Introduction
Neutropenia is a common abnormality in childhood
with an incidence of 12.36% in Beijing from July 2015 to July 2016
(1), and is most often associated
with infections, partcularly respiratory tract infections, skin
soft tissue infections, and inherited bone marrow failure syndromes
(IBMFS) is one of the causes of neutropenia. Fanconi anemia (FA), a
rare autosomal recessive condition, is a hereditary IBMFS that was
first described by Guido Fanconi in 1927 (2). The mortality rate of FA in the United
States and Israel is 1–5 cases for every 1 million people, with a
carrier frequency of ~1 in 300 individuals (3). Although it is often characterized by
congenital malformations, progressive bone marrow failure, abnormal
skin pigmentation patterns and susceptibility to cancer (4), at least 25% of patients with FA have few
or no abnormal symptoms that may aid in its early diagnosis
(5). As a hereditary disease, at
least 16 genes including Fanconi Anemia Complementation Group
(FANC)A, FANCC, FANCG, FANCD1 and FANCD2 have been
identified to cause FA. Among these, mutations in the FANCD1
and FANCD2 genes are the causative factor in 5% of all cases
of this disease (6–12). The present case report describes a
pair of twins whom suffered recurrent upper respiratory tract
infections for >2 years prior to the study. They were identified
to have FA, caused by a double heterozygous mutation of
FANCD2.
Case report
The present study was approved by the Ethical
Committee of Xiangya Hospital (Changsha, Hunan, China). Written
informed consent was obtained from the parents of the twins.
A pair of 4-year-old male mono-twins who suffered a
recurrent upper respiratory tract infection for >2 years
presented to the pediatric outpatient ward of Xiangya Hospital,
Central South University (Changsha, China) on September 15, 2015,
following a routine blood test that had been conducted on the twins
(for a recurrent upper respiratory tract infection lasting >2
years) performed on the same day and had demonstrated abnormal
results (the routine blood test for the elder brother revealed a
white blood cell count (WBC) of 2.6× 109/l (normal
range, 4–9.5×109/l), hemoglobin (HGB) of 118 g/l (normal
range, 120–175 g/l), platelet count (PLT) of 340×1012/l
(normal range, 125–350×109/l), and neutrophil
granulocyte count (NE) of 0.7×109/l (normal range,
1.8–6.3×109/l); the younger brother demonstrated a WBC
of 2.2×109/l, HGB of 110 g/l, PLT of
269×1012/l, and NE of 0.8×109/l). There was
no indication of discomfort from either child. The two patients
were of normal height and weight according to age (13) and had no observable physical
deformities, and physical examination did not reveal anything
significant. Blood samples from the twins were tested repeatedly
(>10 times) every 1 to 2 months over the past 2 years, revealing
WBC of 2.2×109 to 4.3×109/l, HGB from 100 to
120 g/l, PLT from 210×1012 to 314×1012/l and
NE from 0.7×109 to 1×109/l. Other laboratory
test results, including those for immunoglobulins (IgG, IgA, IgM
and IgE), lymphocyte subsets, virus antibodies (herpes simplex
virus, respiratory syncytial virus, adenovirus, Epstein-Barr virus,
coxsackievirus and cytomegalovirus), random blood glucose and
endocrines (sex, thyroid and growth hormones), were not abnormal.
Imaging analysis, including chest radiography, and cardiac and
abdominal ultrasounds, did not reveal any areas of concern. Bone
marrow cell morphology was assessed as follows: The punctured bone
marrow fluid was placed at one end of a slide glass, and one end of
a pushing piece with a flat edge was placed in front of the bone
marrow fluid, and the bone marrow fluid was smoothly pushed forward
to the other end of the slide glass, so that a thin layer of bone
marrow fluid remained on the slide glass, then the slide was
air-dried and fixed with methanol for 5 min at 25°C. Next, the
slide was dyed with Wright-Giemsa stain (Wright stain consisted of
0.1 g Wright's dye and 60 ml methanol, and the Giemsa stain
consisted of 0.5 g Giemsa's dye, 33 ml methanol and 33 ml pure
glycerin, purchased from Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) for 5 min at 25°C prior to observation
with optical microscope (magnification, ×100) to determine the bone
marrow morphology and the degree of bone marrow hyperplasia. The
bone marrow cell morphology test revealed a normal result. In order
to obtain a more accurate diagnosis, next-generation sequencing
(NGS) was performed. Genomic DNA samples obtained from 2 ml
peripheral blood were sheared by sonication. The sheared genomic
DNA was then hybridized using the NimbleGen 2.0 probe sequence
capture array (Roche Molecular Diagnostics, Pleasanton, CA, USA) to
enrich exonic DNA (Joy Orient Translational Medicine Research
Centre, Co., Ltd., Beijing, China). A 1 µg DNA library was mixed
with Buffer BL and GenCap gene panel probe (MyGenostics, Inc.,
Medford, MA, USA), heated at 95°C for 7 min and 65°C for 2 min on a
polymerase chain reaction (PCR) machine. Subsequently, 23 µl of the
65°C pre-warmed Buffer HY (MyGenostics, Inc.) was added to the mix,
and the mixture was held at 65°C for 22 h for hybridization. MyOne
beads (50 µl; Life Technology; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) were washed in 500 µl 1 X binding buffer 3 times
and resuspended in 80 µl 1 X binding buffer. A total of 64 µl 2 X
binding buffer was added to the hybrid mix and transferred to the
tube with 80 µl MyOne beads. The mix was rotated for 1 h at room
temperature on a rotator. The beads were then washed with WB1
buffer at room temperature for 15 min once and WB3 buffer at 65°C
for 15 min 3 times. The bound DNA was then eluted using elution
buffer (1 mol/l Tris-HCl 1 ml, and 0.5 m EDTA 0.2 ml and 100 ml
distilled water). The eluted DNA was amplified for 15 cycles using
the following program: 98°C for 30 sec (1 cycle); 98°C for 25 sec,
65°CC for 30 sec, 72°C for 30 sec (15 cycles); and 72°C for 5 min
(1 cycle). The PCR product was purified using SPRI beads (Beckman
Coulter, Inc., Brea, CA, USA) according to manufacturer's protocol.
