTCTCATCG-3′ (forward),
5′-GTCCCCAGAGTCCGAAAGAT-3′ (reverse); cyclin E,
5′-TTTCTTGAGCAACACCCT-3′ (forward), 5′-GTCACATACGCAAACTGG-3′
(reverse); CDK4, 5′-CTGAGAATGGCTACCTCTCGATATG-3′ (forward),
5′-AGAGTGTAACAACCACGGGTGTAAG-3′ (reverse);
glyceraldehyde-3-phophate dehydrogenase (GAPDH),
5′-CAACGGATTTGGTCGTATT-3′ (forward), 5′-CACAGTCTTCTGGGTGGC-3′
(reverse). The mRNA level associated with each gene was normalized
to that of the internal control, GAPDH, and was quantified using
the −2ΔΔCq method (12).
Statistical analysis
Data were analyzed using Graphpad Prism 5 (Graphpad
Software Inc., La Jolla, CA, USA) and are expressed as the mean ±
standard deviation. Group comparisons of experimental data were
performed using a one-way analysis of variance with post-hoc
Newman-Keuls tests. P<0.05 was considered to indicate a
statistically significant result.
Results
Chaetominine inhibits the cell
viability of K562 and SW1116 cells
The cytotoxic effects of chaetominine on K562 and
SW1116 cells were analyzed using an MTT assay. The IC50
values for chaetominine in K562 and SW116 cells were 33.7±0.2 and
45.9±3.4 nM, respectively. The results demonstrated that
chaetominine inhibited K562 and SW1116 cell viability at
concentrations of 6.25, 12.5, 25, 50 and 100 nM (Fig. 1B). These data were consistent with
those reported in a previous study (11).
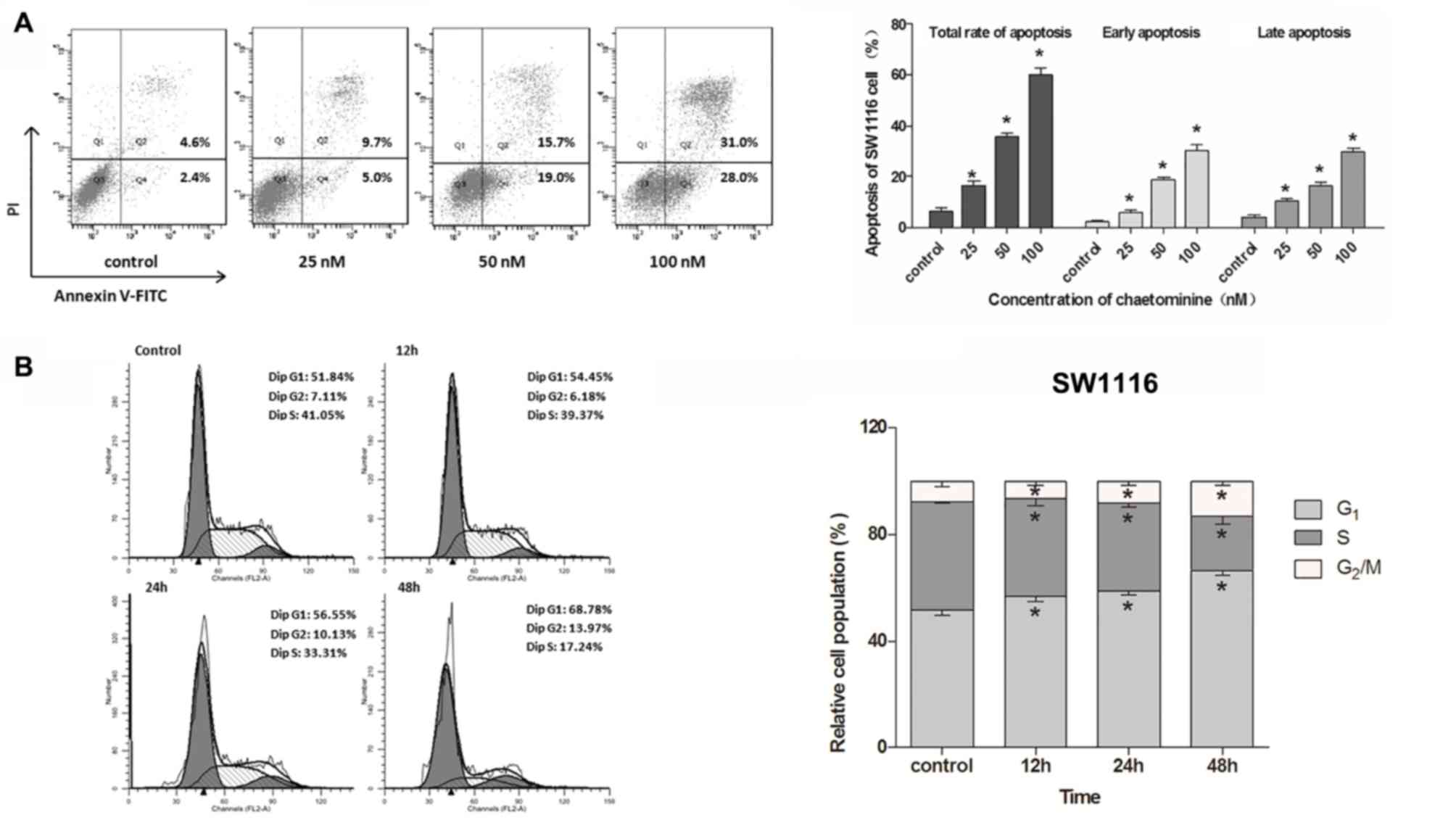
Chaetominine induces apoptosis of
SW1116 cells
A previous study demonstrated that chaetominine
