Breast cancer is the leading cause of
cancer-associated mortality for women. In 2012, 1.7 million people
were diagnosed with breast cancer worldwide, and 521,900 succumbed
to the complications (1). Given the
varied prognosis and response to treatment of patients with breast
cancer, the molecular classification of breast cancer has been
examined to improve the understanding of this disease.
Triple-negative breast cancer (TNBC), which was first reported in
the literature in 2005, is a molecular subset of breast cancer.
TNBC is characterized by the absence of estrogen receptor (ER),
progesterone receptor (PR) and human epidermal growth factor
receptor 2 (HER2) expression (2), and
accounts for 16% of all breast cancer cases (3). Although TNBC tumors display relatively
simple molecular phenotypes, they are inherently heterogeneous. In
particular, TNBCs exhibit varying morphology, gene expression and
signaling pathway activity (4); thus,
they have complicated clinicopathological features, to the
detriment of the prognosis of patients with TNBC. TNBC was compared
with other breast cancer subtypes in a retrospective analysis;
increased risks of distant relapse [hazard ratio (HR), 2.6;
P<0.0001] and mortality (HR, 3.2; P<0.001) in the first five
years were identified (5). As
established endocrine and targeted therapies are ineffective
against TNBC, chemotherapy remains the primary regimen for TNBC
treatment (6). TNBC is highly
sensitive to cytotoxic chemotherapy (7), but this treatment is associated with
relatively low rates of pathological response (8,9). Thus,
developing a more effective therapeutic method for patients with
TNBC is necessary.
With this objective, a number of studies have
investigated the molecular classification of TNBC to determine
optimal individualized therapy strategies for TNBC. Lehmann et
al (4) indicated that TNBCs could
be classified into the following subtypes according to gene
expression profiles: Basal-like subtypes 1 and 2, immunomodulatory
subtype, mesenchymal subtype, mesenchymal stem-like subtype, and
luminal androgen receptor (AR) subtype. Alternatively, Burstein
et al (10) reported that
TNBCs could be divided into four subtypes:
Basal-like/immune-suppressed subtype, basal-like/immune-activated
subtype, mesenchymal subtype and luminal/AR subtype. Patients with
specific tumor molecular abnormalities treated with molecularly
matched targeted therapy respond better to therapy compared with
those treated with non-matched targeted therapy (11). In the present review, the molecular
markers and signaling pathways frequently dysregulated in TNBCs,
and the targeted therapies in clinical trials and preclinical
studies, will be summarized.
RTKs are important components of signal transduction
pathways in the regulation of proliferation, and are associated
with two downstream signaling pathways in particular: The
Ras/mitogen-activated protein kinase (MAPK) pathway, and the
phosphoinositide 3-kinase (PI3K)/AKT/mechanistic target of
rapamycin (mTOR) pathway. The RTKs include epidermal growth factor
receptor (EGFR), vascular endothelial growth factor receptor
(VEGFR) 1–3, platelet-derived growth factor receptor (PDGFR) α/β,
insulin-like growth factor receptor (IGFR), fibroblast growth
factor receptor (FGFR), c-Met, and transforming growth factor
receptor-β (TGFR-β), all of which are potential targets for TNBC
therapy (4,12–16). EGFR
dysregulation is the most commonly identified in TNBC tumors;
60–80% of TNBC tumors demonstrate EFGR overexpression (17,18).
However, the applicability of anti-RTK drugs against TNBC are
limited on account of biochemical multiplicity and toxicity
(19). For example, lapatinib, a dual
EGFR and HER2 TK inhibitor, is ineffective in patients with TNBC,
although it is clinically effective against HER2-positive breast
cancer. The mechanism of lapatinib resistance in TNBC may be
associated with interleukin-6 expression (20). The inhibition of Src homology
phosphotyrosyl phosphatase 2 (SHP2), an important molecule in
EGFR/FGFR1/c-Met signaling (21), was
reported to suppress TNBC tumorigenesis and metastasis in
vitro (22), indicating the
potential anti-tumor efficiency of RTK inhibitors in TNBC
treatment. A number of RTK inhibitors have also exhibited promising
anticancer therapeutic efficacy in a clinical setting. For example,
bevacizumab is an anti-VEGF monoclonal antibody. In a single-arm
and phase II multicenter study of bevacizumab, docetaxel, and
carboplatin-based neoadjuvant treatment for patients with stage
II/III TNBC, the results demonstrated a relatively high
pathological complete response rate (42%) with a low risk of
adverse events (23); additionally,
adding bevacizumab to neoadjuvant chemotherapy regimens improved
the pathological complete response rate among patients with TNBC
(39.3 vs. 27.9%; P=0.003) (24).
The Ras/MAPK pathway promotes cell proliferation,
cell differentiation and angiogenesis (25). Ras family members, including H-Ras,
K-Ras and N-Ras, can be activated by RTKs to transmit growth
signals from the cell membrane to the nucleus via a series of
phosphorylated proteins, including Raf, MAPK kinase 1 (MEK) and
extracellular signal-regulated kinases (ERK) 1/2 (26). Although the frequency of mutations in
the Ras/MAPK signaling pathway is <2% in TNBC, copy number
variations of certain genes from the Ras/MAPK pathway have been
demonstrated to be associated with TNBC (26). For example, the overexpression of ERK
is associated with a higher mortality rate in patients with TNBC
(27). The MEK inhibitor selumetinib
inhibited the motility and invasiveness of the MDA-MB-231 and
SUM149 TNBC cell lines in vitro. In addition, selumetinib
had a significant effect on the prevention of lung metastasis in a
TNBC-bearing mouse xenograft model (25). These findings may provide evidence of
the applicability of MEK inhibitors in TNBC treatment. However,
certain genetic defects along the Ras/MAPK pathway, including the
loss of negative regulators of MAPK signaling, such as phosphatase
and tensin homolog (PTEN) and certain regulatory micro (mi)RNAs,
such as the let-7 family, are also proposed to serve an important
role in TNBC development (26). The
Ras/MAPK pathway negatively regulates tumor immunogenicity by
affecting the process of tumor antigen presentation in TNBC cells;
compared with solo therapy, combining MEK inhibition and programmed
death-1 (PD-1)/programmed death ligand 1 (PD-L1) immune checkpoint
inhibitors increased the therapeutic efficiency in a murine
syngeneic TNBC model (28).
PI3K family members are activated by RTKs. Activated
PI3Ks phosphorylate phosphatidylinositol-4,5-diphosphate
(PIP2) to phosphatidylinositol-3,4,5-triphosphate
(PIP3), resulting in the downstream phosphorylation of
AKT (29). Phosphorylated AKT then
activates mTOR, a serine/threonine protein kinase, through the
intermediary tuberous sclerosis complex 1/2 to promote protein
synthesis and cell growth (30,31). PTEN
serves an important tumor suppressor role in the process by
inhibiting the dephosphorylation of PIP3 to
PIP2 (32). The
PI3K/AKT/mTOR pathway mediates a range of processes, including cell
growth, survival and migration, and tumor formation and
angiogenesis (33). The dysregulation
of the PI3K/AKT/mTOR pathway occurs frequently in TNBC.
PI3KCα-activating mutations are observed in 23.7% of TNBC patients
(34), and PTEN loss mutations,
including promoter silencing and functional suppression, are
detected in 25–30% of TNBC cases (32,35,36). With
regards to outcome, the hyperactivation of AKT and mTOR are
associated with the poor prognosis of patients with TNBC, and based
on success in preclinical experiments, dual inhibition of these
molecules may represent a promising strategy for TNBC treatment
(37–39).
