Introduction
Celecoxib, a cyclooxygenase-2 (COX-2)-specific
inhibitor, reduces the risk of colorectal cancer, exhibiting
anti-proliferative and chemoprophylactic effects on this cancer
(1,2).
Treatment with celecoxib induces the upregulation of endoplasmic
reticulum (ER) chaperones, and promotes tumor cell death in
vitro and in vivo through the ER stress response
(3,4).
This ER stress induces the nuclear phosphorylation and activation
of p53, leading to ER stress-induced cell death in MCF-7 and HeLa
cells (5). The co-treatment of
p53-deficient colon cancer cells with zerumbone and celecoxib also
induces ER stress and the transactivation of death receptor 5 (DR5)
(6). The underlying molecular
mechanisms by which celecoxib inhibits each cancer type have yet to
be completely characterized. Therefore, it is necessary to
investigate the downstream signaling pathways induced by treatment
with celecoxib for clinical applications, and to examine whether it
is more efficacious to treat cancer with a combination of drugs,
rather than celecoxib alone.
The proteasome inhibitor bortezomib is a promising
candidate for the treatment of hematological and solid cancer types
(7). Bortezomib induces the unfolded
protein response (UPR) to a limited extent, whereas the induction
of binding immunoglobulin protein (BiP) and CCAAT/enhancer binding
protein homologous protein (CHOP) by an ER stress-inducing agent is
attenuated following exposure to this drug (8). Bortezomib activates downstream targets
of p53, including p21, p53-upregulated modulator of apoptosis
(PUMA) and Bcl-2-associated X (Bax); however, the induction of
apoptosis by bortezomib is not affected by the deletion of p53 in
colon cancer cells (9). Autophagy can
protect cells from apoptotic stimuli, including growth factor
deprivation and ER stress (10,11).
Autophagy may also induce cell death, as the components of the
autophagic and apoptotic machinery are interconnected and shared
(12). The inhibition of
cisplatin-induced autophagy by bortezomib has been shown to enhance
the chemotherapeutic efficacy of cisplatin in ovarian cancer
(13). The autophagy inhibitor
3-methyladenine (3-MA) enhances celecoxib-induced apoptosis in
human colon cancer cells (14). On
the basis of these reports, the effect and underlying mechanism of
bortezomib or celecoxib on the induction of p53- and
ER-stress-associated apoptosis in cancer cells remain
controversial. Furthermore, the role of autophagy in cancer cells
is complex and highly cell-type-dependent.
Despite the established connections between
bortezomib or celecoxib treatment with ER stress or autophagy, it
has yet to be determined whether combination treatment with
celecoxib and bortezomib can improve the efficacy of treatment in
colon cancer treatment by further promoting ER
stress/autophagy-associated cell death. The present study focused
on the development of novel chemotherapy combinations containing
celecoxib and bortezomib for the treatment of colon cancer; it
investigated whether the order of administration was critical for
the induction of ER stress or stimulation of autophagy-associated
cell death in colon cancer cells. In addition, the present study
attempted to identify the role of p53 in the ER stress-mediated
autophagy signaling pathway following the combination of celecoxib
with bortezomib in HCT-116 and p53−/− HCT-116 cells.
Materials and methods
Cell lines and reagents
The HCT-116, HCT-8 and HT-29 human colorectal cancer
cell lines were purchased from the American Type Culture Collection
(Manassas, VA, USA). p53−/− HCT-116 cells were kindly
provided by Professor Bert Vogelstein (Johns Hopkins University,
Baltimore, MD, USA). All cells were maintained in RPMI-1640 medium
(Corning Incorporated, Corning, NY, USA) supplemented with 10% FBS
(HyClone; GE Healthcare, Chicago, IL, USA), streptomycin and
glutamine at 37°C in 5% CO2. Bortezomib was purchased
from LC Laboratories (Woburn, MA, USA). Celecoxib was obtained from
Selleck Chemicals (Houston, TX, USA). SP600125, a c-Jun N-terminal
kinase (JNK) inhibitor, SB203580, a p38-mitogen-activated protein
kinase (MAPK) inhibitor and salubrinal, an ER stress inhibiter,
were purchased from Calbiochem (Merck KGaA, Darmstadt, Germany).
BAPTA-AM and 3-MA were obtained from Sigma-Aldrich (Merck KGaA).
Pifithrin (PFT)-α was purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). To inhibit the activation of JNK or
p38-mitogen-activated protein kinase, cells were pre-treated with
SP600125 (20 µM) or SB203580 (10 µM) for 2 h at 37°C. To block
endoplasmic reticulum stress, cells were pre-incubated with
salubrinal (2 µM) for 1 h at 37°C. To inhibit the autophagic
signal, cells were pre-treated with 3-MA (10 mM) for 2 h at 37°C.
To block the expression of p53, cells were pre-exposed to PTF-α (50
µM) for 1 h at 37°C. To inhibit the effect of Ca2+,
cells were pre-treated with BAPTA-AM (2 µM) for 30 min at 37°C.
Cell viability assay with AlamarBlue
and Cell Counting kit-8 (CCK-8)
HCT-116, HCT-8 or HT-29 cells (2×104
cells/well) were seeded in RPMI-1640 containing 10% FBS in 96-well
plates. Cells were pre-treated with bortezomib (20 nM) for 6 h and
then treated with celecoxib (20 µM) for an additional 18 h. For
comparison, cells were treated either with bortezomib (100 nM) or
celecoxib (80 µM) alone or co-treated (20 nM bortezomib + 20 µM
celecoxib) for 24 h. Finally, cells pre-exposed to celecoxib (20
µM) for 6 h were then treated with bortezomib (20 nM) for 18 h.
Cell viability was measured using an AlamarBlue
assay (Serotec; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
AlamarBlue was added (10% by volume) to each well, and the relative
fluorescence unit (RFU) values were determined 7 h later using a
SpectraMax M2e Multi-Detection Microplate Reader (excitation, 530
nm; emission, 590 nm; Molecular Devices, LLC, Sunnyvale, CA, USA).
Experiments were performed in triplicate, and RFU values were
expressed as the mean ± standard deviation (SD) of three
replicates.
Cell viability was also measured using a CCK-8 assay
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol. Briefly, HCT-116, HCT-8 or HT-29 cells (2×104
cells/well) in 96-well plates were pre-treated with celecoxib (20
µM) for 6 h and then with bortezomib (20 nM) for an additional 18
h. For comparison, cells were treated with celecoxib (80 µM). At 24
h, the cells were stained with 10 µl CCK-8 dye in 90 µl of culture
medium for 2 h at 37°C. The absorbance was measured at 450 nm.
Analysis of apoptosis by flow
cytometry
HCT-116 and p53−/− HCT-116 cells
(1×105 cells/ml) were cultured in 6-well plates and
pre-treated with 20 nM bortezomib for 6 h and then with 20 µM
celecoxib for an additional 18 h. For comparison, cells were
treated either with 100 nM bortezomib or 80 µM celecoxib alone or
co-treated (20 nM bortezomib + 20 µM celecoxib) for 24 h. Finally,
cells were pre-exposed to 20 µM celecoxib for 6 h and then treated
with 20 nM bortezomib for 18 h.
The percentages of cells undergoing apoptosis were
determined by flow cytometry using fluorescein isothiocyanate
(FITC)-labeled annexin-V and 7-aminoactinomycin (7-AAD) (both BD
Biosciences, San Diego, CA, USA). Cells were harvested, rinsed with
PBS, and resuspended in 100 µl of 1X annexin-V binding buffer (BD
Biosciences). A total of 3 µl annexin-V-FITC and 3 µl 7-AAD were
added, and cells were incubated at room temperature for 15 min in
the dark, with gentle vortexing. The stained cells were analyzed
using a FACSCalibur flow cytometer equipped with CellQuestpro
software version 5.1 (BD Biosciences). Dot plot graphs were
produced to quantify the percentage of viable cells
(annexin-V−/7-AAD−), early-stage apoptotic
cells (Annexin-V+/7-AAD−), late-stage
apoptotic cells (annexin-V+/7-AAD+) and
necrotic cells (Annexin-V−/7-AAD+).
Quantification of cytosolic and
mitochondrial Ca2+ levels
Cytosolic Ca2+ levels were determined
using the fluorescent dye Fluo3-AM (5 µM; log mode in FITC setting;
Molecular Probes; Thermo Fisher Scientific, Inc.). Mitochondrial
Ca2+ levels were determined using the fluorescent dye
Rhod2-AM (1 µM; Molecular Probes; Thermo Fisher Scientific, Inc.).
Cells were incubated with the fluorescent dyes for 20 min at 37°C,
washed with calcium-free Dulbecco's PBS, and analyzed by flow
cytometry. For certain experiments, cells were pretreated with
BAPTA-AM (2 µM) for 30 min.