induces apoptotic cell death in K562 cells (4). Following these results, the present
study utilized flow cytometry to assess whether chaetominine
induces cell apoptosis in SW1116 cells. The apoptosis rates were
determined using dual staining with Annexin V-FITC and PI. It was
revealed that chaetominine significantly increased early and late
apoptosis rates compared with the control group (Fig. 2A). The total apoptosis rate gradually
increased from 6.4% in the control group to 16.4, 35.6 and 60.0% in
cells incubated with 25, 50 and 100 nM chaetominine, respectively.
These results suggested that chaetominine induces apoptotic cell
death in SW1116 cells.
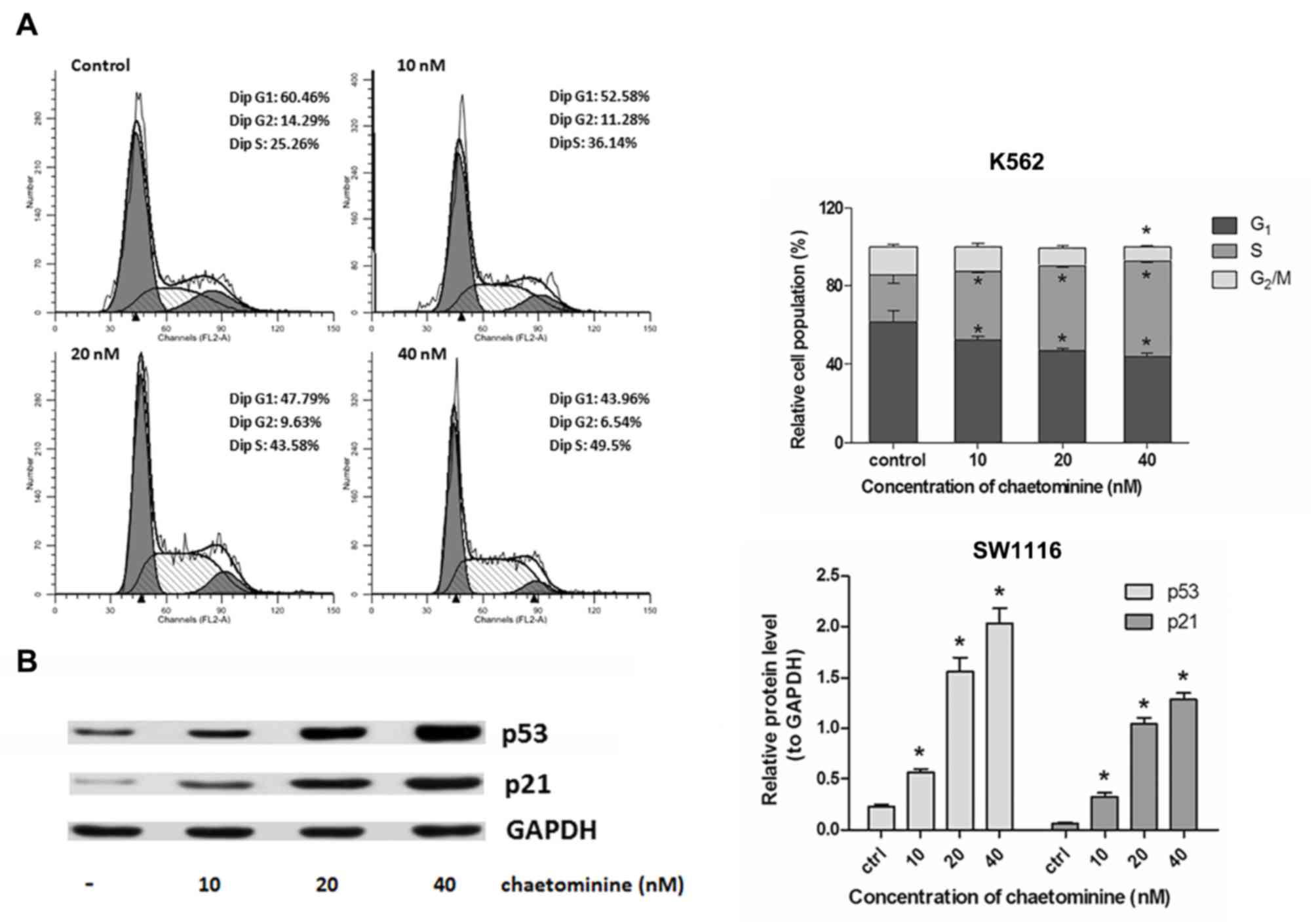
Chaetominine results in G1
or S phase cell cycle arrest
To elucidate whether the cell cycle was involved in
the inhibition of cell growth following chaetominine treatment,
K562 and SW1116 cells were treated with chaetominine to facilitate
the detection of cell cycle phase distribution. The results
demonstrated that chaetominine treatment resulted in an
accumulation of G1-phase SW1116 cells (control, 51.84%;
12 h, 54.45%; 24 h, 56.55%; and 48 h, 68.78%) in a time-dependent
manner (Fig. 2B). In addition,
chaetominine caused an increase in the number of S-phase K562 cells
(Fig. 3A; control, 25.26%; 10 nM,
36.14%; 20 nM, 43.54%; and 40 nM, 49.5%). This occurred in a
dose-dependent manner. These results indicated that cell cycle