The mTOR inhibitor everolimus has been approved by
the US Food and Drug Administration (FDA), and can be combined with
the aromatase inhibitor exemestane for patients with metastatic
homologous recombination (HR)-positive breast cancer (40). The therapeutic effect of everolimus
has been confirmed for patients with TNBC; mTOR activation may lead
to platinum therapy resistance (41),
and so everolimus combined with carboplatin has been proposed as an
effective therapy for patients with metastatic TNBC (42). Phase I trials have also demonstrated
that patients with metastatic TNBC who received a combination of
chemotherapy and PI3K/AKT/mTOR inhibitor-based targeted therapy had
a significantly prolonged median PFS time compared with patients
who did not receive the targeted therapy (43). Another study reported that PI3K
inhibition causes HR impairment and increased sensitivity to
poly(ADP-ribose) polymerase (PARP) inhibition in TNBC without
breast cancer associated (BRCA) 1/2 mutations. Therefore, PI3K
inhibition can improve the therapeutic efficiency of PARP
inhibition in BRCA-wild type TNBC (44). Based on this observation, a clinical
trial with BKM120 (buparlisib) and olaparib was initiated, as
presented in Table I (45–67).
A high level of crosstalk between the Ras/MAPK and
PI3K/AKT/mTOR pathways has been detected in basal-like breast
cancer models (68), and an approach
that inhibits both pathways may be feasible. However, the high
toxicity levels of such an approach are concerning (69), so a greater understanding of the
cross-talk mechanisms between pathways is required before effective
targeted therapy of this type with improved tolerability can be
developed.
EMT is an essential biological process that also
assists the migration and invasion of malignant tumor cells. Thus,
elucidating the molecular mechanism of the EMT process and its
association with the occurrence, development and metastasis of
cancer is of great significance.
During EMT, epithelial cells lose the expression of
E-cadherin and acquire mesenchymal markers, including vimentin
(70). There is evidence to indicate
that a number of RTKs, including EGFR, IGF1R, hepatocyte growth
factor receptor and c-Met, non-RTKs, including Src, and embryonic
transcription factors, including Twist and Slug (71–77), can
induce EMT. Diverse signaling pathways, including MAPKs, PI3K and
nuclear factor-κB, also promote EMT (76,78). Other
pathways, including Notch and Wnt/β-catenin signaling pathways, are
also associated with EMT (79). Tumor
cells undergoing EMT may acquire stem cell-like phenotypes and
migratory abilities (80). Previous
studies have indicated that the genes involved in EMT and
conversion to the cancer stem cell phenotype are activated in TNBC
(4,81). Additional studies have demonstrated
that EMT may induce the resistance to chemotherapy and radiotherapy
(82), and may thus have potential as
a therapeutic target in TNBC.
In preclinical trials, miRNAs have been demonstrated
to regulate tumor EMT and metastasis by inhibiting the expression
of certain genes (83). For example,
a previous study indicated that miR-200b-3p and miR-200b-5p
synergize to suppress TNBC cell migration by inhibiting EMT
(84). This finding may provide a
novel strategy for clinical treatment. Furthermore, protein
tyrosine kinase 6 (PTK6) is an intracellular non-receptor kinase
that can promote EMT and regulate the metastasis of TNBC cells by
modulating E-cadherin expression. PTK6 inhibition may prevent the
metastasis of TNBC cells, and thus exhibits clinical potential for
improving the treatment strategies for patients with mesenchymal
TNBC (85). Considering the
association of EMT with breast cancer stem cells (80), aldehyde dehydrogenase 1 (ALDH1)
inhibitors were proposed as a therapeutic alternative in TNBC
therapy, targeting the characteristic ALDH1 phenotypic marker of
breast cancer stem cells (86,87). A
preclinical study confirmed that LBH589, a histone deacetylase
inhibitor, could inhibit the metastasis of TNBC cells by partially
reversing EMT (88).
Aberrant regulation of the Wnt signaling pathway
serves an important role in tumorigenesis (89). This regulation is associated with EMT
and self-renewal in breast cancer, as it regulates the
transcription factors Twist and Slug (90,91).
During tumorigenesis, the Wnt ligand binds to
Frizzled (Fz), a seven-pass transmembrane surface receptor, and its
co-receptor, low-density lipoprotein receptor-related 5/6 (LRP5/6),
to form a Wnt-Fz-LRP6 complex. The combination of this complex with
the protein Dishevelled can elicit the phosphorylation of LRP6 and
the recruitment of the axin complex, which is composed of axin,
anaphase-promoting complex, casein kinase 1 and glycogen synthase
kinase 3. These events ultimately stabilize β-catenin, which is
degraded by the axin complex without Wnt. The β-catenin protein is
transferred to the nucleus where it activates the transcription of
Wnt target genes (92).
In addition to the Wnt pathway, the TGF-β/Smad
pathway can also regulate cell proliferation, invasion, apoptosis
and metastasis, and induce EMT, and thus has potential in targeted
strategies against TNBC (97). When
secreted from cells, TGF-β remains as an inactive, latent
homodimeric polypeptide (98). TGF-β
can be activated by hydrolyzing the latent complex. The TGF-β then
binds to and activates its receptors, TGF-β receptor type II
(TβRII) and I (TβRI), which are transmembrane serine/threonine
kinases. This induces two TβRI and two TβRII molecules to form a
heterotetramer, and TβRII triggers the cross-phosphorylation of
TβRI, allowing the activation of substrate Smad proteins (99). Smads include receptor-regulated Smads
(R-Smads), common mediator Smads (Co-Smads) and inhibitory Smads
(I-Smads). Among the Smad types, Smad-2 and Smad-3, as R-Smads, are
the direct substrates of TβRI. The activated R-Smads combine with
Smad-4, a Co-Smad, where they induce the activation of specific
genes. Inversely, Smad-7, as an I-Smad, can inactivate the
TGF-β/Smad pathway by disrupting the combination of R-Smad with
TβRI (100).
Smad-2 or Smad-3 overexpression, when combined with
Smad-4, can induce EMT. Inversely, the reduced expression of
Smad-2, Smad-3 and Smad-4, or the overexpression of Smad-7, can
constrain EMT (101). Clinical
evidence suggests that ~40% of human breast cancer tumors have high
TGFβ1, TGFβ2, Smad-3 and Smad-4 expression levels, although the
positive TGFβ gene signature occurs primarily in ER-positive breast
tumors and lung metastases (102).
In addition, a high TGF-β1 expression level was detected in TNBC
cells (MDA231, Hs578T, HCC1806) compared with non-TNBC (BT474,
ZR75-1, SKBR3) (103). Treatment
with zerumbone may inhibit the tumorigenicity of TNBC cells by
suppressing the TGFβ/Smad pathway, and thus has a potential as a
targeted drug for TNBC. LY2109761, another selective TβRI/II dual
inhibitor, can also suppress the invasion and motility of TNBC
cells (103). The antidiabetic agent
metformin can hinder the TGF-β/Smad pathway by disrupting the
activation of Smad-2 and Smad-3 in TNBC (104). This finding may offer a novel
perspective for the clinical treatment of TNBC patients.
Although chemotherapy is the central treatment
methodology against TNBC, patients with TNBC are likely to
eventually develop drug resistance and disease recurrence, which is
contributed to by cancer stem-like cells. The TβRI inhibitor
LY2157299 can constrain the development of stem-like cells,
indicating the potential combination of chemotherapy and TGF-β
targeted drugs in clinical trials (105).
ER and PR are widely accepted molecular markers in
the occurrence, development and prognosis of breast cancer. TNBC
tumors lack ER and PR expression. An association between TNBC and
AR, another hormone receptor, has been observed (4). Lehmann et al (4) classified cases of TNBC according to gene
expression profiling, in which a luminal AR subtype, featuring
increased gene expression in the AR signaling pathway, was
identified. In another study of 593 TNBC cases, a luminal AR
subtype was also identified based on the expression profiles and
histopathological features of primary TNBC tumors (106).
As a member of the steroid hormone receptor family,
AR is expressed in ~77% of breast cancer tumors (107), and serves an important role in
regulating cell proliferation (108). Testosterone, particularly
dihydrotestosterone, is the main activator of AR (109). Subsequent to ligand binding, AR is
usually bound to chaperone proteins, including heat shock proteins,
before forming a homodimer. The homodimer translocates to the
nucleus and promotes the transcription of target genes (110).