Western blotting
Cells were washed in PBS and lysed in NP-40 buffer
(Elpis Biotech, Inc., Daejeon, Korea) supplemented with a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). Protein
phosphorylation states were preserved through the addition of
phosphatase inhibitors (Cocktail II; Sigma-Aldrich; Merck KGaA) to
the NP-40 buffer. Protein concentrations were determined using a
BCA assay kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Proteins (10 µg/lane) were resolved through SDS-PAGE (12%)
and transferred to a nitrocellulose membrane (EMD Millipore,
Billerica, MA, USA). Membranes were blocked with 5% skimmed milk
for 1 h at room temperature prior to western blot analysis. The
following primary antibodies were used: Caspase-3 (cat. no. 9665;
1:1,000), caspase-9 (cat. no. 9502; 1:1,000), poly (ADP-ribose)
polymerase (PARP; cat. no. 9542; 1:1,000), β-actin (cat. no. 4967;
1:1,000), Bcl-2 (cat. no. 2870; 1:1,000), Bax (cat. no. 2772;
1:1,000), Bcl-2 homologous antagonist/killer (Bak; cat. no. 6947;
1:1,000), Mcl-1 (cat. no. 4572; 1:1,000), Puma (cat. no. 4976;
1:1,000), survivin (cat. no. 2808; 1:1,000), p53 (cat. no. 2524;
1:1,000), phosphorylated (p)-extracellular-related kinase (ERK)1/2
(Thr202/Tyr204; cat. no. 9101; 1:1,000),
ERK1/2 (Tyr925; cat. no. 9102; 1:1,000), p-p38-MAPK
(Thr180/Tyr182; cat. no. 9211; 1:1,000),
p38-MAPK (cat. no. 9212; 1:1,000), p-JNK
(Thr183/Tyr185; cat. no. 4671; 1:1,000), JNK
(cat. no. 9258; 1:1,000), Beclin-1 (cat. no. 3495; 1:1,000),
microtubule-associated protein 1A/1B-light chain 3 (LC3)-I/II (cat.
no. 4108; 1:1,000). These antibodies were purchased from Cell
Signaling Technology (Beverly, MA, USA), and CHOP (Santa Cruz
Biotechnology, Inc.). Chemiluminescence was detected using an ECL
kit (Advansta, Inc., Menlo Park, CA, USA) and a multiple Gel DOC
system (Fujifilm, Tokyo, Japan). The membrane was probed with
primary antibodies overnight at 4°C, followed by the application of
the following secondary antibodies for 1 h at room temperature:
Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (cat.
no. K0211589; 1:3,000) or HRP-conjugated goat anti-rabbit IgG (cat.
no. K0211708; 1:3,000; both KOMABiotech, Seoul, Korea).
Confocal microscopy
Cells were permeabilized with permeabilization
buffer (0.1% saponin in PBS), incubated with primary antibodies
against LC3-I/II (cat. no. 4108; 1:200; Cell Signaling Technology)
and lysosomal associated membrane protein 1 (LAMP1; cat. no. 4108;
1:100; Santa Cruz Biotechnology) for 30 min at 4°C, and incubated
with FITC-conjugated goat anti-rabbit IgG (cat. no. F9887; 1:400)
or PE-conjugated secondary goat anti-mouse IgG (cat. no. P9287;
1:400) (both Sigma-Aldrich; Merck KGaA) for 20 min at 4°C. Cells
were mounted using Dako fluorescent mounting medium (Agilent
Technologies, Inc., Santa Clara, CA, USA) and observed with a
confocal laser scanning microscope at ×400 magnification. Images
were acquired using Confocal Microscopy Software version 3.0 (Carl
Zeiss AG, Oberkochen, Germany).
Statistical analysis
Data are expressed as the mean ± SD. Statistical
analysis was conducted using one-way analysis of variance (ANOVA)
using SigmaPlot software (version 10.0; Systat Software, Inc., San
Jose, CA, USA). Bonferroni post hoc analysis was performed
following ANOVA for multiple comparisons. P<0.05 was considered
to indicate a statistically significant difference.
Results
Sequential treatment with celecoxib
and bortezomib enhances the apoptotic signaling pathway through the
up-regulation of p53, JNK, and p38-MAPK expression
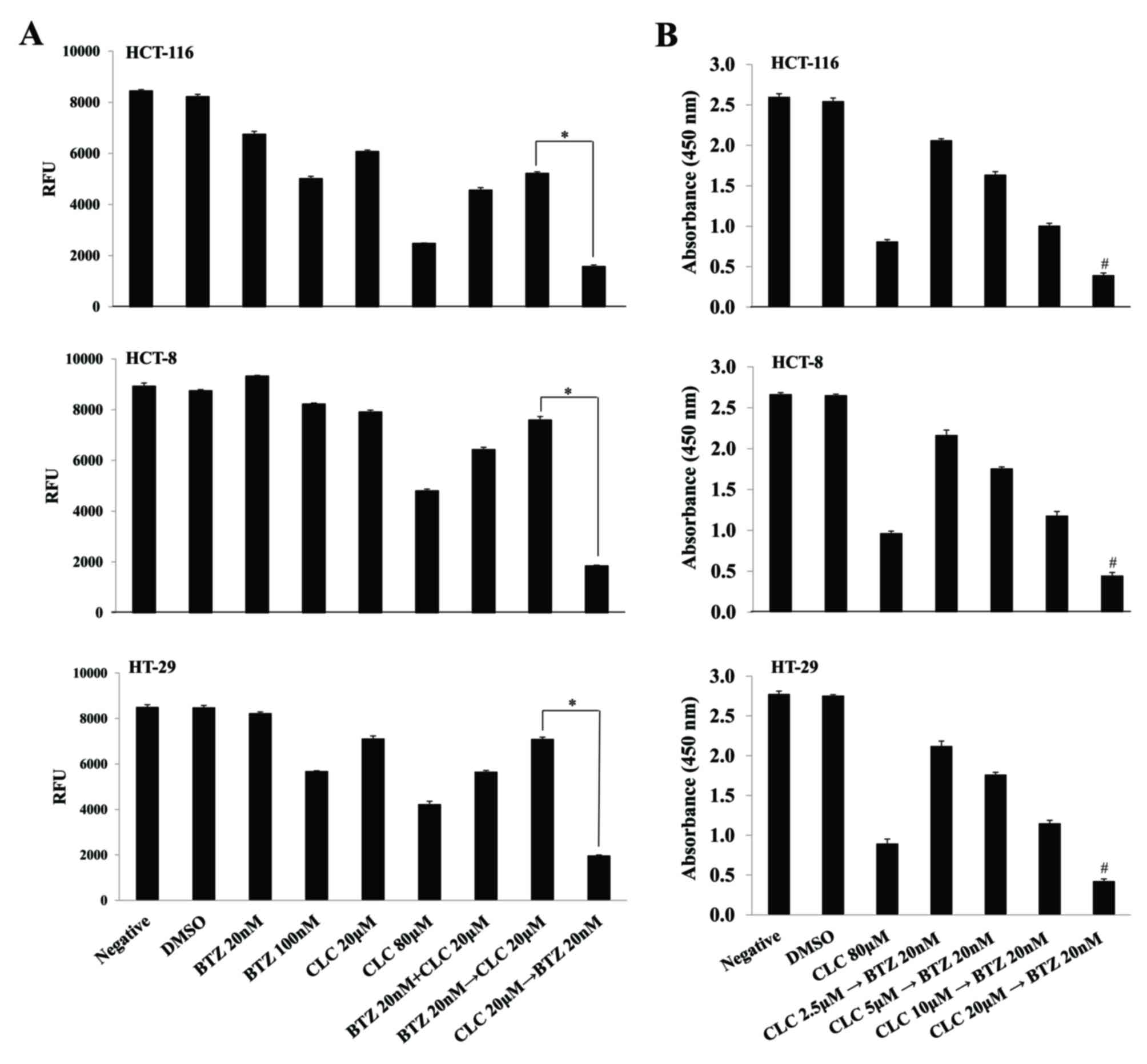
Although sub-toxic doses of each drug in isolation
(bortezomib 20 nM; celecoxib 20 µM) slightly suppressed cell
viability, the effect of the treatment with bortezomib (20 nM) and
celecoxib (20 µM) together on cell survival depended on the order
in which the drugs were applied. When colon cancer cells were
treated with celecoxib (CLC) then bortezomib (BTZ) (CLC 20 µM then
BTZ 20 nM), cell viability was significantly decreased compared
with the group treated with bortezomib and then celecoxib (BTZ 20
nM then CLC 20 µM) (P<0.01, CLC 20 µM then BTZ 20 nM vs. BTZ 20
nM then CLC 20 µM) (Fig. 1A). The
viability of colon cancer cells sequentially treated with celecoxib
and bortezomib also decreased in a celecoxib dose-dependent manner.
Notably, the effect of consecutive treatment with celecoxib and
bortezomib on cell viability differed significantly depending on
the doses of celecoxib (P<0.01, CLC 2.5 µM then BTZ 20 nM vs.
CLC 20 µM then BTZ 20 nM) (Fig.
1B).