arrest was a mechanism used by chaetominine to induce cytotoxicity
in K562 and SW1116 cells. In addition, chaetominine treatment
resulted in cell cycle arrest at the G1 stage (SW1116)
or S stage (K562) depending on which cell type was analyzed.
Chaetominine influences the expression
of ATM/Chk2/p53/p21 in SW1116 cells
In order to elucidate the molecular mechanism
associated with G1-phase arrest exhibited by
chaetominine treated SW1116 cells, the signaling proteins involved
in the regulation cascade of the cell cycle were evaluated using
western blotting. p53 induces cell cycle arrest at the
G1-stage, resulting in the activation of p21, which
promotes the inhibition of CDKs (2).
The results demonstrated that the expression of p53 and p21 were
upregulated in SW1116 cells following incubation with chaetominine
(Fig. 3B). This upregulation occurred
in a time-dependent manner. The association between ATM/Chk2 and
chaetominine was assessed. It was demonstrated that chaetominine
increased the phosphorylation and accumulation of ATM and Chk2,
respectively. However, treatment did not notably affect the
expression of ATM (Fig. 4A). The
significant phosphorylation of ATM is accompanied by the
upregulation of Chk2 expression and demonstrates that pATM is
associated with Chk1 regulation, which assists in checkpoint
regulation (13). It was determined
that chaetominine modulates the ATM/Chk2/p53/p21 signaling pathway,
which is an important effector of G1-phase arrest
(7,13).