Anti-androgens are the most common drugs for
treating AR-positive cancer, including AR-positive TNBC. In recent
research, bicalutamide (150 mg daily) was prescribed to 424
patients with AR-positive, ER and PR-negative metastatic breast
cancer. The six-month clinical beneficial rate was 19% [95%
confidence interval (CI), 7–39%], and the median progression-free
survival time was 12 weeks (95% CI, 11–22 weeks). These findings
provided evidence in favor of the application of anti-androgen
therapy in ER/PR-negative and AR-positive breast cancer (111). In a phase II clinical trial of
enzalutamide, another AR antagonist, considerable beneficial
therapeutic efficiency was demonstrated in AR-positive TNBC
patients, indicating the potential efficacy of anti-AR therapy in
clinical settings (clinicaltrials.gov: NCT01889238).
A previous study indicated that the concomitant
administration of the anti-androgen bicalutamide with a EGFR,
PDGFRβ or Erk1/2 inhibitor significantly decreased the AR
expression level when compared with the single administration of
the inhibitors (112). Another study
reported that activating PIK3CA mutations are enriched in
AR-positive TNBCs, potentially providing a basis for the
concomitant use of AR antagonists with PI3K/mTOR inhibitors
(4). In addition, enhanced
therapeutic effects of bicalutamide combined with the PI3K
inhibitor GDC-0941 (pictilisib) or the mTOR inhibitor GDC-0980
(apitolisib) have also been also observed in MDA-MB-453 and CAL-148
luminal AR subtype cell xenografts, indicating that the inhibitors
targeting the PI3K/mTOR signaling pathway also notably decrease the
amount of AR protein (113). It was
concluded that AR expression can be regulated
post-transcriptionally by activating the RTK, PI3K or Erk1/2
signaling pathways (113). The
combination of anti-androgen therapy with targeted therapy against
the RTK, PI3K and Erk1/2 signaling pathways may be a promising
alternative treatment for AR-positive TNBC.
The DNA damage response has recently attracted
considerable attention in cancer research. Three major pathways
operate in this process: DNA repair mechanisms that remove DNA
lesions, cell cycle checkpoints that prevent the growth of cells
with DNA damage and apoptotic pathways that eliminate cells with
irreparable DNA lesions (114).
Among the various types of DNA damage, DNA double-strand breaks
(DSB) are of particular interest on account of their function in
genomic instability, which promotes tumorigenesis (115). DSB repair is accomplished through
the HR or non-homologous end-joining pathways (116). BRCA1 and BRCA2 are critical genes
modulating DSB repair through HR (117).
TNBC is commonly associated with BRCA1/2 mutations.
A germline mutation in BRCA1 or BRCA2 is present in ~15% of
patients with TNBC; TNBC cases account for 70% of cases of breast
cancer with BRCA1 mutation, and 16–23% of those with BRCA2 mutation
(118). TNBC also has similar
clinical and pathological features as breast cancer with a BRCA1/2
mutation. For instance, the patients are more likely to be young
and present with a high grade and lymph node invasion ratio
(119,120). In addition, BRCA1/2 mutations have
been confirmed to be indicators of a poor TNBC prognosis (121). In a study of 182 women with TNBC,
>50% were carrying inherited BRCA1 mutations, thus demonstrating
the close association between TNBC and BRCA1 (122).
Polyadenosine 5′-diphosphoribose produced by PARP
enzymes serves an important role in the repair of DNA damage
(123). Where there is a defect in
DNA repair genes, such as BRCA in TNBC, a PARP inhibitor may be a
desirable choice for therapy. In an open-label phase II clinical
study, results indicated that the combination of PARP inhibitor
iniparib and conventional chemotherapy drugs, including gemcitabine
and carboplatin, could produce significant clinical benefit rates
(from 34 to 56%; P=0.01) and a high overall response rate (from 32
to 52%; P=0.02) in patients with TNBC. The median progression-free
and overall survival times were also prolonged, with an extension
from 3.6 to 5.9 months (progression HR, 0.59; P=0.01) and from 7.7
to 12.3 months (mortality HR, 0.57; P=0.01), respectively (124). In addition, in a phase I trial of
the oral PARP inhibitor olaparib for metastatic TNBC, 7 (37%) out
of 19 patients receiving olaparib in combination with weekly
paclitaxel had confirmed partial responses (125). Data regarding the safety of PARP
inhibitor treatment for advanced TNBC and/or BRCA-mutated breast
cancer from clinical trials is summarized in Table I; the side effects of PARP inhibitors
are well tolerated. Interestingly, in the TNBC patients with BRCA
downregulation, the sensitivity to PARP inhibitor treatment was
enhanced when combined with PI3K inhibition, as the blockade of
PI3K impaired HR, inducing sensitization to PARP inhibitors
(44), providing a theoretical basis
for the combination of PI3K with PARP inhibitors for these
patients.
In addition to the aforementioned BRCA1/2 genes,
other breast cancer predisposition genes, including RAD51D, MRE11A,
checkpoint kinase 2, mutL homolog 1, mutS homolog 6 and partner and
localizer of BRCA2, have been confirmed to be associated with the
development and progression of TNBC (126). It was previously reported that the
cytoplasmic expression and lack of nuclear expression of RAD51 were
associated with TN phenotypes, the aberrant expression of BRCA1 and
a poor prognosis for patients (127), indicating that RAD51 may be a
promising biomarker for selecting patients who are suitable for
treatment with DNA-damaging agents.
An increasing number of studies have demonstrated
that the tumor microenvironment, particularly the immune
microenvironment, is associated with the development and
progression of breast cancer (128).
Lehmann et al (4) and Burstein
et al (10) reported that a
subtype of TNBC displayed upregulated immunological responses,
immune cell markers and immune transcription factors, implying the
dysregulation of immune pathways in TNBC, and that the
immunotherapeutic approach may be a valuable treatment strategy for
patients with TNBC (4,10).
High levels of stromal lymphocytic infiltration was
confirmed to be associated with improved TNBC prognosis in two
adjuvant phase III trials. Results of one trial indicated that
compared with the patients who had a lower level of stromal
lymphocytic infiltration, patients with high lymphocytic
infiltration had a 14% reduced risk of recurrence (P=0.02), an 18%
reduced risk of distant recurrence (P=0.04) and a 19% reduced risk
of mortality (P=0.01) for every 10% increase in stromal lymphocytic
infiltration (129). Another phase
III randomized adjuvant breast cancer trial reported similar
results, indicating that an increase in lymphocytic infiltration
was associated with an improved prognosis in node-positive and
ER-negative/HER2-negative breast cancer, regardless of chemotherapy
(130). In addition, the presence of
tumor-infiltrating lymphocytes predicted a better clinical response
to neoadjuvant chemotherapy (131).
Therefore, the existence of tumor-infiltrating lymphocytes can be
regarded as a prognostic parameter for the prediction of the
response to clinical treatment of patients with TNBC.
Tumor cells can evade the recognition and
destruction by the host immune system through the immune checkpoint
system; blocking the immune checkpoint system is a promising
treatment for achieving effective antitumor immunity. PD-1 is a
well-established immune checkpoint protein and cell-surface
receptor that disrupts the T-cell response by triggering inhibitory
pathways (132). Notably, PD-1 is
expressed in 20% of TNBC tumors (133). PD-L1 is the ligand of PD-1, and is
expressed in 58.6% of TNBC tumors (66,134).
PD-L2 is expressed on the surface of dendritic cells, macrophages,
mast cells and B cells (135).
Antibodies to inhibit PD-1 signaling are currently being assessed
for clinical use. For example, pembrolizumab, a monoclonal antibody
against PD-1, has demonstrated an overall response rate of 18.5% in
a phase Ib study (n=32) of patients with PD-L1-positive TNBCs
(66). Furthermore, a preliminary
phase I trial study suggested that the monoclonal anti-PD-L1
antibody MPDL3280A prolonged progression-free survival time and
produced durable therapeutic effects in certain patients with TNBC
(136).