Sequential treatment with celecoxib followed by
bortezomib synergistically increased the apoptotic cell death of
colon cancer cells (P<0.01, co-treatment with bortezomib and
celecoxib vs. celecoxib→bortezomib; P<0.005,
bortezomib→celecoxib vs. celecoxib→bortezomib; Fig. 2A), which was associated with the
activation and cleavage of caspase-9, caspase-3 and PARP (Fig. 2B). Furthermore, colon cancer cells
treated with celecoxib followed by bortezomib exhibited decreased
Bcl-2 expression, and increased Bax, p53 and PUMA expression,
compared with any other single or combination treatment (Fig. 2C). The expression of p-JNK and
p-p38-MAPK, which are required for the initiation of apoptosis, was
induced in the HCT-116 cells sequentially treated with celecoxib
followed by bortezomib; however, the expression of p-ERK was
downregulated (Fig. 2D). With
pre-exposure to SB203580 (a p38-MAPK inhibitor) or SP600125 (a JNK
inhibitor), the activation of MAPKs and caspases in the colon
cancer cells by treatment with celecoxib followed by bortezomib was
inhibited (Fig. 2D). These data
suggest that sequential treatment with celecoxib and bortezomib
enhances the synergistic induction of apoptotic death in human
colon cancer cells in a manner that depends on the order in which
the drugs are applied.
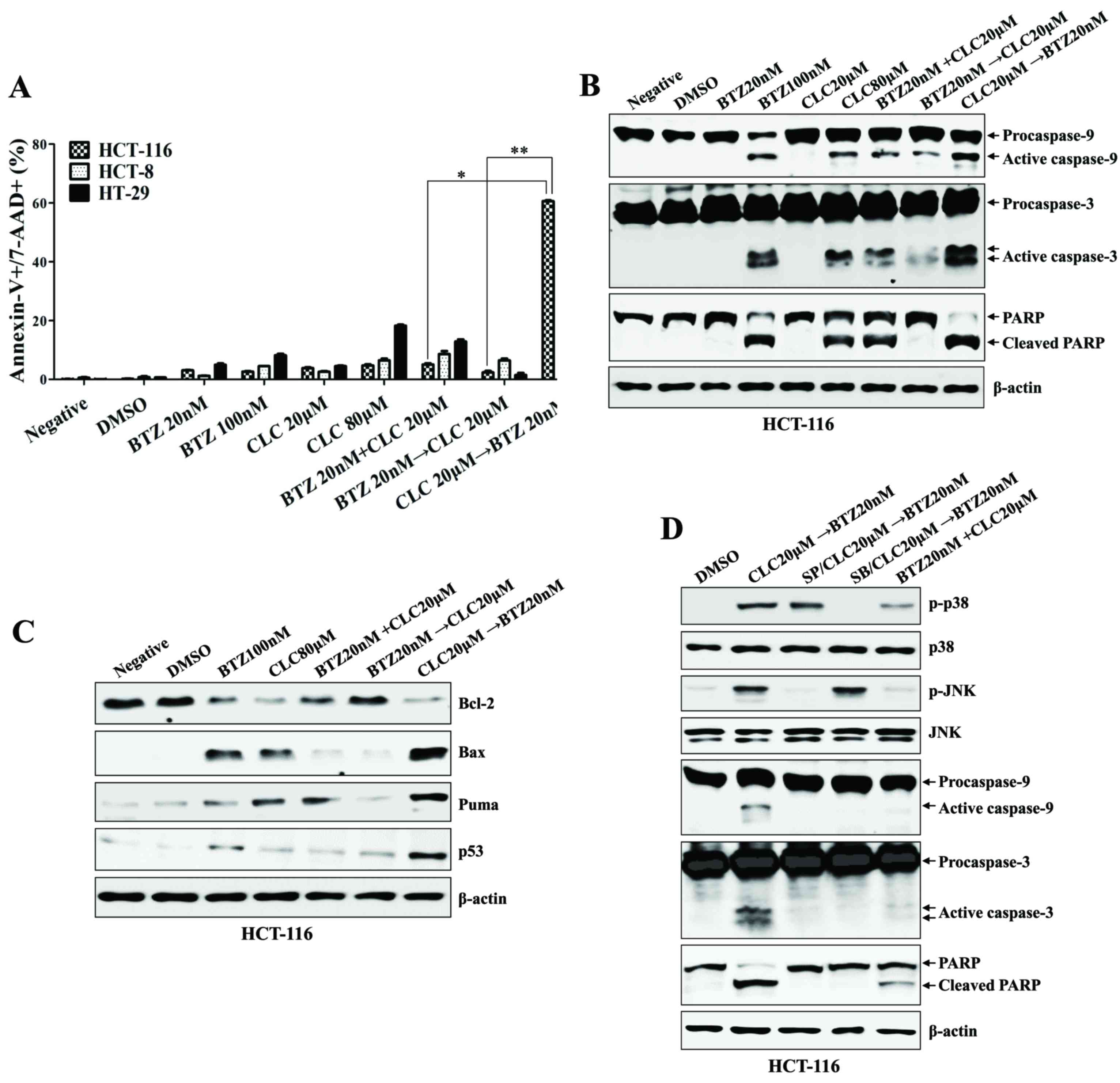 | Figure 2.Sequential treatment with CLC and BTZ
enhances apoptotic death of colon cancer cells. HCT-116 cells were
treated with BTZ or CLC individually or in combination. (A)
Annexin-V/7-AAD staining was used to estimate the rate of
apoptosis. The number of late-stage apoptotic cells
(annexin-V+/7-AAD+) was calculated by flow
cytometry. *P<0.01; **P<0.005. Cells from each indicated
condition were harvested and blotted with the indicated antibodies,
including (B) caspases (procaspase-9, active caspase-9,
procaspase-3, active caspase-3 and PARP (full-length PARP, cleaved
PARP), and (C) Bcl-2, Bax, PUMA and p53. β-actin served as an
internal control. (D) To inhibit the activation of JNK or
p38-mitogen-activated protein kinase, cells were pre-treated with
SP600125 (20 µM) or SB203580 (10 µM) for 2 h. The expression of
caspase-9, caspase-3 and cleaved PARP were detected by western
blotting. The results are representative of three independent
experiments. CLC, celecoxib; BTZ, bortezomib; 7-AAD,
7-aminoactinomycin D; PARP, poly (ADP-ribose) polymerase; negative,
non-treated cell; DMSO, dimethyl sulfoxide control; p-,
phosphorylated; JNK, c-Jun N-terminal kinase; PUMA, p53-upregulated
modulator of apoptosis. |
Sequential treatment with celecoxib
followed by bortezomib induces apoptosis in colon cancer cells
through ER stress-mediated calcium translocation
Whether treatment with celecoxib followed by
bortezomib stimulated ER stress-mediated mitochondrial dysfunction,
in which the translocation of calcium from the ER induces
mitochondria-dependent apoptosis (15), was investigated next. The treatment of
cells with celecoxib followed by bortezomib resulted in the
increased induction of CHOP, compared with cells co-treated with
alternative drug combinations, or with bortezomib followed by
celecoxib (Fig. 3A). Pre-exposure to
salubrinal (an ER stress inhibitor) of the colon cancer cells
inhibited the expression of p53 and CHOP, and prevented the
cleavage of caspase-9, caspase-3 and PARP subsequent to treatment
with celecoxib followed by bortezomib (Fig. 3B). Treatment with celecoxib followed
by bortezomib, and high doses of celecoxib alone, increased the
cytosolic and mitochondrial Ca2+ levels in colon cancer
cells; pre-treatment with salubrinal inhibited this effect
(Fig. 3C and D). The data
collectively suggest that ER stress-mediated apoptosis of colon
cancer cells treated with celecoxib followed by bortezomib is
associated, at least partially, with increases in cytosolic and
mitochondrial Ca2+.