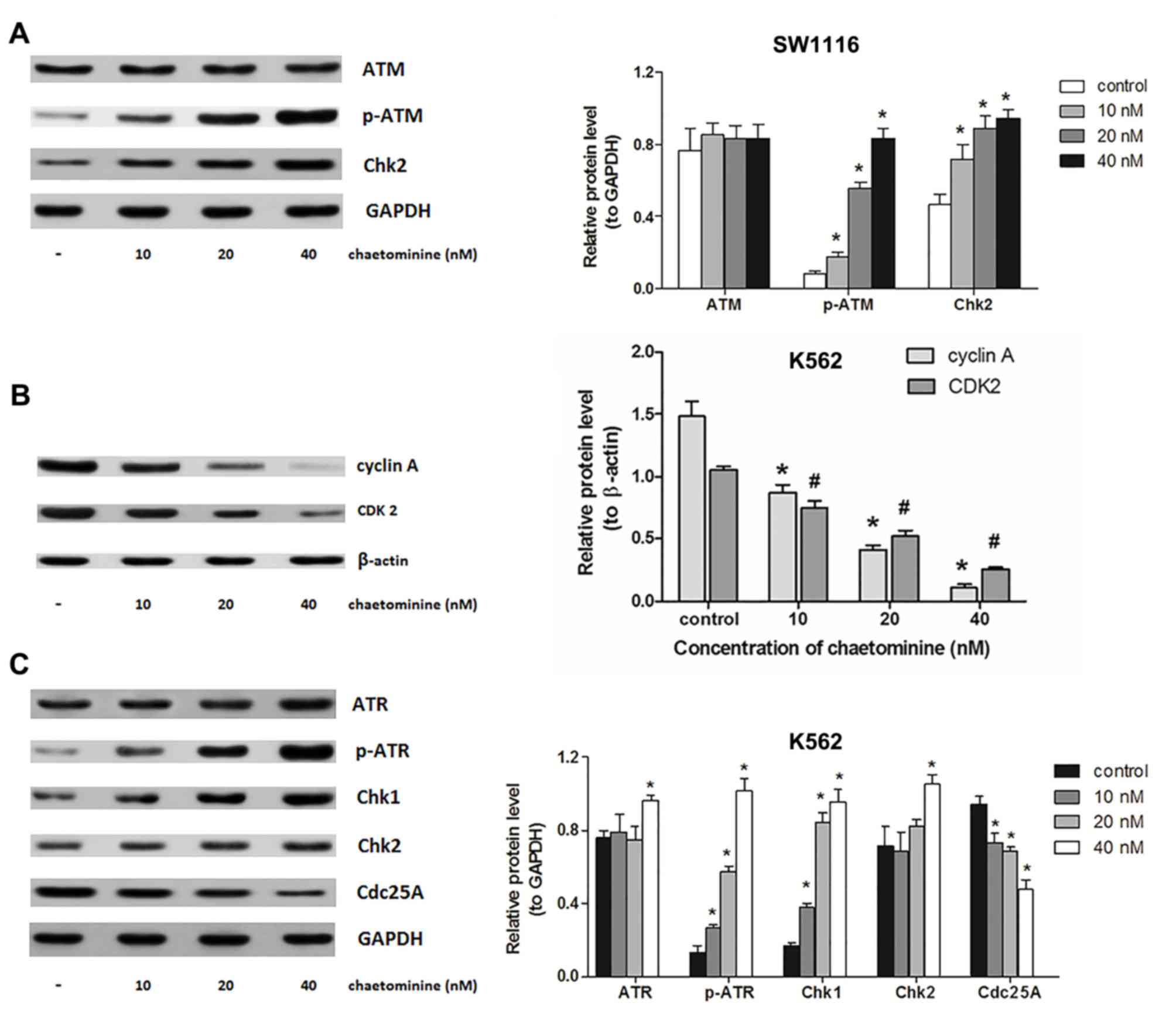 | Figure 4.Modulation of signal molecules
involved in chaetominine-induced cell cycle arrest. SW1116 and K562
cells were exposed to 0, 10, 20 and 40 nM chaetominine for 24 h.