In addition, there is an association between the
immune response and the Ras/MAPK pathway in TNBC. A study has
indicated that the Ras/MAPK pathway negatively regulated antitumor
immunity by affecting antigen presentation, including that of
MHC-I, MHC-II and PD-1, and it was verified that a combination of
MEK inhibition and PD-1/PD-L1 antibodies increased the effect of
treatment in murine syngeneic tumor models (26). An additional study identified that an
oncolytic viral therapy (NV1066) eliminated >70% of the cells
from all the TNBC cell lines tested by day 7, and effectively
reduced the tumor size compared with control treatment groups (57
vs. 438 mm; P=0.002) through downregulating the Ras/MAPK pathway
(137).
TNBC is attracting increasing attention on account
of its unique clinical pathology and molecular features.
Chemotherapy remains the exclusive effective systemic treatment for
patients with TNBC. These patients exhibit varied therapeutic
responses and prognoses. Thus, individualized treatment and
prognostic analysis in patients with TNBC can be difficult,
particularly when this depends on conventional clinical and
pathological features, including the histological grade, primary
tumor size, lymph node metastasis status and ER/PR/HER2 expression.
With the emergence of targeted therapy, screening more reliable
molecular markers is imperative, and comprehensively understanding
the signaling pathways that regulate biological behaviors may
facilitate the establishment of a precise molecular classification
for TNBC and effective therapeutic regimens.
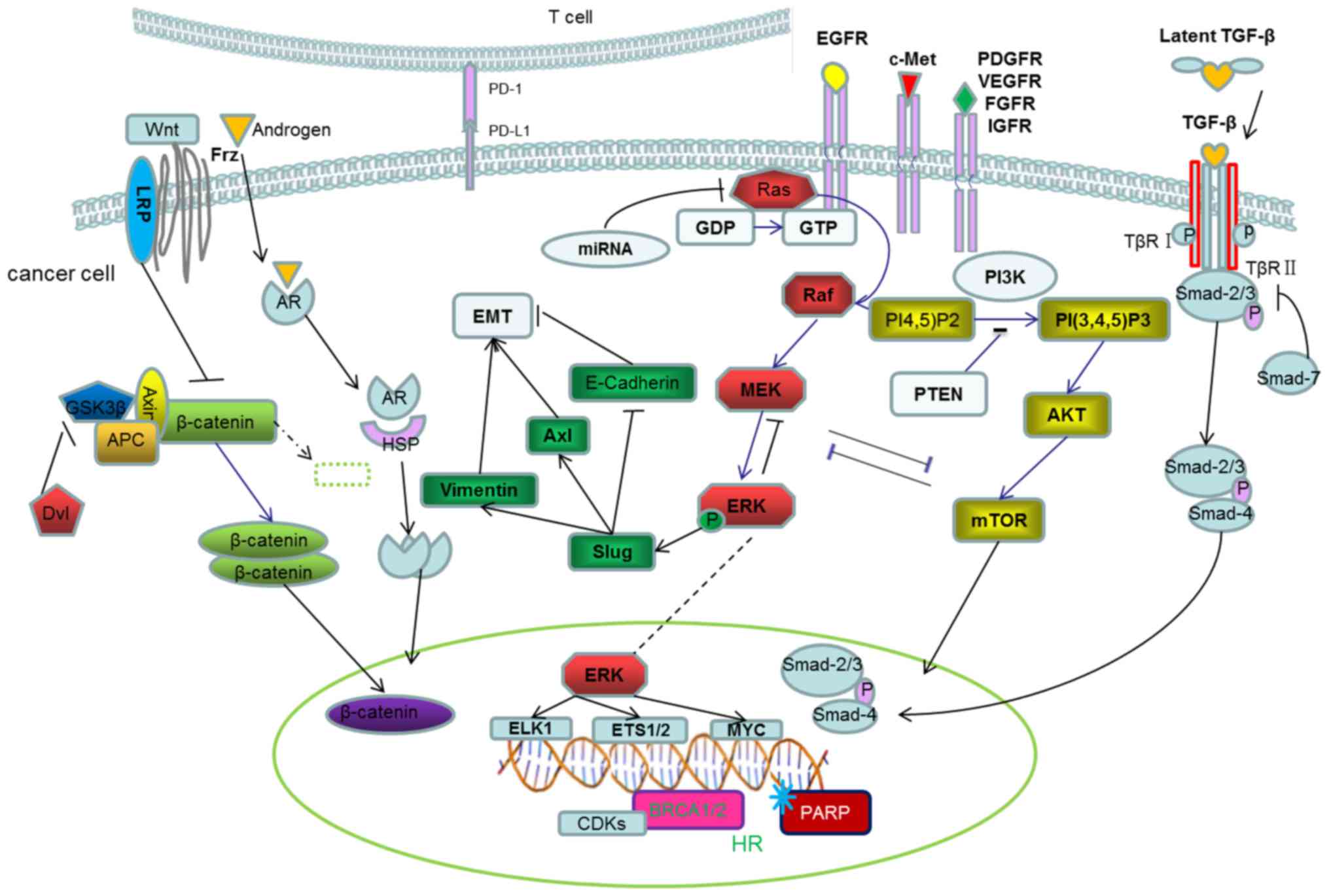
In the present review, TNBC-associated tumorigenic
signaling pathways were summarized in five categories, specifically
RTKs and downstream signaling pathways, epithelial-to-mesenchymal
transition and associated pathways, immunoregulatory tumor
microenvironment, DNA damage repair pathways, and AR and
coordinating pathways. These pathways are illustrated in Fig. 1 to demonstrate the interactions across
the entire network, and the relevant drugs against specific
pathways are summarized in Table I.
Enhancing the understanding of the molecular heterogeneity of TNBCs
can lay the foundation for the individualized treatment of patients
with TNBC.
It is worth noting that a study concerning the
efficacy of a number of biological agents, including bevacizumab,
sunitinib, sorafenib, lapatinib, iniparib and cetuximab, in
metastatic TNBCs indicated that a significant PFS improvement was
obtained following treatment with bevacizumab or cetuximab,
indicating the importance of targeted therapy. Regrettably, the
impact of the other agents in the study on patient survival was not
significant (138), possibly due to
concurrent abnormalities occurring in a number of the patients. If
that is correct, the concurrent inhibition of tumorigenic pathways
may inhibit the cancer process (45).
Therefore, the understanding of the signaling crosstalk and
feedback among the TNBC-associated tumorigenic signaling pathways
needs to be improved to allow effective treatments with tolerable
side effects to be developed.
Not applicable.
This work was supported by the Key Projects in the
National Science & Technology Pillar Program (grant nos.
2015BAl12B12 and 2015BAl12B15), National Natural Science Foundation
of China (grant nos. 81472473 and 81272360) and Tianjin Municipal
Commission of Science & Technology Key Research Program (grant
no. 13ZCZCSY20300).
Not applicable.
NW led the review's designing, consulting and
writing. JL and JY led the conception and review of the paper.
JZhang, JZhao, KM, JuZ, and ZJ made contributions to the collection
of relevant literature. All authors have read and approved the
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenton JD, Carey LA, Ahmed AA and Caldas
C: Molecular classification and molecular forecasting of breast
cancer: Ready for clinical application? J Clin Oncol. 23:7350–7360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shah SP, Roth A, Goya R, Oloumi A, Ha G,
Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al: The clonal
and mutational evolution spectrum of primary triple-negative breast
cancers. Nature. 486:395–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gluz O, Liedtke C, Gottschalk N, Pusztai
L, Nitz U and Harbeck N: Triple-negative breast cancer-current
status and future directions. Ann Oncol. 20:1913–1927. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berry DA, Cirrincione C, Henderson IC,
Citron ML, Budman DR, Goldstein LJ, Martino S, Perez EA, Muss HB,
Norton L, et al: Estrogen-receptor status and outcomes of modern
chemotherapy for patients with node-positive breast cancer. JAMA.