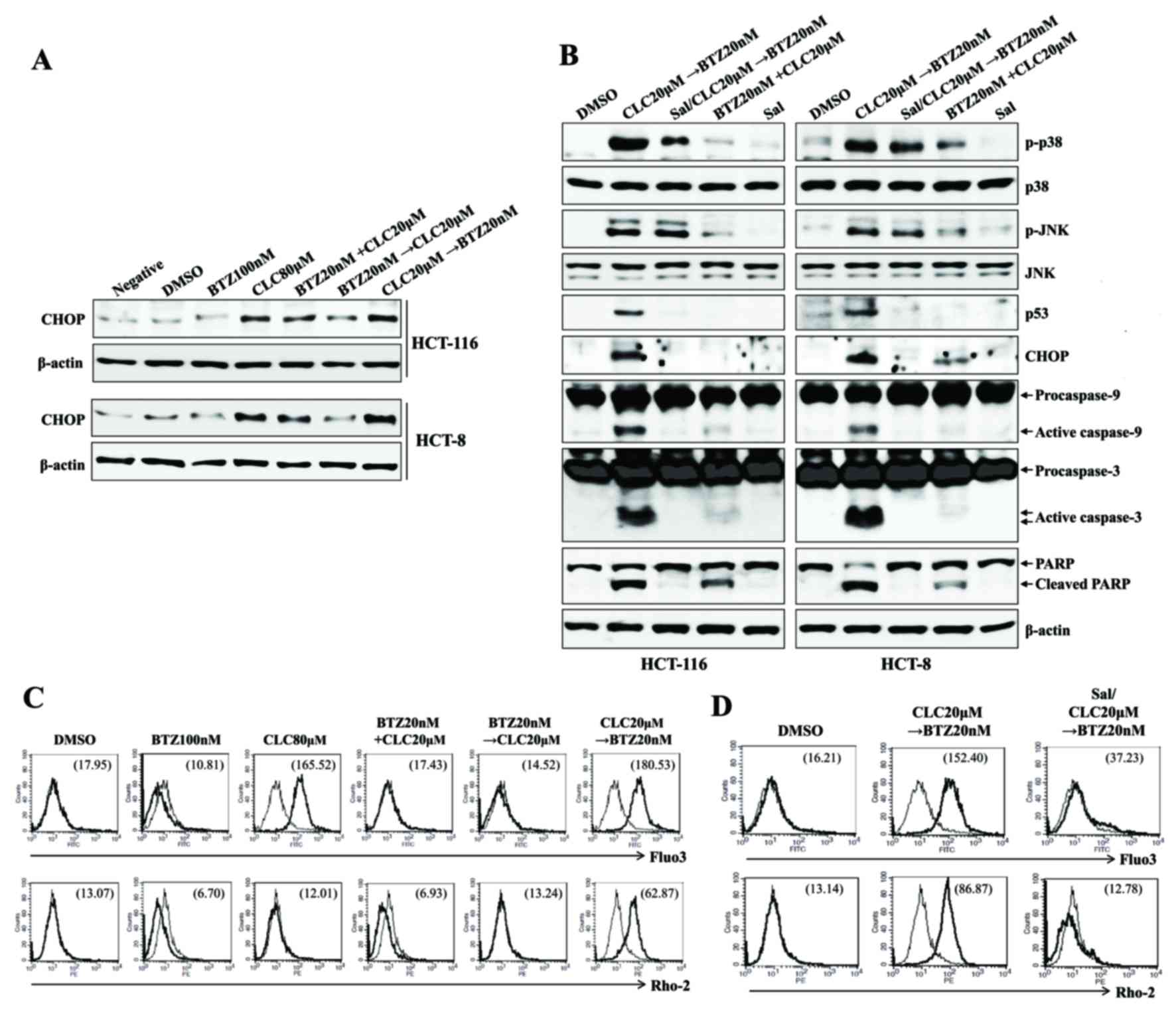 | Figure 3.Sequential treatment with CLC and BTZ
induces apoptosis in colon cancer cells through ER stress-mediated
calcium translocation. HCT-116 cells were treated with BTZ or CLC
alone or in combination. (A) Cells treated as indicated were
blotted with antibodies against CHOP. (B) Cells treated as
indicated were blotted with a range of antibodies. β-actin served
as an internal control. To inhibit endoplasmic reticulum stress,
cells were pre-incubated with Sal (2 µM) for 1 h. (C) Cytosolic and
(D) mitochondrial Ca2+ levels in HCT-116 cells were
determined using Fluo3-AM and Rhod2-AM, respectively. To block
endoplasmic reticulum stress, cells were pre-incubated with Sal (2
µM) for 1 h. The results are representative of three independent
experiments. CLC, celecoxib; BTZ, bortezomib; neg, non-treated
cell; CHOP, CCAAT-enhancer-binding protein homologous protein; Sal,
salubrinal; DMSO, dimethyl sulfoxide control; PARP, poly
(ADP-ribose) polymerase. |
Treatment with celecoxib followed by
bortezomib induces ER stress-mediated autophagy-associated cell
death in colon cancer cells
Whether treatment with celecoxib followed by
bortezomib was also connected to autophagy-associated cell death
was examined. Treatment with celecoxib followed by bortezomib
induced the expression of Beclin-1 and autophagosome-associated
LC3-I/II proteins, which are associated with autophagy-associated
cell death (Fig. 4A); however,
pre-treatment with salubrinal decreased the expression of Beclin-1
and LC3-I/II by treatment with celecoxib followed by bortezomib
(Fig. 4B). Although pre-exposure to
the autophagy inhibitor 3-MA inhibited the autophagy-associated
cell death of colon cancer cells subsequent to treatment with
celecoxib followed by bortezomib, the expression levels of CHOP,
p53 and p-MAPKs remained upregulated (Fig. 4C). Furthermore, treatment of the cells
with 3-MA had no effect on the levels of cytosolic or mitochondrial
Ca2+ subsequent to treatment with celecoxib followed by
bortezomib (Fig. 4D). These data
suggest that ER stress-associated apoptotic signaling in colon
cancer cells leads to the induction of autophagy-associated cell
death subsequent to treatment with celecoxib followed by
bortezomib.
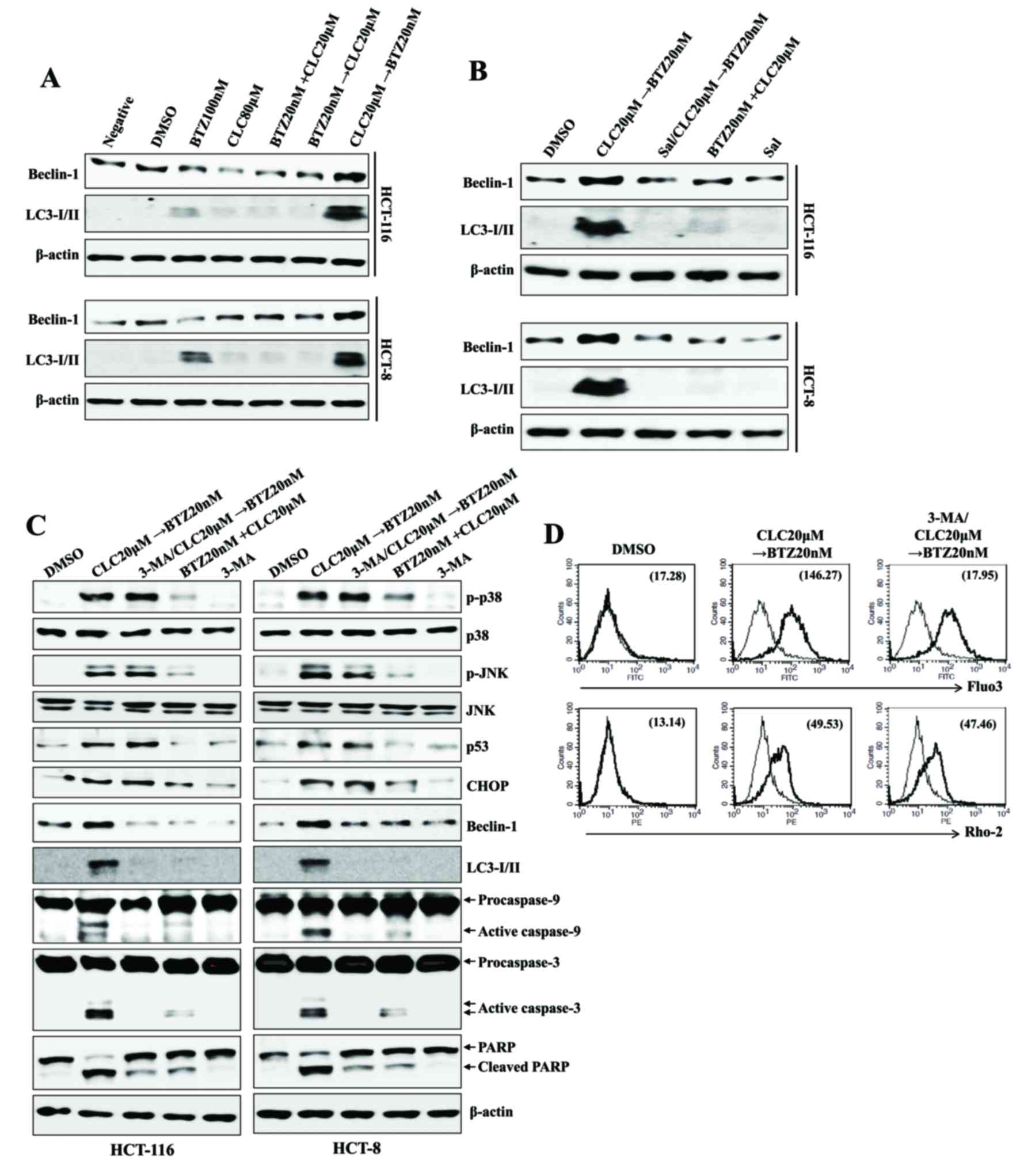 | Figure 4.Sequential treatment with CLC and BTZ
provokes ER stress-mediated autophagy-associated cell death in
colon cancer cells. HCT-116 cells were treated with BTZ or CLC
alone or in combination. (A-C) Total lysates of cells under each
indicated condition were harvested and blotted with the indicated
antibodies. β-actin served as an internal control. (B) To block ER
stress, cells were pre-incubated with salubrinal (2 µM) for 1 h.