(A) Protein levels following western blotting using antibodies
against ATM, p-ATM (Ser1981) and Chk2. The analysis was conducted
on protein extracts from SW1116 cells. *P<0.05 vs. respective
control group. (B) Results of western blotting following incubation
with antibodies against cyclin A and CDK2 of K562 cells. The
protein expression levels were analyzed following densitometric
analysis and results were compared to the β-actin loading control.
Representative results for each experiment are presented in A and
B. Data are presented as the mean ± standard deviation (n=3)
*P<0.05 vs. cyclin A controls; #P<0.05 vs. CDK2
controls. (C) Protein levels following western blotting using
antibodies against ATR, p-ATR (Ser428), Chk1, Chk2 and cdc25A in
K562 cells. The bar graphs represent a densitometric analysis
conducted to determine protein expression relative to the internal
reference protein, GAPDH. Representative blots from three
independent experiments are presented and data are presented as the
mean ± standard deviation (n=3). *P<0.05 vs. respective control
group. ATM, ataxia telangiectasia mutated; ATR, ATM and
Rad3-related; p-ATR, phosphorylated ATR; CDK2, cyclin-dependent
kinase 2; Chk, checkpoint kinase; cdc25A, cell division cycle
25A. |
Chaetominine alters the expression of
ATR/Chk1/cdc25A in K562 cells
The mechanism by which chaetominine induces S-phase
arrest in K562 cells was further assessed. Western blotting was
utilized to determine if the effects elicited by chaetominine were
associated with the alteration of cell-cycle regulatory kinases.
Cyclin A is a critical component involved in the regulation of cell
cycle progression, which becomes functionally active once bound to
CDK2. This association subsequently allows cells to continue
through the S-phase (14). Based on
previous data, the present study determined the expression levels
of cyclin A, CDK2 and upstream proteins including cdc25A, Chk1/2
and ATR/p-ATR in K562 cells. The results of the present study
demonstrated that cyclin A and CDK2 expression were significantly
reduced by chaetominine compared with the control (Fig. 4B). In addition, K562 cells expressed
significantly higher levels of Chk1 (P<0.05 at 10, 20 and 40 µM)
and Chk2 (P<0.05 at 40 µM) following treatment with chaetominine
compared with the control (Fig. 4C).
Furthermore, a detectable alteration in Chk2 levels was only
observed following 40 nM chaetominine treatment, while the
expression of Chk1 changed significantly even at the lowest
concentration (10 nM) of chaetominine (Fig. 4C). Chk1 protein levels were 5.7-fold
greater following 40 nM chaetominine treatment compared with
controls, while the Chk2 protein levels had increased by 1.5-fold
when the same treatment was applied. The level of cdc25A was
marginally attenuated following 40 nM chaetominine treatment (to
~50% that of the control group; Fig.
4C). The results of the present study suggest that an elevation
in p-ATR expression occurred following treatment with chaetominine.
These results suggest that chaetominine treatment upregulated p-ATR
and Chk1 protein levels and downregulated cdc25A, cyclin A and
CDK2, and these proteins are reported to participate in the
initiation of S-phase arrest (14,15).