295:1658–1667. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nabholtz JM, Abrial C, Mouret-Reynier MA,
Dauplat MM, Weber B, Gligorov J, Forest AM, Tredan O, Vanlemmens L,
Petit T, et al: Multicentric neoadjuvant phase II study of
panitumumab combined with an anthracycline/taxane-based
chemotherapy in operable triple-negative breast cancer:
Identification of biologically defined signatures predicting
treatment impact. Ann Oncol. 25:1570–1577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Torrisi R, Balduzzi A, Ghisini R, Rocca A,
Bottiglieri L, Giovanardi F, Veronesi P, Luini A, Orlando L, Viale
G, et al: Tailored preoperative treatment of locally advanced
triple negative (hormone receptor negative and HER2 negative)
breast cancer with epirubicin, cisplatin, and infusional
fluorouracil followed by weekly paclitaxel. Cancer Chemother
Pharmacol. 62:667–672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Burstein MD, Tsimelzon A, Poage GM,
Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK,
Hilsenbeck SG, Chang JC, et al: Comprehensive genomic analysis
identifies novel subtypes and targets of triple-negative breast
cancer. Clin Cancer Res. 21:1688–1698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tsimberidou AM, Iskander NG, Hong DS,
Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra
R, et al: Personalized medicine in a phase I clinical trials
program: The MD Anderson cancer center initiative. Clin Cancer Res.
18:6373–6383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jovanović B, Beeler JS, Pickup MW, Chytil
A, Gorska AE, Ashby WJ, Lehmann BD, Zijlstra A, Pietenpol JA and
Moses HL: Transforming growth factor beta receptor type III is a
tumor promoter in mesenchymal-stem like triple negative breast
cancer. Breast Cancer Res. 16:R692014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Litzenburger BC, Creighton CJ, Tsimelzon
A, Chan BT, Hilsenbeck SG, Wang T, Carboni JM, Gottardis MM, Huang
F, Chang JC, et al: High IGF-IR activity in triple-negative breast
cancer cell lines and tumorgrafts correlates with sensitivity to
anti-IGF-IR therapy. Clin Cancer Res. 17:2314–2327. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharpe R, Pearson A, Herrera-Abreu MT,
Johnson D, Mackay A, Welti JC, Natrajan R, Reynolds AR, Reis-Filho
JS, Ashworth A and Turner NC: FGFR signaling promotes the growth of
triple-negative and basal-like breast cancer cell lines both in
vitro and in vivo. Clin Cancer Res. 17:5275–5286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spanheimer PM, Lorenzen AW, De Andrade JP,
Kulak MV, Carr JC, Woodfield GW, Sugg SL and Weigel RJ: Receptor
tyrosine kinase expression predicts response to sunitinib in breast
cancer. Ann Surg Oncol. 22:4287–4294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan S, Jiao X, Zou H and Li K: Prognostic
significance of c-Met in breast cancer: A meta-analysis of 6010
cases. Diagn Pathol. 10:622015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Burness ML, Grushko TA and Olopade OI:
Epidermal growth factor receptor in triple-negative and basal-like
breast cancer: Promising clinical target or only a marker? Cancer
J. 16:23–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Siziopikou KP, Ariga R, Proussaloglou KE,
Gattuso P and Cobleigh M: The challenging estrogen
receptor-negative/progesterone receptor-negative/HER-2-negative
patient: A promising candidate for epidermal growth factor
receptor-targeted therapy? Breast J. 12:360–362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bayraktar S and Glück S: Molecularly
targeted therapies for metastatic triple-negative breast cancer.
Breast Cancer Res Treat. 138:21–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsiao YC, Yeh MH, Chen YJ, Liu JF, Tang CH
and Huang WC: Lapatinib increases motility of triple-negative
breast cancer cells by decreasing miRNA-7 and inducing
Raf-1/MAPK-dependent interleukin-6. Oncotarget. 6:37965–37978.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Agazie YM and Hayman MJ: Molecular
mechanism for a role of SHP2 in epidermal growth factor receptor
signaling. Mol Cell Biol. 23:7875–7886. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matalkah F, Martin E, Zhao H and Agazie
YM: SHP2 acts both upstream and downstream of multiple receptor
tyrosine kinases to promote basal-like and triple-negative breast
cancer. Breast Cancer Res. 18:22016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim HR, Jung KH, Im SA, Im YH, Kang SY,
Park KH, Lee S, Kim SB, Lee KH, Ahn JS, et al: Multicentre phase II
trial of bevacizumab combined with docetaxel-carboplatin for the
neoadjuvant treatment of triple-negative breast cancer (KCSG
BR-0905). Ann Oncol. 24:1485–1490. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gerber B, Loibl S, Eidtmann H, Rezai M,
Fasching PA, Tesch H, Eggemann H, Schrader I, Kittel K, Hanusch C,
et al: Neoadjuvant bevacizumab and anthracycline-taxane-based
chemotherapy in 678 triple-negative primary breast cancers; results
from the geparquinto study (GBG 44). Ann Oncol. 24:2978–2984. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bartholomeusz C, Xie X, Pitner MK, Kondo
K, Dadbin A, Lee J, Saso H, Smith PD, Dalby KN and Ueno NT: MEK
inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung
metastasis in a triple-negative breast cancer xenograft model. Mol
Cancer Ther. 14:2773–2781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Giltnane JM and Balko JM: Rationale for
targeting the Ras/MAPK pathway in triple-negative breast cancer.
Discov Med. 17:275–283. 2014.PubMed/NCBI
|
|
27
|
Bartholomeusz C, Gonzalez-Angulo AM, Liu
P, Hayashi N, Lluch A, Ferrer-Lozano J and Hortobágyi GN: High ERK
protein expression levels correlate with shorter survival in
triple-negative breast cancer patients. Oncologist. 17:766–774.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Loi S, Dushyanthen S, Beavis PA, Salgado
R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV,
et al: RAS/MAPK activation is associated with reduced
tumor-infiltrating lymphocytes in triple-negative breast cancer:
Therapeutic cooperation between MEK and PD-1/PD-L1 immune
checkpoint inhibitors. Clin Cancer Res. 22:1499–1509. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rameh LE and Cantley LC: The role of
phosphoinositide 3-kinase lipid products in cell function. J Biol
Chem. 274:8347–8350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baselga J: Targeting the
phosphoinositide-3 (PI3) kinase pathway in breast cancer.
Oncologist. 16 Suppl 1:S12–S19. 2011. View Article : Google Scholar
|
|
31
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Meric-Bernstam F and Gonzalez-Angulo AM:
Targeting the mTOR signaling network for cancer therapy. J Clin
Oncol. 27:2278–2287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cossu-Rocca P, Orrù S, Muroni MR, Sanges
F, Sotgiu G, Ena S, Pira G, Murgia L, Manca A, Uras MG, et al:
Analysis of PIK3CA mutations and activation pathways in triple
negative breast cancer. PLoS One. 10:e01417632015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cancer Genome Atlas Network, .
Comprehensive molecular portraits of human breast tumours. Nature.
490:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Korkaya H, Paulson A, Charafe-Jauffret E,
Ginestier C, Brown M, Dutcher J, Clouthier SG and Wicha MS:
Regulation of mammary stem/progenitor cells by
PTEN/Akt/beta-catenin signaling. PLoS Biol. 7:e10001212009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shrivastava S, Kulkarni P, Thummuri D,
Jeengar MK, Naidu VG, Alvala M, Redddy GB and Ramakrishna S:
Piperlongumine, an alkaloid causes inhibition of PI3K/Akt/mTOR
signaling axis to induce caspase-dependent apoptosis in human
triple-negative breast cancer cells. Apoptosis. 19:1148–1164. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chin YR, Yoshida T, Marusyk A, Beck AH,
Polyak K and Toker A: Targeting Akt3 signaling in triple-negative
breast cancer. Cancer Res. 74:964–973. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Montero JC, Esparis-Ogando A, Re-Louhau
MF, Seoane S, Abad M, Calero R, Ocaña A and Pandiella A: Active
kinase profiling, genetic and pharmacological data define mTOR as
an important common target in triple-negative breast cancer.