(C) To inhibit the autophagic signal, cells were pre-treated with
3-MA (10 mM) for 2 h. (D) Cytosolic and mitochondrial
Ca2+ levels in HCT-116 cells were determined using
Fluo3-AM and Rhod2-AM, respectively. The results are representative
of three independent experiments. CLC, celecoxib; BTZ, bortezomib;
ER, endoplasmic reticulum; 3-MA, 3-methyladenine; negative,
non-treated cell; LC3-I/II, microtubule-associated protein
1A/1B-light chain 3; DMSO, dimethyl sulfoxide control; PARP, poly
(ADP-ribose) polymerase; p-, phosphorylated; JNK, c-Jun N-terminal
kinase; CHOP, CCAAT-enhancer-binding protein homologous
protein. |
p53 expression regulates ER
stress-mediated autophagy-associated cell death following treatment
with celecoxib followed by bortezomib
ER stress is associated with the induction of p53
expression (16), and the level of
cytoplasmic p53 serves a critical role in regulating ER
stress-associated Ca2+ homeostasis (17). Treatment with celecoxib followed by
bortezomib induced the expression of p53 and increased cytosolic
and mitochondrial Ca2+ levels. Next, it was investigated
whether the increased expression of p53 by treatment with celecoxib
followed by bortezomib was responsible for the ER
stress/autophagy-associated cell death due to the induction of
apoptosis. Pre-treatment with PFT-α, a selective p53 inhibitor, not
only prevented the increases in CHOP, Beclin-1 and LC-3I/II
expression (Fig. 5A), but also
reduced the cleavage of caspase-9, caspase-3 and PARP in colon
cancer cells treated with celecoxib followed by bortezomib
(Fig. 5B). When PFT-α was applied in
cells treated with celecoxib followed by bortezomib, intracellular
and mitochondrial levels of Ca2+ were reduced (Fig. 5C), and the co-localization of LAMP1
and LC3-I/II was inhibited, compared with the cells treated with
celecoxib followed by bortezomib (Fig.
5D). These data suggest that the expression of p53 in colon
cancer cells serves a role in inducing ER
stress/autophagy-associated cell death subsequent to treatment with
celecoxib followed by bortezomib.
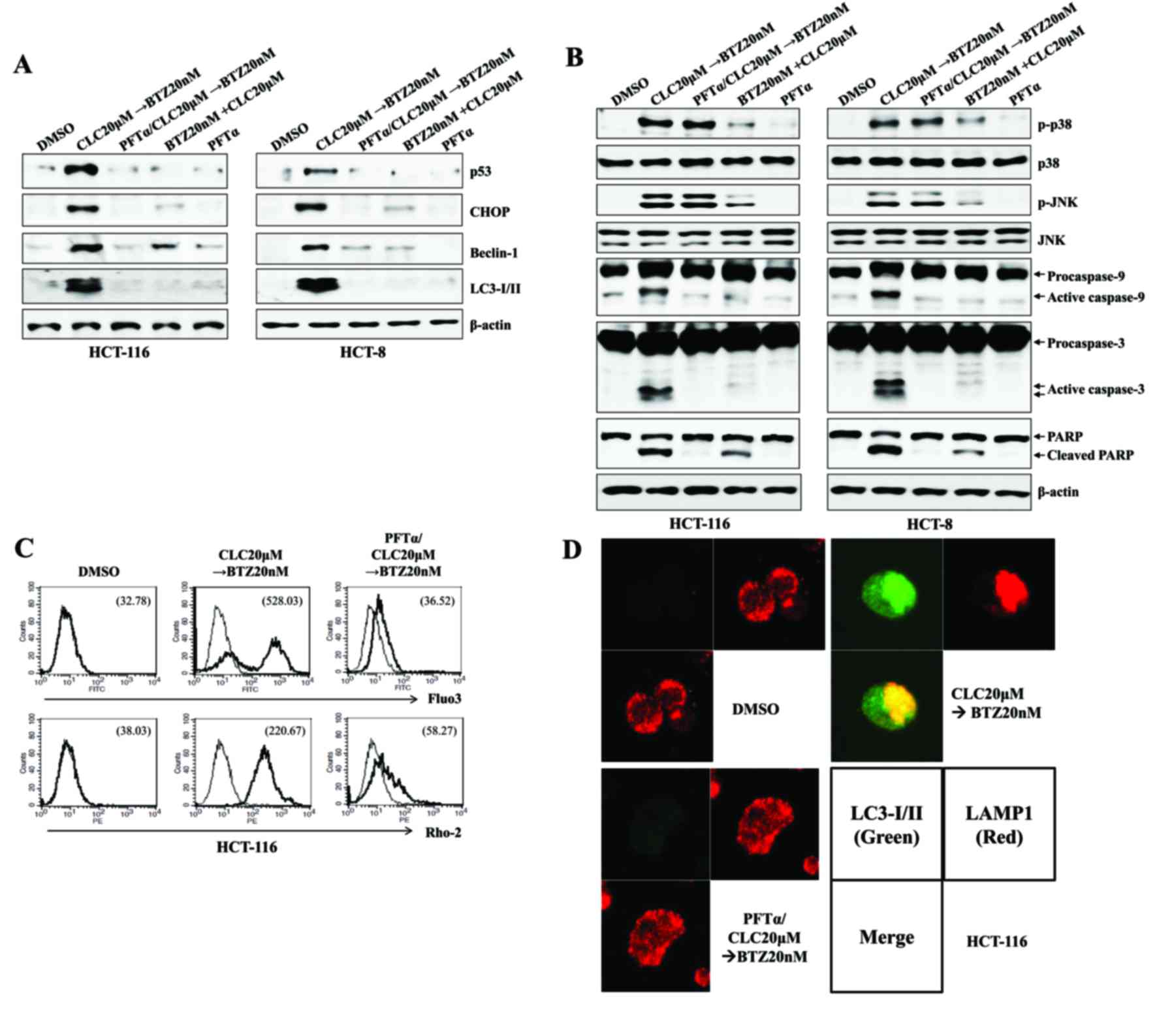 | Figure 5.The expression of p53 is dispensable
for the endoplasmic reticulum stress-mediated autophagy-associated
cell death of colon cancer cells treated sequentially with CLC and
BTZ. HCT-116 or HCT-8 cells were pre-incubated with PTF-α (50 µM)
for 1 h. (A and B) Cells in each indicated condition were harvested
and blotted with the indicated antibodies. β-actin served as the
internal control. (C) Cytosolic and mitochondrial Ca2+
levels in HCT-116 cells were determined using Fluo3-AM and
Rhod2-AM, respectively. (D) Subcellular distribution of LC3-I/II
and LAMP1 in HCT-116 cells. Cells were observed under a confocal
microscope (magnification, ×400). Green fluorescence indicates
LC3-I/II, and red fluorescence indicates LAMP1. The results are
representative of three independent experiments. CLC, celecoxib;
BTZ, bortezomib; PFT-α, pifithrin-α; LC3-I/II,
microtubule-associated protein 1A/1B-light chain 3; LAMP1,
lysosomal-associated membrane protein 1; DMSO, dimethyl sulfoxide
control; CHOP, CCAAT-enhancer-binding protein homologous protein;
p-, phosphorylated; JNK, c-Jun N-terminal kinase; PARP, poly
(ADP-ribose) polymerase. |
Treatment with celecoxib followed by
bortezomib induces the autophagy-associated cell death of colon
cancer cells even in the absence of p53 expression
Whether the treatment with celecoxib followed by
bortezomib affects ER stress-mediated autophagy-associated cell
death in p53−/− colon cancer cells was then
investigated. The treatment of p53−/− HCT-116 cells with
celecoxib followed by bortezomib induced the phosphorylation of
p38-MAPK and JNK and the expression of activated caspase-9 and
caspase-3 (Fig. 6A). Furthermore, the
induction of Beclin-1 and LC-3I/II expression was observed in
p53−/− HCT-116 cells treated with celecoxib followed by
bortezomib, although the induction of CHOP was not detected
(Fig. 6B). Notably, changes to the
levels of Bcl-2, Bax and PUMA expression, which are induced by p53,
were not observed (Fig. 6B). Although
no Ca2+ increase was detected in the cytoplasm or
mitochondria of p53−/− HCT-116 cells (Fig. 6C), fusion of the autophagosomes and
the lysosomes was detected subsequent to treatment with celecoxib
followed by bortezomib (Fig. 6D).
These data suggest that treatment with celecoxib followed by
bortezomib may cause ER stress-mediated autophagy-associated cell
death independent of p53 expression.