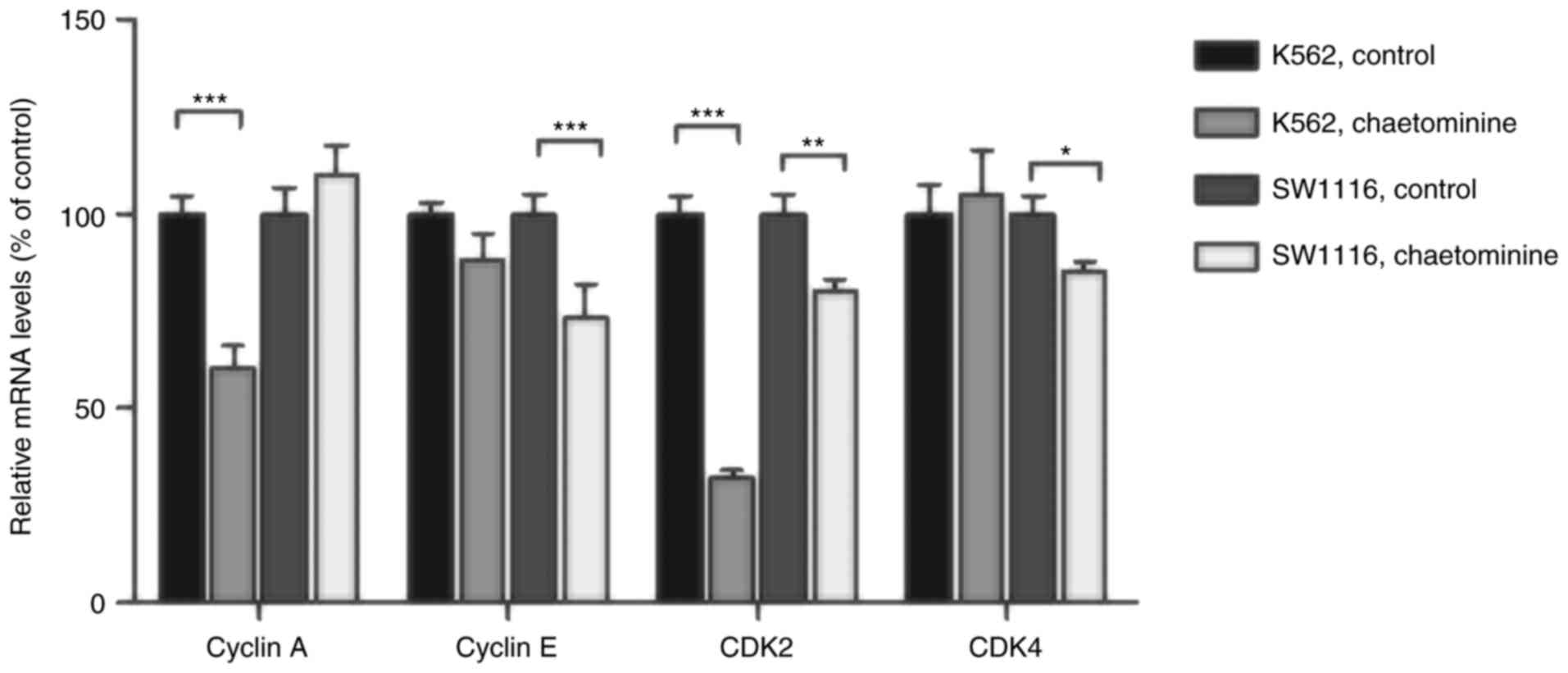
Chaetominine treatment affects cell
cycle regulator cyclin A/E and CDK2/4 mRNA levels
To assess the role of direct regulators of cell
cycle arrest in K562 and SW1116 cells, the mRNA levels of specific
genes were determined using RT-qPCR. The results demonstrated that
the mRNA levels of cyclin A and CDK2 in K562 cells were
significantly decreased following incubation with 40 nM
chaetominine (Fig. 5). However,
minimal changes in the mRNA levels of cyclin E or CDK4 were
observed, when compared with the control groups. These results are
consistent with the cyclin A and CDK2 expression variation trends
observed in chaetominine-treated K562 cells in the present study.
Conversely, chaetominine resulted in the downregulation of cyclin
E, CDK2 and CDK4 in SW1116 cells. However, the same effect was not
observed in cyclin A. These results suggested that chaetominine
treatment altered the mRNA levels of cyclin E, CDK2 and CDK4 in
SW1116 cells as described in Fig.
6.
Discussion
Among natural anti-cancer alkaloids, those derived
from marine metabolites and their derivatives may be a vital
resource for chemotherapeutic discovery due to a low effective
dosage and increased selectivity (2,16).
Previous studies pertaining to these benefits have served a crucial
role in the investigation of anticancer candidates in natural
products. To further aid in these investigations, the present study
utilized the fungal metabolite chaetominine isolated from a culture
of Aspergillus fumigatus CY018 (3). This compound exhibited cytotoxic effects
in two human cancer cell lines (K562 and SW1116 cells) when used in
nanomolar concentrations. However, the effects of chaetominine on
the regulation of the cell cycle, which is an important component
involved in controlling cellular proliferation, are not well
understood. The current study was performed to investigate the
relative contribution of chaetominine to the molecular mechanisms
associated with cell cycle regulation in K562 and SW1116 cells.