Oncogene. 33:148–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baselga J, Campone M, Piccart M, Burris HA
III, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun
F, et al: Everolimus in postmenopausal hormone-receptor-positive
advanced breast cancer. N Engl J Med. 366:520–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Beuvink I, Boulay A, Fumagalli S,
Zilbermann F, Ruetz S, O'Reilly T, Natt F, Hall J, Lane HA and
Thomas G: The mTOR inhibitor RAD001 sensitizes tumor cells to
DNA-damaged induced apoptosis through inhibition of p21
translation. Cell. 120:747–759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singh J, Novik Y, Stein S, Volm M, Meyers
M, Smith J, Omene C, Speyer J, Schneider R, Jhaveri K, et al: Phase
2 trial of everolimus and carboplatin combination in patients with
triple negative metastatic breast cancer. Breast Cancer Res.
16:R322014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ganesan P, Moulder S, Lee JJ, Janku F,
Valero V, Zinner RG, Naing A, Fu S, Tsimberidou AM, Hong D, et al:
Triple-negative breast cancer patients treated at MD Anderson
Cancer Center in phase I trials: Improved outcomes with combination
chemotherapy and targeted agents. Mol Cancer Ther. 13:3175–3184.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ibrahim YH, Garcia-Garcia C, Serra V, He
L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J,
et al: PI3K inhibition impairs BRCA1/2 expression and sensitizes
BRCA-proficient triple-negative breast cancer to PARP inhibition.
Cancer Discov. 2:1036–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schuler M, Awada A, Harter P, Canon JL,
Possinger K, Schmidt M, De Grève J, Neven P, Dirix L, Jonat W, et
al: A phase II trial to assess efficacy and safety of afatinib in
extensively pretreated patients with HER2-negative metastatic
breast cancer. Breast Cancer Res Treat. 134:1149–1159. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bernsdorf M, Ingvar C, Jörgensen L, Tuxen
MK, Jakobsen EH, Saetersdal A, Kimper-Karl ML, Kroman N, Balslev E
and Ejlertsen B: Effect of adding gefitinib to neoadjuvant
chemotherapy in estrogen receptor negative early breast cancer in a
randomized phase II trial. Breast Cancer Res Treat. 126:463–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mailliez A, Baldini C, Van JT, Servent V,
Mallet Y and Bonneterre J: Nasal septum perforation: A side effect
of bevacizumab chemotherapy in breast cancer patients. Br J Cancer.
103:772–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Scott AJ, Messersmith WA and Jimeno A:
Apatinib: A promising oral antiangiogenic agent in the treatment of
multiple solid tumors. Drugs Today (Barc). 51:223–229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hong DS, Garrido-Laguna I, Ekmekcioglu S,
Falchook GS, Naing A, Wheler JJ, Fu S, Moulder SL, Piha-Paul S,
Tsimberidou AM, et al: Dual inhibition of the vascular endothelial
growth factor pathway: A phase 1 trial evaluating bevacizumab and
AZD2171 (cediranib) in patients with advanced solid tumors. Cancer.
120:2164–2173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tolaney SM, Tan S, Guo H, Barry W, Van
Allen E, Wagle N, Brock J, Larrabee K, Paweletz C, Ivanova E, et
al: Phase II study of tivantinib (ARQ 197) in patients with
metastatic triple-negative breast cancer. Invest New Drugs.
33:1108–1114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tolaney SM, Ziehr DR, Guo H, Ng MR, Barry
WT, Higgins MJ, Isakoff SJ, Brock JE, Ivanova EV, Paweletz CP, et
al: Phase II and biomarker study of cabozantinib in metastatic
triple-negative breast cancer patients. Oncologist. 22:25–32. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Soria JC, DeBraud F, Bahleda R, Adamo B,
Andre F, Dienstmann R, Delmonte A, Cereda R, Isaacson J, Litten J,
et al: Phase I/IIa study evaluating the safety, efficacy,
pharmacokinetics, and pharmacodynamics of lucitanib in advanced
solid tumors. Ann Oncol. 25:2244–2251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu YL, Zhang LI, Trandafir L, Dong T,
Duval V, Hazell K and Xu B: Phase I study of the Pan-PI3K inhibitor
buparlisib in adult chinese patients with advanced solid tumors.
Anticancer Res. 36:6185–6194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Juric D, Krop I, Ramanathan RK, Wilson TR,
Ware JA, Sanabria Bohorquez SM, Savage HM, Sampath D, Salphati L,
Lin RS, et al: Phase I dose-escalation study of taselisib, an oral
PI3K inhibitor, in patients with advanced solid tumors. Cancer
Discov. 7:704–715. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tamura K, Hashimoto J, Tanabe Y, Kodaira
M, Yonemori K, Seto T, Hirai F, Arita S, Toyokawa G, Chen L, et al:
Safety and tolerability of AZD5363 in Japanese patients with
advanced solid tumors. Cancer Chemother Pharmacol. 77:787–795.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Doi T, Tamura K, Tanabe Y, Yonemori K,
Yoshino T, Fuse N, Kodaira M, Bando H, Noguchi K, Shimamoto T and
Ohtsu A: Phase 1 pharmacokinetic study of the oral pan-AKT
inhibitor MK-2206 in Japanese patients with advanced solid tumors.
Cancer Chemother Pharmacol. 76:409–416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Saura C, Roda D, Roselló S, Oliveira M,
Macarulla T, Pérez-Fidalgo JA, Morales-Barrera R, Sanchis-García
JM, Musib L, Budha N, et al: A first-in-human phase I study of the
ATP-competitive AKT inhibitor ipatasertib demonstrates robust and
safe targeting of AKT in patients with solid tumors. Cancer Discov.
7:102–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pascual T, Apellániz-Ruiz M, Pernaut C,
Cueto-Felgueroso C, Villalba P, Álvarez C, Manso L, Inglada-Pérez
L, Robledo M, Rodríguez-Antona C and Ciruelos E: Polymorphisms
associated with everolimus pharmacokinetics, toxicity and survival
in metastatic breast cancer. PLoS One. 12:e01801922017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chiu JW, Hotte SJ, Kollmannsberger CK,
Renouf DJ, Cescon DW, Hedley D, Chow S, Moscow J, Chen Z, Perry M,
et al: A phase I trial of ANG1/2-Tie2 inhibitor trebaninib (AMG386)
and temsirolimus in advanced solid tumors (PJC008/NCI 9041). Invest
New Drugs. 34:104–111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Schwartzberg LS, Yardley DA, Elias AD,
Patel M, LoRusso P, Burris HA, Gucalp A, Peterson AC, Blaney ME,
Steinberg JL, et al: A Phase I/Ib study of enzalutamide alone and
in combination with endocrine therapies in women with advanced
breast cancer. Clin Cancer Res. 23:4046–4054. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Drew Y, Ledermann J, Hall G, Rea D,
Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D,
et al: Phase 2 multicentre trial investigating intermittent and
continuous dosing schedules of the poly(ADP-ribose) polymerase
inhibitor rucaparib in germline BRCA mutation carriers with
advanced ovarian and breast cancer. Br J Cancer. 114:e212016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
O'Shaughnessy J, Schwartzberg L, Danso MA,
Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M,
Richards P, et al: Phase III study of iniparib plus gemcitabine and
carboplatin versus gemcitabine and carboplatin in patients with
metastatic triple-negative breast cancer. J Clin Oncol.