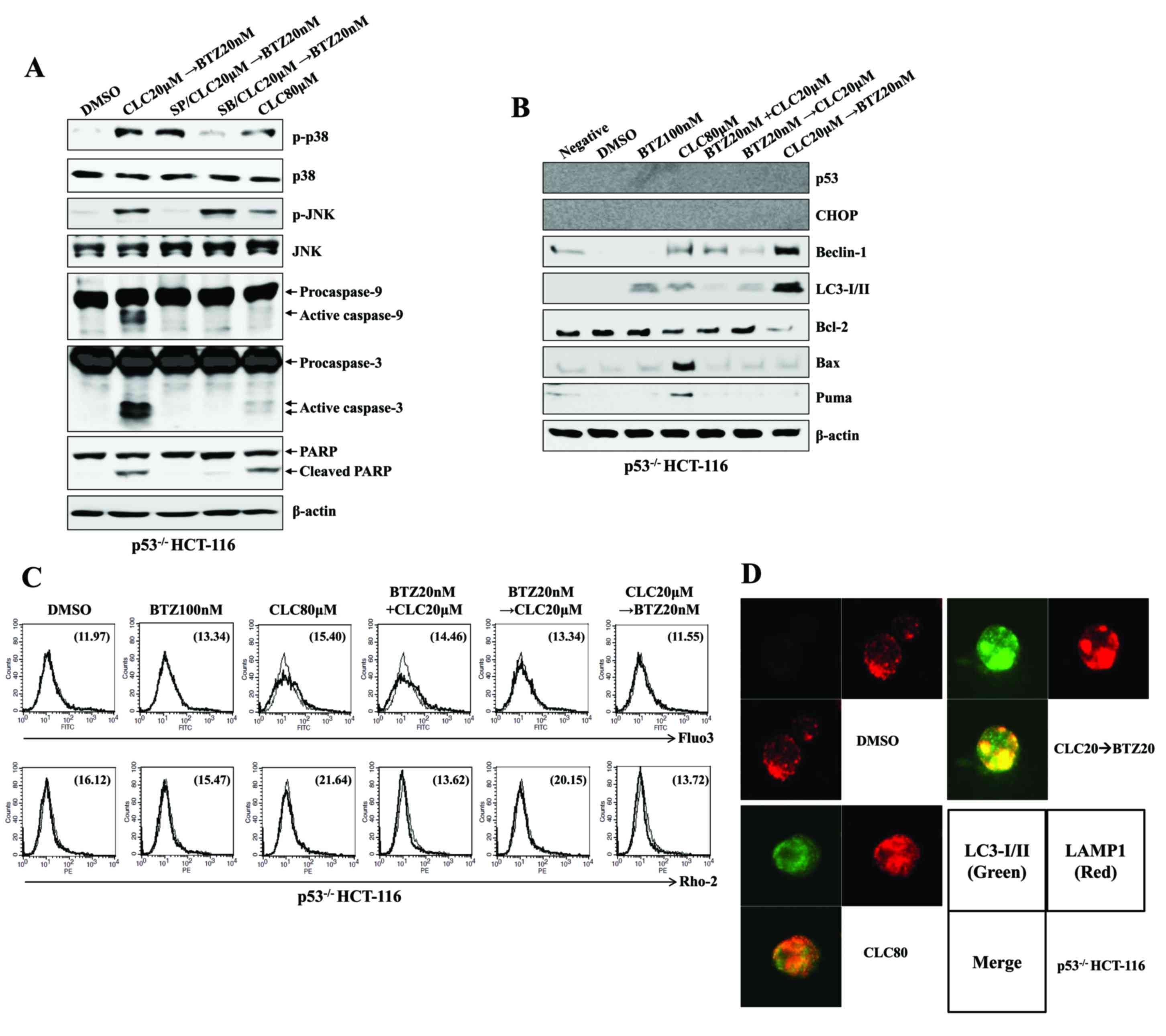 | Figure 6.Sequential treatment with CLC and BTZ
induces the autophagy-associated cell death of p53−/−
colon cancer cells. p53−/− HCT-116 cells were treated
with BTZ or CLC alone or in combination. (A and B) Total cell
lysates of each condition were harvested and blotted with the
indicated antibodies. β-actin served as an internal control. (C)
Cytosolic and mitochondrial Ca2+ levels in HCT-116 cells
were determined using Fluo3-AM and Rhod2-AM, respectively. (D)
Subcellular distribution of LC3-I/II and LAMP1 in p53−/−
HCT-116 cells. Cells were observed under a confocal microscope
(magnification, ×400). Green fluorescence indicates LC3-I/II, and
red fluorescence indicates LAMP1. The results are representative of
three independent experiments. CLC, celecoxib; BTZ, bortezomib;
LC3-I/II, microtubule-associated protein 1A/1B-light chain 3;
LAMP1, lysosomal-associated membrane protein 1; DMSO, dimethyl
sulfoxide control; p-, phosphorylated; JNK, c-Jun N-terminal
kinase; PARP, poly (ADP-ribose) polymerase; negative, non-treated
cells; CHOP, CCAAT-enhancer-binding protein homologous protein;
Bax; Bcl-associated X; PUMA, p53-upregulated modulator of
apoptosis. |
The expression of p53 regulates
intracellular calcium release from the ER to enhance
autophagy-associated cell death
As treatment with celecoxib followed by bortezomib
increased the autophagy-associated cell death of colon cancer cells
regardless of p53 expression, the role of, and association between,
intracellular Ca2+ and p53 expression in the ER
stress-induced autophagy-associated cell death of colon cancer
cells was investigated. The p53-expressing HCT-116 colon cancer
cells were more susceptible to apoptotic cell death than
p53−/− HCT-116 colon cancer cells following treatment
with celecoxib followed by bortezomib (P<0.05, p53-expressing
HCT-116 compared with p53-null HCT-116; Fig. 7A). In addition, treatment with
BAPTA-AM, a calcium chelator, failed to inhibit apoptosis or the
cleavage of caspase-3 in p53−/− HCT-116 cells subsequent
to treatment with celecoxib followed by bortezomib (#P<0.05,
celecoxib→bortezomib in p53-expressing HCT-116 vs. pretreatment
with BAPTA-AM, and then celecoxib→bortezomib in p53-expressing
HCT-116; ##P<0.001, pretreatment with BAPTA-AM, and then
celecoxib→bortezomib in p53-expressing HCT-116 vs. pretreatment
with BAPTA-AM, and then celecoxib→bortezomib in p53-null HCT-116;
Fig. 7B). Exposure to BAPTA-AM not
only reduced the expression of CHOP, Beclin-1 and LC3-I/II, but
also prevented the cleavage of caspase-3 in p53-expressing HCT-116
colon cancer cells treated with celecoxib followed by bortezomib
(Fig. 7C); however, treatment with
BAPTA-AM failed to inhibit the activation of Beclin-1 and LC3-I/II
and prevent the cleavage of caspase-3 in p53−/− HCT-116
cells subsequent to treatment with celecoxib followed by bortezomib
(Fig. 7C and D). These data suggest
that p53 expression is critical for promoting autophagy-associated
cell death through the regulation of intracellular Ca2+
released from the ER.