The present study determined that chaetominine
markedly inhibited cell growth in K562 (IC50; 34 nM) and
SW1116 (IC50; 46 nM) cells. These results are consistent
with those of previous studies, which indicated that chaetominine
may be a candidate for anti-cancer treatment (11,15). The
results of the present study, including the dose-dependent increase
in apoptosis rate and a sub-G1 peak in the SW1116 cells
treated with 40 nM chaetominine for 24 h, demonstrate that the
cytotoxic role of chaetominine in SW1116 and K562 cells is
associated with the induction of apoptosis. However, no obvious
sub-G1 peak following exposure to higher concentrations
of chaetominine resulted in an inability to collect all the
fragments released by dead cells. The cell cycle is an important
regulatory mechanism associated with cell growth and proliferation
(7,8).
A variety of anti-cancer agents target the abnormal growth of cells
by disrupting cell cycle progression and/or inducing apoptosis
(5). In the present study, SW1116
cells underwent G1-phase cell cycle arrest, while K562
cells underwent S-phase arrest following chaetominine treatment.
These results suggest that the role of chaetominine in cancer cell
death is dependent on cell cycle arrest and apoptosis.
Cyclins and CDKs have been demonstrated to function
in the direct regulation of the cell cycle (9). RT-qPCR analysis utilized in the present
study demonstrated that chaetominine-induced cell cycle arrest in
K562 cells was associated with the downregulation of cyclin A and
CDK2. In SW1116 cells, this occurrence was accompanied by a
decrease in cyclin E, CDK2, and to a lesser extent, CDK4. CDK2
activity is restricted to the G1-S phase of the cell
cycle and results in the binding of different cyclin partners.
Cyclin E/CDK2 and CDK4 are recruited for the transition from the
G1-phase to the S phase, while cyclin A/CDK2 is utilized for
progression through the S-phase. Cyclin A and cyclin E
overexpression is commonly observed in leukemia and colon cancer
cells, respectively (14). This
overexpression may be indicative of the different types of cell
cycle arrest observed in the two cancer cell lines utilized in the
present study following treatment with chaetominine. Further
studies are required to elucidate whether chaetominine acts as a
pharmacological inhibitor of CDKs, with a potent anti-cancer
activity.
Cell cycle checkpoint pathways are crucial
regulatory machineries involved in the determination of cellular
responses to cancer therapy (8).
ATM/Chk2 and ATR/Chk1 signaling modules are involved in the control
of checkpoint networks and promote delays in the cell cycle at the
G1, S or G2 phase (7,13). p53 is
a key substrate of the ATM/Chk2 module and also acts as a
pro-apoptotic protein (17). The
accumulation of p53 results in the activation of p21, which
inactivates the cyclin E/CDK2 complex, subsequently leading to
G1-phase arrest. This is consistent with results of the
present study. It was demonstrated that SW1116 cells expressed
higher levels of p-ATM, Chk2, p53 and p21 following chaetominine
treatment and that these molecules were important mediators in the
arrest of cells at the G1-phase (13,18). The
ATR/Chk2 module elicits its effects following an increase in Chk2
activity, which causes the attenuation of cdc25, cyclin A and CDK2,
thus leading to S-phase blockade (19). In K562 cells, these effects resulted
in an increase in the expression of p-ATR and Chk2 following
incubation with chaetominine. Conversely, the protein level of
cdc25A, cyclin A and CDK2 decreased in chaetominine-treated K562
cells. However, Chk1 protein levels remained relatively stable
following incubation with the same compound. These results are
consistent with those of the present study, suggesting that
chaetominine affected the ATR-Chk2-cdc25A-cyclin A/CDK2 signaling
pathway. This also occurred independently of p53, facilitating the
occurrence of cell cycle arrest during the S-phase (17,19).
In conclusion, the present study demonstrated that
chaetominine causes the inhibition of cell growth through apoptosis
and cell cycle arrest in K562 and SW1116 cells. The current study
also revealed the mechanism of action that underpins cell cycle
arrest following chaetominine treatment. However, it remains
unclear whether the cellular response to chaetominine is actually
associated with DNA damage caused by ATM or ATR signal initiation
(20). Furthermore, all of the
experiments conducted in the present study were performed in
vitro. Further in vivo studies are therefore required to
fully characterize the effects associated with this compound.