32:3840–3847. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nishikawa T, Matsumoto K, Tamura K,
Yoshida H, Imai Y, Miyasaka A, Onoe T, Yamaguchi S, Shimizu C,
Yonemori K, et al: Phase 1 dose-escalation study of single-agent
veliparib in Japanese patients with advanced solid tumors. Cancer
Sci. 108:1834–1842. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
de Bono J, Ramanathan RK, Mina L, Chugh R,
Glaspy J, Rafii S, Kaye S, Sachdev J, Heymach J, Smith DC, et al:
Phase I, dose-escalation, two-part trial of the PARP inhibitor
talazoparib in patients with advanced germline BRCA1/2 mutations
and selected sporadic cancers. Cancer Discov. 7:620–629. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Robson M, Im SA, Senkus E, Xu B, Domchek
SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al:
Olaparib for metastatic breast cancer in patients with a germline
BRCA mutation. N Engl J Med. 377:523–533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nanda R, Chow LQ, Dees EC, Berger R, Gupta
S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, et al:
Pembrolizumab in patients with advanced triple-negative breast
cancer: Phase Ib KEYNOTE-012 study. J Clin Oncol. 34:2460–2467.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lee JM, Cimino-Mathews A, Peer CJ, Zimmer
A, Lipkowitz S, Annunziata CM, Cao L, Harrell MI, Swisher EM,
Houston N, et al: Safety and clinical activity of the programmed
death-ligand 1 inhibitor durvalumab in combination with poly
(ADP-Ribose) polymerase inhibitor olaparib or vascular endothelial
growth factor receptor 1–3 inhibitor cediranib in women's cancers:
A dose-escalation, phase I study. J Clin Oncol. 35:2193–2202. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hoeflich KP, O'Brien C, Boyd Z, Cavet G,
Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, et
al: In vivo antitumor activity of MEK and phosphatidylinositol
3-kinase inhibitors in basal-like breast cancer models. Clin Cancer
Res. 15:4649–4664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Britten CD: PI3K and MEK inhibitor
combinations: Examining the evidence in selected tumor types.
Cancer Chemother Pharmacol. 71:1395–1409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nieto MA: Epithelial plasticity: A common
theme in embryonic and cancer cells. Science. 342:12348502013.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Al Moustafa AE, Achkhar A and Yasmeen A:
EGF-receptor signaling and epithelial-mesenchymal transition in
human carcinomas. Front Biosci (Schol Ed). 4:671–684. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Guarino M: Src signaling in cancer
invasion. J Cell Physiol. 223:14–26. 2010.PubMed/NCBI
|
|
74
|
Hung CM, Kuo DH, Chou CH, Su YC, Ho CT and
Way TD: Osthole suppresses hepatocyte growth factor (HGF)-induced
epithelial-mesenchymal transition via repression of the
c-Met/Akt/mTOR pathway in human breast cancer cells. J Agric Food
Chem. 59:9683–9690. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sivakumar R, Koga H, Selvendiran K,
Maeyama M, Ueno T and Sata M: Autocrine loop for IGF-I receptor
signaling in SLUG-mediated epithelial-mesenchymal transition. Int J
Oncol. 34:329–338. 2009.PubMed/NCBI
|
|
77
|
Vincent-Salomon A and Thiery JP: Host
microenvironment in breast cancer development:
Epithelial-mesenchymal transition in breast cancer development.
Breast Cancer Res. 5:101–106. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
78
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Foroni C, Broggini M, Generali D and Damia
G: Epithelial-mesenchymal transition and breast cancer: Role,
molecular mechanisms and clinical impact. Cancer Treat Rev.
38:689–697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Prat A, Parker JS, Karginova O, Fan C,
Livasy C, Herschkowitz JI, He X and Perou CM: Phenotypic and
molecular characterization of the claudin-low intrinsic subtype of
breast cancer. Breast Cancer Res. 12:R682010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Singh R and Mo YY: Role of microRNAs in
breast cancer. Cancer Biol Ther. 14:201–212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Rhodes LV, Martin EC, Segar HC, Miller DF,
Buechlein A, Rusch DB, Nephew KP, Burow ME and Collins-Burow BM:
Dual regulation by microRNA-200b-3p and microRNA-200b-5p in the
inhibition of epithelial-to-mesenchymal transition in
triple-negative breast cancer. Oncotarget. 6:16638–16652. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ito K, Park SH, Nayak A, Byerly JH and
Irie HY: PTK6 inhibition suppresses metastases of triple-negative
breast cancer via SNAIL-Dependent E-cadherin regulation. Cancer
Res. 76:4406–4417. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rhodes LV, Tate CR, Segar HC, Burks HE,
Phamduy TB, Hoang V, Elliott S, Gilliam D, Pounder FN, Anbalagan M,
et al: Suppression of triple-negative breast cancer metastasis by
pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT
master regulators. Breast Cancer Res Treat. 145:593–604. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kahn M: Can we safely target the WNT
pathway? Nat Rev Drug Discov. 13:513–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Conacci-Sorrell M, Simcha I, Ben-Yedidia
T, Blechman J, Savagner P and Ben-Ze'ev A: Autoregulation of
E-cadherin expression by cadherin-cadherin interactions: The roles
of beta-catenin signaling, Slug, and MAPK. J Cell Biol.
163:847–857. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Howe LR, Watanabe O, Leonard J and Brown
AM: Twist is up-regulated in response to Wnt1 and inhibits mouse
mammary cell differentiation. Cancer Res. 63:1906–1913.
2003.PubMed/NCBI
|
|
92
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Dey N, Barwick BG, Moreno CS,
Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C,
Kerstann KF, Sledge GW Jr, et al: Wnt signaling in triple negative
breast cancer is associated with metastasis. BMC Cancer.
13:5372013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li Y, Li PK, Roberts MJ, Arend RC, Samant
RS and Buchsbaum DJ: Multi-targeted therapy of cancer by
niclosamide: A new application for an old drug. Cancer Lett.
349:8–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Londoño-Joshi AI, Arend RC, Aristizabal L,
Lu W, Samant RS, Metge BJ, Hidalgo B, Grizzle WE, Conner M,
Forero-Torres A, et al: Effect of niclosamide on basal-like breast
cancers. Mol Cancer Ther. 13:800–811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Koval A, Ahmed K and Katanaev VL:
Inhibition of Wnt signalling and breast tumour growth by the
multi-purpose drug suramin through suppression of heterotrimeric G
proteins and Wnt endocytosis. Biochem J. 473:371–381. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nam JS, Suchar AM, Kang MJ, Stuelten CH,
Tang B, Michalowska AM, Fisher LW, Fedarko NS, Jain A, Pinkas J, et
al: Bone sialoprotein mediates the tumor cell-targeted
prometastatic activity of transforming growth factor beta in a
mouse model of breast cancer. Cancer Res. 66:6327–6335. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zu X, Zhang Q, Cao R, Liu J, Zhong J, Wen
G and Cao D: Transforming growth factor-β signaling in tumor
initiation, progression and therapy in breast cancer: An update.
Cell Tissue Res. 347:73–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Katz LH, Li Y, Chen JS, Muñoz NM, Majumdar
A, Chen J and Mishra L: Targeting TGF-β signaling in cancer. Expert
Opin Ther Targets. 17:743–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Valcourt U, Kowanetz M, Niimi H, Heldin CH
and Moustakas A: TGF-beta and the Smad signaling pathway support
transcriptomic reprogramming during epithelial-mesenchymal cell
transition. Mol Biol Cell. 16:1987–2002. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kim S, Lee J, Jeon M, Lee JE and Nam SJ:
Zerumbone suppresses the motility and tumorigenecity of triple
negative breast cancer cells via the inhibition of TGF-β1 signaling
pathway. Oncotarget. 7:1544–1558. 2016.PubMed/NCBI
|
|
104
|
Wahdan-Alaswad R, Harrell JC, Fan Z,
Edgerton SM, Liu B and Thor AD: Metformin attenuates transforming
growth factor beta (TGF-β) mediated oncogenesis in mesenchymal
stem-like/claudin-low triple negative breast cancer. Cell Cycle.