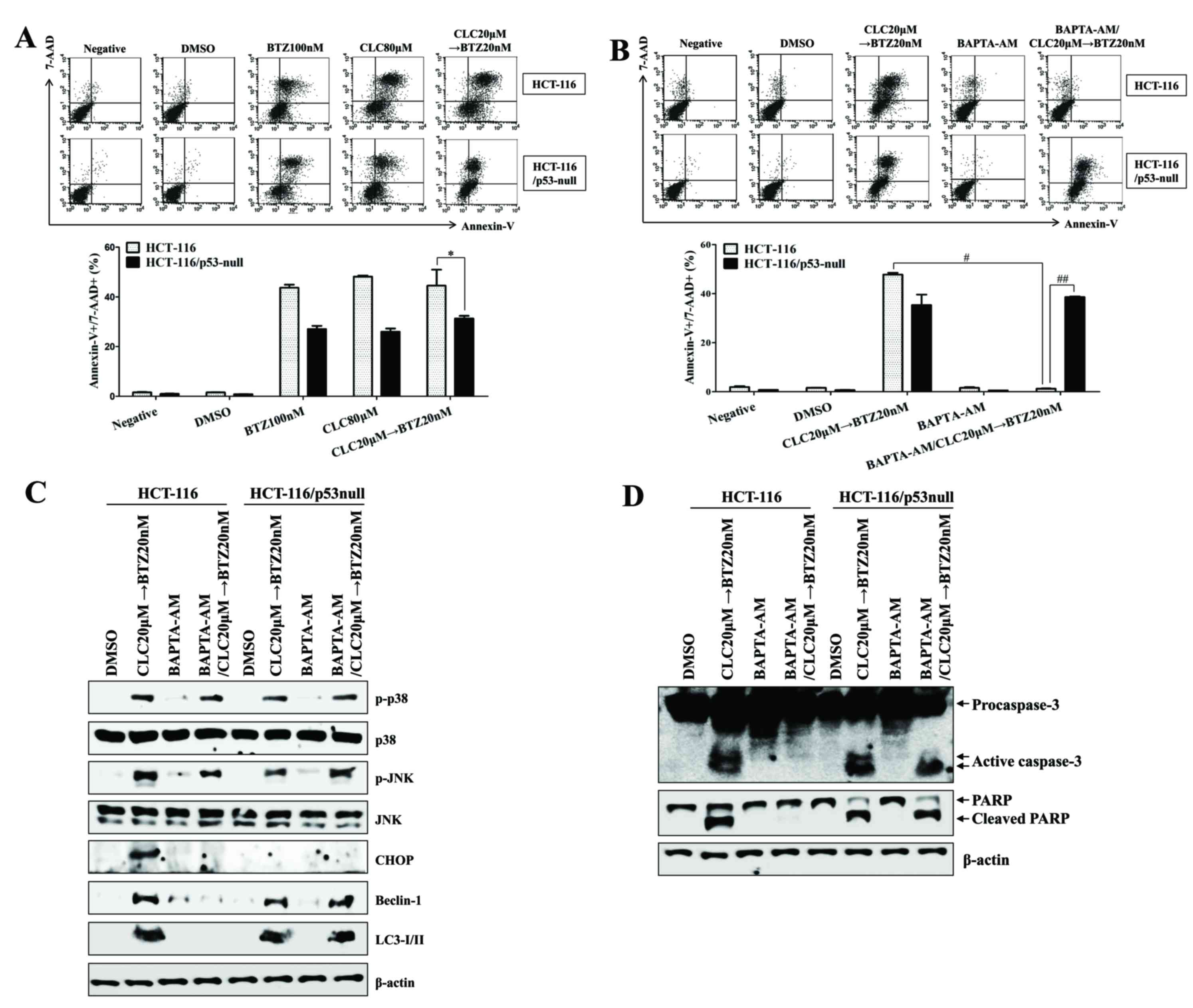 | Figure 7.The expression of p53 is critical for
endoplasmic reticulum stress-mediated autophagy-associated cell
death through the regulation of intracellular calcium. HCT-116 or
p53−/− HCT-116 cells were treated with BTZ or CLC alone
or in combination. (A and B) The percentage of apoptotic cells was
estimated by annexin-V/7-AAD staining. Data are representative of
three independent experiments. The number of late-stage apoptotic
cells (annexin-V+/7-AAD+) was calculated by
flow cytometry. *P<0.05; #P<0.05 and
##P<0.001. (B-D) To block the effect of
Ca2+, HCT-116 or p53−/− HCT-116 cells were
pre-incubated with BAPTA-AM (2 µM) for 30 min. (C and D) Total cell
lysates of each condition were harvested and immunoblotted with the
indicated antibodies. β-actin served as an internal control. The
results are representative of three independent experiments. BTZ,
bortezomib; CLC, celecoxib; 7-AAD, 7-aminoactinomycin D; negative,
non-treated cells; DMSO, dimethyl sulfoxide control; p-,
phosphorylated; JNK, c-Jun N-terminal kinase; CHOP,
CCAAT-enhancer-binding protein homologous protein; LC3-I/II,
microtubule-associated protein 1A/1B-light chain 3; PARP, poly
(ADP-ribose) polymerase. |
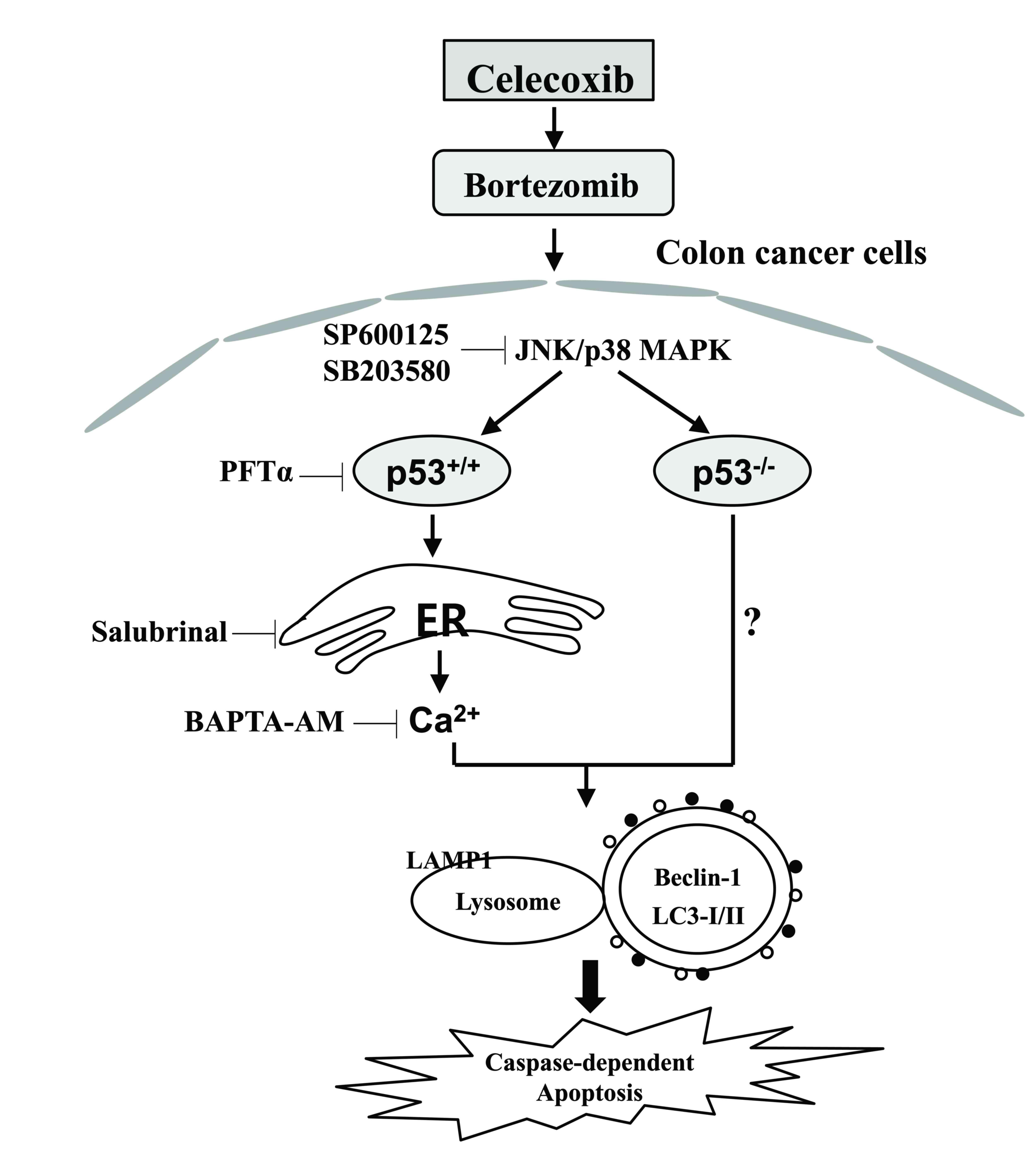
Discussion
Bortezomib and celecoxib or dimethyl-celecoxib (DMC)
as a novel drug combination enhance apoptotic cell death through
the upregulation of ER stress in addition to the activation of JNK
in glioblastoma (18). Pre-treatment
with bortezomib enhances the rate of apoptosis induced by treatment
with celecoxib through the induction of cell cycle arrest and ER
stress (19). In the present study,
the anticancer activity was enhanced by the treatment with
celecoxib followed by bortezomib; this was associated with
autophagy-associated cell death, regardless of p53 expression.
Furthermore, the expression of p53 was required for ER
stress-mediated Ca2+ translocation to promote
autophagy-associated cell death following treatment with celecoxib
followed by bortezomib (Fig. 8).
The anticancer activity of celecoxib has been
demonstrated in various animal tumor models and may potentially be
useful for the treatment of colorectal cancer and other tumor types
(20,21). Although celecoxib toxicity in tumor
cells occurs by toxic precipitation of increased doses of the drug
(22), celecoxib also causes ER
stress-induced apoptosis in a COX-2-independent manner (14,23).
However, the long-term use of high-dose celecoxib is associated
with severe gastrointestinal and cardiovascular adverse effects
(24). Additionally, the combination
treatment of colorectal cancer cells with celecoxib and other
chemotherapeutic agents induces autophagy-mediated drug resistance
(14). Although bortezomib exhibits a
unique mechanism for anticancer activity, it also blocks the
anticancer effect of the ER stress-inducing agents thapsigargin and
tunicamycin (8). The induction of
autophagy and the ER stress response was demonstrated to protect
MCF7 breast cancer cells from bortezomib-induced cell death
(25). On the basis of these results,
the limited effectiveness of celecoxib and bortezomib against
cancer cells has led to a search for therapeutic combination
regimens with other agents.
Persistent or intense ER stress induces different
effects on normal ER function and induces cell death or apoptosis
(26). The disturbance of autophagy
also renders cells vulnerable to ER stress, and autophagy
activation has been linked to cell death through the regulation of
ER stress (11,27). In the present study, the combination
of celecoxib followed by bortezomib was the most efficient regimen
for the induction of ER stress-mediated autophagy-associated cell
death, compared with the groups treated with single drugs alone or
with other co-treatment regimens. Although the inhibition of
autophagy by 3-MA suppressed the generation of autophagolysosomes,
leading to autophagy-associated cell death, 3-MA had no effect on
the inhibition of CHOP and p53 expression in colon cancer cells
sequentially treated with celecoxib and bortezomib. Exposure to
celecoxib followed by bortezomib induced the generation of
autophagolysosomes in p53−/− HCT-116 colon cancer cells.
These results suggest that ER stress is not a critical regulation
step for stimulating autophagy-associated cell death in colon
cancer cells following the serial treatment with celecoxib and
bortezomib.