Nevertheless, chaetominine may serve as an important
chemotherapeutic agent with several putative clinical
applications.
Acknowledgements
The present study was supported by the National High
Technology Research and Development Program of China (grant no.
2013AA092901) and partially financed by the Fundamental Research
Funds for the Central Universities (grant no. WF1113010) and the
National Special Fund for State Key Laboratory of Bioreactor
Engineering (grant no. 2060204).
References
|
1
|
Hussain S, Fareed S, Ansari S and Khan S:
Marine natural products: A lead for anti-cancer. Indian J Geo Mar
Sci. 41:27–39. 2012.
|
|
2
|
Imperatore C, Aiello A, D'Aniello F,
Senese M and Menna M: Alkaloids from marine invertebrates as
important leads for anticancer drugs discovery and development.
Molecules. 19:20391–20423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu YH, Zhu YX, Jiao RH, Tan RX, Yao LY and
Hu WW: Method and medium for producing Fumigaclavine C by
Aspergillus fumigatus Fermentation. China patent, CN 103849663 B.
Filed January 10, 2014; issued May 4. 2016.
|
|
4
|
Yao J, Jiao R, Liu C, Zhang Y, Yu W, Lu Y
and Tan R: Assessment of the cytotoxic and apoptotic effects of
chaetominine in a human leukemia cell line. Biomol Ther (Seoul).
24:147–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mahata S, Bharti AC, Shukla S, Tyagi A,
Husain SA and Das BC: Berberine modulates AP-1 activity to suppress
HPV transcription and downstream signaling to induce growth arrest
and apoptosis in cervical cancer cells. Mol Cancer. 10:392011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li S, Lei Y, Jia Y, Li N, Wink M and Ma Y:
Piperine, a piperidine alkaloid from Piper nigrum re-sensitizes
P-gp, MRP1 and BCRP dependent multidrug resistant cancer cells.
Phytomedicine. 19:83–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kerzendorfer C and O'Driscoll M: Human DNA
damage response and repair deficiency syndromes: Linking genomic
instability and cell cycle checkpoint proficiency. DNA Repair
(Amst). 8:1139–1152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sánchez-Martínez C, Gelbert LM, Lallena MJ
and de Dios A: Cyclin dependent kinase (CDK) inhibitors as
anticancer drugs. Bioorg Med Chem Lett. 25:3420–3435. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Poi MJ, Knobloch TJ, Sears MT, Uhrig LK,
Warner BM, Weghorst CM and Li J: Coordinated expression of
cyclin-dependent kinase-4 and its regulators in human oral tumors.
Anticancer Res. 34:3285–3292. 2014.PubMed/NCBI
|
|
11
|
Jiao RH, Xu S, Liu JY, Ge HM, Ding H, Xu
C, Zhu HL and Tan RX: Chaetominine, a cytotoxic alkaloid produced
by endophytic Chaetomium sp. IFB-E015. Org Lett. 8:5709–5712. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yam CH, Fung TK and Poon RY: Cyclin A in
cell cycle control and cancer. Cell Mol Life Sci. 59:1317–1326.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Berthet C, Raj K, Saudan P and Beard P:
How adeno-associated virus Rep78 protein arrests cells completely
in S phase. Proc Natl Acad Sci USA. 102:13634–13639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kshirsagar UA: Recent developments in the
chemistry of quinazolinone alkaloids. Org Biomol Chem.
13:9336–9352. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wahl GM and Carr AM: The evolution of
diverse biological responses to DNA damage: Insights from yeast and
p53. Nat Cell Biol. 3:E277–E286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang J, Ghio AJ, Gao M, Wei K, Rosen GD
and Upadhyay D: Ambient particulate matter induces alveolar
epithelial cell cycle arrest: Role of G1 cyclins. FEBS Lett.
581:5315–5320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bartek J, Lukas C and Lukas J: Checking on
DNA damage in S phase. Nat Rev Mol Cell Biol. 5:792–804. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zuco V, Benedetti V and Zunino F: ATM- and
ATR-mediated response to DNA damage induced by a novel
camptothecin, ST1968. Cancer Lett. 292:186–196. 2010. View Article : Google Scholar : PubMed/NCBI
|