15:1046–1059. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bhola NE, Balko JM, Dugger TC, Kuba MG,
Sánchez V, Sanders M, Stanford J, Cook RS and Arteaga CL: TGF-β
inhibition enhances chemotherapy action against triple-negative
breast cancer. J Clin Invest. 123:1348–1358. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Purrington KS, Visscher DW, Wang C,
Yannoukakos D, Hamann U, Nevanlinna H, Cox A, Giles GG,
Eckel-Passow JE, Lakis S, et al: Genes associated with
histopathologic features of triple negative breast tumors predict
molecular subtypes. Breast Cancer Res Treat. 157:117–131. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Collins LC, Cole KS, Marotti JD, Hu R,
Schnitt SJ and Tamimi RM: Androgen receptor expression in breast
cancer in relation to molecular phenotype: Results from the nurses'
health study. Mod Pathol. 24:924–931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liao DJ and Dickson RB: Roles of androgens
in the development, growth, and carcinogenesis of the mammary
gland. J Steroid Biochem Mol Biol. 80:175–189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hickey TE, Robinson JL, Carroll JS and
Tilley WD: Minireview: the androgen receptor in breast tissues:
Growth inhibitor, tumor suppressor, oncogene? Mol Endocrinol.
26:1252–1267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gucalp A and Traina TA: Triple-negative
breast cancer: Role of the androgen receptor. Cancer J. 16:62–65.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gucalp A, Tolaney S, Isakoff SJ, Ingle JN,
Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, et al:
Phase II trial of bicalutamide in patients with androgen
receptor-positive, estrogen receptor-negative metastatic breast
cancer. Clin Cancer Res. 19:5505–5512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lehmann BD, Bauer JA, Schafer JM,
Pendleton CS, Tang L, Johnson KC, Chen X, Balko JM, Gómez H,
Arteaga CL, et al: PIK3CA mutations in androgen receptor-positive
triple negative breast cancer confer sensitivity to the combination
of PI3K and androgen receptor inhibitors. Breast Cancer Res.
16:4062014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Cuenca-López Md, Montero JC, Morales JC,
Prat A, Pandiella A and Ocana A: Phospho-kinase profile of triple
negative breast cancer and androgen receptor signaling. BMC Cancer.
14:3022014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Maugeri-Saccà M, Bartucci M and De Maria
R: DNA damage repair pathways in cancer stem cells. Mol Cancer
Ther. 11:1627–1636. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Pierce AJ, Stark JM, Araujo FD, Moynahan
ME, Berwick M and Jasin M: Double-strand breaks and tumorigenesis.
Trends Cell Biol. 11:S52–S59. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Powell SN and Kachnic LA: Roles of BRCA1
and BRCA2 in homologous recombination, DNA replication fidelity and
the cellular response to ionizing radiation. Oncogene.
22:5784–5791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bouwman P and Jonkers J: The effects of
deregulated DNA damage signalling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Stevens KN, Vachon CM and Couch FJ:
Genetic susceptibility to triple-negative breast cancer. Cancer
Res. 73:2025–2030. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Atchley DP, Albarracin CT, Lopez A, Valero
V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN and Arun BK: Clinical
and pathologic characteristics of patients with BRCA-positive and
BRCA-negative breast cance. J Clin Oncol. 26:4282–4288. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Goodwin PJ, Phillips KA, West DW, Ennis M,
Hopper JL, John EM, O'Malley FP, Milne RL, Andrulis IL, Friedlander
ML, et al: Breast cancer prognosis in BRCA1 and BRCA2 mutation
carriers: An international prospective breast cancer family
registry population-based cohort study. J Clin Oncol. 30:19–26.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Stoppa-Lyonnet D, Ansquer Y, Dreyfus H,
Gautier C, Gauthier-Villars M, Bourstyn E, Clough KB, Magdelénat H,
Pouillart P, Vincent-Salomon A, et al: Familial invasive breast
cancers: Worse outcome related to BRCA1 mutations. J Clin Oncol.
18:4053–4059. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Rummel S, Varner E, Shriver CD and
Ellsworth RE: Evaluation of BRCA1 mutations in an unselected
patient population with triple-negative breast cancer. Breast
Cancer Res Treat. 137:119–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
O'Shaughnessy J, Osborne C, Pippen JE,
Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM and Bradley C:
Iniparib plus chemotherapy in metastatic triple-negative breast
cancer. N Engl J Med. 364:205–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Dent RA, Lindeman GJ, Clemons M, Wildiers
H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL and
Carmichael J: Phase I trial of the oral PARP inhibitor olaparib in
combination with paclitaxel for first- or second-line treatment of
patients with metastatic triple-negative breast cancer. Breast
Cancer Res. 15:R882013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ollier M, Radosevic-Robin N, Kwiatkowski
F, Ponelle F, Viala S, Privat M, Uhrhammer N, Bernard-Gallon D,
Penault-Llorca F, Bignon YJ and Bidet Y: DNA repair genes
implicated in triple negative familial non-BRCA1/2 breast cancer
predisposition. Am J Cancer Res. 5:2113–2126. 2015.PubMed/NCBI
|
|
127
|
Alshareeda AT, Negm OH, Aleskandarany MA,
Green AR, Nolan C, TigHhe PJ, Madhusudan S, Ellis IO and Rakha EA:
Clinical and biological significance of RAD51 expression in breast
cancer: A key DNA damage response protein. Breast Cancer Res Treat.
159:41–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Loi S, Michiels S, Salgado R, Sirtaine N,
Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V,
Desmedt C, et al: Tumor infiltrating lymphocytes are prognostic in
triple negative breast cancer and predictive for trastuzumab
benefit in early breast cancer: Results from the FinHER trial. Ann
Oncol. 25:1544–1550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Adams S, Gray RJ, Demaria S, Goldstein L,
Perez EA, Shulman LN, Martino S, Wang M, Jones VE and Saphner TJ:
Prognostic value of tumor-infiltrating lymphocytes in
triple-negative breast cancers from two phase III randomized
adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin
Oncol. 32:2959–2966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Loi S, Sirtaine N, Piette F, Salgado R,
Viale G, Van Eenoo F, Rouas G, Francis P, Crown JP, Hitre E, et al:
Prognostic and predictive value of tumor-infiltrating lymphocytes
in a phase III randomized adjuvant breast cancer trial in
node-positive breast cancer comparing the addition of docetaxel to
doxorubicin with doxorubicin-based chemotherapy: BIG 02–98. J Clin
Oncol. 31:860–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Adams S, Goldstein LJ, Sparano JA, Demaria
S and Badve SS: Tumor infiltrating lymphocytes (TILs) improve
prognosis in patients with triple negative breast cancer (TNBC).
Oncoimmunology. 4:e9859302015. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Criscitiello C and Curigliano G:
Immunotherapy of breast cancer. Prog Tumor Res. 42:30–43. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Mittendorf EA, Philips AV, Meric-Bernstam
F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM,
Akcakanat A, et al: PD-L1 expression in triple-negative breast
cancer. Cancer Immunol Res. 2:361–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Dong H, Strome SE, Salomao DR, Tamura H,
Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al:
Tumor-associated B7-H1 promotes T-cell apoptosis: A potential
mechanism of immune evasion. Nat Med. 8:793–800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Topalian SL, Hodi FS, Brahmer JR,
Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD,
Sosman JA, Atkins MB, et al: Safety, activity, and immune
correlates of anti-PD-1 antibody in cancer. N Engl J Med.
366:2443–2454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Immunotherapy slows TNBC progression.
Cancer Discov. 5:5702015. View Article : Google Scholar
|
|
137
|
Gholami S, Chen CH, Gao S, Lou E, Fujisawa
S, Carson J, Nnoli JE, Chou TC, Bromberg J and Fong Y: Role of MAPK
in oncolytic herpes viral therapy in triple-negative breast cancer.
Cancer Gene Ther. 21:283–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Bramati A, Girelli S, Torri V, Farina G,
Galfrascoli E, Piva S, Moretti A, Dazzani MC, Sburlati P and La
Verde NM: Efficacy of biological agents in metastatic
triple-negative breast cancer. Cancer Treat Rev. 40:605–613. 2014.
View Article : Google Scholar : PubMed/NCBI
|