Intracellular Ca2+ released due to ER
stress as a result of various stimuli is absorbed by mitochondria.
Once calcium is taken into the mitochondria, it causes the
activation of the apoptotic pathway (28). Giorgi et al (17) demonstrated that wild-type cytoplasmic
p53 at the ER binds to Ca2+ pumps to modulate
Ca2+ homeostasis and mitochondrial function. In the
present study, sequential treatment with celecoxib and bortezomib
also promoted ER stress-mediated apoptosis through Ca2+
translocation and changes in the intracellular and mitochondrial
Ca2+ concentration in p53-expressing HCT-116 cells.
Calcium chelation with BAPTA-AM decreased the expression levels of
CHOP and inhibited autophagy-associated cell death in wild-type
HCT-116 colon cancer cells. Although p53-expressing HCT-116 cells
were more sensitive to autophagy-associated cell death than
p53−/− HCT-116 cells, the treatment of p53−/−
HCT-116 cells with celecoxib followed by bortezomib also induced
apoptosis and the expression of Beclin-1 and LC3-I/II, but not
CHOP. These results suggest that the sequential drug combination
can cause apoptosis in a p53-independent manner, although for full
effectiveness, p53 expression is required to induce the release of
Ca2+ from the ER.
The overexpression of Bcl-2 inhibits Ca2+
transfer from the ER to the mitochondria and prevents apoptosis
(29). Bcl-2 proteins have been shown
to inhibit autophagy by disrupting Bcl-2/Bcl-XL-Beclin-1
complexes (30); however, it remains
unclear how Bcl-2-family proteins are associated with
autophagy-associated cell death in the absence of p53 expression.
In the present study, only Bcl-2 was decreased in p53−/−
HCT-116 colon cells sequentially treated with celecoxib and
bortezomib. On the basis of these results, further studies are
required to investigate how the combined treatment of celecoxib
followed by bortezomib induces autophagy-mediated cell death in the
absence of p53 expression.
Taken together, the results of the present study
provide information about the optimal order for anticancer drug
administration and the role of calcium and p53 on ER
stress-mediated autophagy-associated cell death in colon cancer
cells subsequent to treatment with celecoxib followed by
bortezomib.
Acknowledgements
The present study was supported by a 2016 Inje
University research grant (grant no. 20170021).
References
|
1
|
Arico S, Pattingre S, Bauvy C, Gane P,
Barbat A, Codogno P and Ogier-Denis E: Celecoxib induces apoptosis
by inhibiting 3-phosphoinositide-dependent protein kinase-1
activity in the human colon cancer HT-29 cell line. J Biol Chem.
277:27613–27621. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lynch PM, Ayers GD, Hawk E, Richmond E,
Eagle C, Woloj M, Church J, Hasson H, Patterson S, Half E and Burke
CA: The safety and efficacy of celecoxib in children with familial
adenomatous polyposis. Am J Gastroenterol. 105:1437–1443. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsutsumi S, Namba T, Tanaka KI, Arai Y,
Ishihara T, Aburaya M, Mima S, Hoshino T and Mizushima T: Celecoxib
upregulates endoplasmic reticulum chaperones that inhibit
celecoxib-induced apoptosis in human gastric cells. Oncogene.
25:1018–1029. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsutsumi S, Gotoh T, Tomisato W, Mima S,
Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M and Mizushima
T: Endoplasmic reticulum stress response is involved in
nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death
Differ. 11:1009–1016. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin WC, Chuang YC, Chang YS, Lai MD, Teng
YN, Su IJ, Wang CC, Lee KH and Hung JH: Endoplasmic reticulum
stress stimulates p53 expression through NF-κB activation. PLoS
One. 7:e391202012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Edagawa M, Kawauchi J, Hirata M, Goshima
H, Inoue M, Okamoto T, Murakami A, Maehara Y and Kitajima S: Role
of activating transcription factor 3 (ATF3) in endoplasmic
reticulum (ER) stress-induced sensitization of p53-deficient human
colon cancer cells to tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL)-mediated apoptosis through
up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib.
J Biol Chem. 289:21544–21561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ludwig H, Khayat D, Giaccone G and Facon
T: Proteasome inhibition and its clinical prospects in the
treatment of hematologic and solid malignancies. Cancer.
104:1794–1807. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nawrocki ST, Carew JS, Pino MS, Highshaw
RA, Dunner K Jr, Huang P, Abbruzzese JL and McConkey DJ: Bortezomib
sensitizes pancreatic cancer cells to endoplasmic reticulum
stress-mediated apoptosis. Cancer Res. 65:11658–11666. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu J, Tiwari S, Steiner P and Zhang L:
Differential apoptotic response to the proteasome inhibitor
Bortezomib [VELCADE, PS-341] in Bax-deficient and p21-deficient
colon cancer cells. Cancer Biol Ther. 2:694–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kao C, Chao A, Tsai CL, Chuang WC, Huang
WP, Chen GC, Lin CY, Wang TH, Wang HS and Lai CH: Bortezomib
enhances cancer cell death by blocking the autophagic flux through
stimulating ERK phosphorylation. Cell Death Dis. 5:e15102014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang S and Sinicrope FA:
Celecoxib-induced apoptosis is enhanced by ABT-737 and by
inhibition of autophagy in human colorectal cancer cells.
Autophagy. 6:256–269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deniaud A, el dein Sharaf O, Maillier E,
Poncet D, Kroemer G, Lemaire C and Brenner C: Endoplasmic reticulum
stress induces calcium-dependent permeability transition,
mitochondrial outer membrane permeabilization and apoptosis.
Oncogene. 27:285–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Lee B and Lee AS: Endoplasmic
reticulum stress-induced apoptosis: Multiple pathways and
activation of p53-up-regulated modulator of apoptosis (PUMA) and
NOXA by p53. J Biol Chem. 281:7260–7270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giorgi C, Bonora M, Sorrentino G,
Missiroli S, Poletti F, Suski JM, Ramirez Galindo F, Rizzuto R, Di
Virgilio F, Zito E, et al: p53 at the endoplasmic reticulum
regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci
USA. 112:1779–1784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kardosh A, Golden EB, Pyrko P, Uddin J,
Hofman FM, Chen TC, Louie SG, Petasis NA and Schönthal AH:
Aggravated endoplasmic reticulum stress as a basis for enhanced
glioblastoma cell killing by bortezomib in combination with
celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer
Res. 68:843–851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JE, Lee JI, Jin DH, Lee WJ, Park GB,
Kim S, Kim YS, Wu TC, Hur DY and Kim D: Sequential treatment of HPV
E6 and E7-expressing TC-1 cells with bortezomib and celecoxib
promotes apoptosis through p-p38 MAPK-mediated downregulation of
cyclin D1 and CDK2. Oncol Rep. 31:2429–2437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Altorki NK, Keresztes RS, Port JL, Libby
DM, Korst RJ, Flieder DB, Ferrara CA, Yankelevitz DF, Subbaramaiah
K, Pasmantier MW and Dannenberg AJ: Celecoxib, a selective
cyclo-oxygenase-2 inhibitor, enhances the response to preoperative
paclitaxel and carboplatin in early-stage non-small-cell lung
cancer. J Clin Oncol. 21:2645–2650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Masferrer JL, Leahy KM, Koki AT, Zweifel
BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ and
Seibert K: Antiangiogenic and antitumor activities of
cyclooxygenase-2 inhibitors. Cancer Res. 60:1306–1311.
2000.PubMed/NCBI
|
|
22
|
Sacchetti A: Cancer cell killing by
Celecoxib: Reality or just in vitro precipitation-related artifact?
J Cell Biochem. 114:1434–1444. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho HY, Thomas S, Golden EB, Gaffney KJ,
Hofman FM, Chen TC, Louie SG, Petasis NA and Schönthal AH: Enhanced
killing of chemo-resistant breast cancer cells via controlled
aggravation of ER stress. Cancer Lett. 282:87–97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Solomon SD, McMurray JJ, Pfeffer MA,
Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E and
Bertagnolli M: Adenoma Prevention with Celecoxib (APC) Study
Investigators: Cardiovascular risk associated with celecoxib in a
clinical trial for colorectal adenoma prevention. N Engl J Med.
352:1071–1080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Milani M, Rzymski T, Mellor HR, Pike L,
Bottini A, Generali D and Harris AL: The role of ATF4 stabilization
and autophagy in resistance of breast cancer cells treated with
Bortezomib. Cancer Res. 69:4415–4423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: A matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cuervo AM: Autophagy: In sickness and in
health. Trends Cell Biol. 14:70–77. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oakes SA, Scorrano L, Opferman JT, Bassik
MC, Nishino M, Pozzan T and Korsmeyer SJ: Proapoptotic BAX and BAK
regulate the type 1 inositol trisphosphate receptor and calcium
leak from the endoplasmic reticulum. Proc Natl Acad Sci USA.
102:105–110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pinton P, Giorgi C and Pandolfi PP: The
role of PML in the control of apoptotic cell fate: A new key player
at ER-mitochondria sites. Cell Death Differ. 18:1450–1456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar
|