The libraries were first tested for size distribution and
concentration using the Agilent Bioanalyzer 2100 with the 2100
Bioanalyzer Expert software (Agilent Technologies, Inc., Santa
Clara, CA, USA). The samples were then sequenced on an Illumina
Hiseq2500 with HiSeq Control software (HCS2.0; Illumina, Inc., San
Diego, CA, USA) and analyzed using DNASTAR software (version 5.0;
DNASTAR, Inc., Madison, WI, USA). A total of 2 parallel reactions
were performed for each sample, and Protein structure prediction
software (v1.1, downloaded from J. Craig Venter Institute, La
Jolla, CA, USA) was used to predict the functional effect of amino
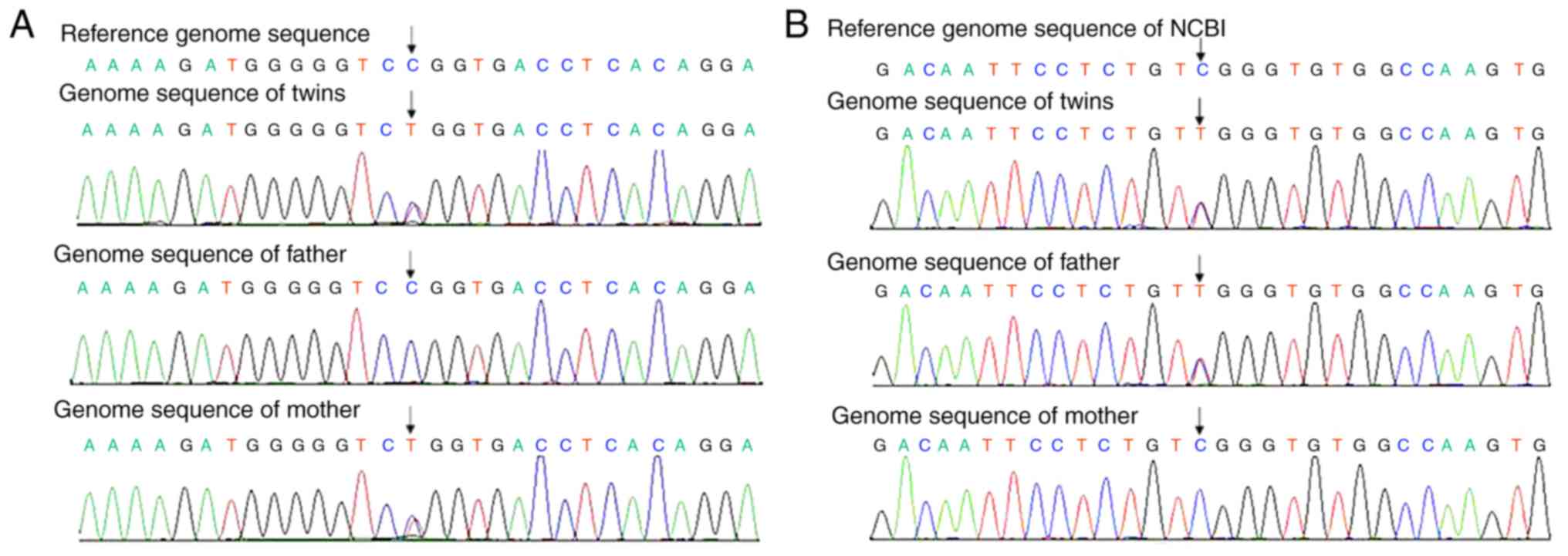
acid substitutions and indels. NGS revealed double heterozygous
mutations in the FCND2 gene in the two patients (Tables I–III;
Fig. 1). Additional diagnostic
evidence obtained by chromosome breakage analysis. Chromosome
culture was performed using the standardized method of the
International Atomic Energy Agency 405 technical report (14). Mitomycin C (0, 50 and 100 µg/l; Kyowa
Hakko Bio Co., Ltd, Tokyo, Japan) was added after 24 h and cells
were cultured for 48 h at 37°C, harvested, added a potassium
chloride hypotonic (0.188%) solution for 20 min at 37°C, pre-fixed
with glacial acetic acid-methanol fixed fluid (consisting of
glacial acetic acid and methanol at a ratio of 1:3) for 15 min at
room temperature. A total of 1–2 drops was dropped onto slides from
40–50 cm and dried naturally prior to observation using a light
microscope (magnification, ×100). The number of chromosome
aberration cells and the chromosome breakage number were counted
under the microscope, and the chromosome aberration rate and
breakage rate was analyzed. The breakage rate in 100 cells was
defined as (the number of broken chromosomes/100) ×100%, aberration
rate was defined as (the number of cells which appeared to exhibit
broken chromosomes/100) ×100%. The chromosome aberration rate and
breakage rate of the experimental group were considered to be
positive if they were higher compared with that of the control
group. A total of 100 mitotic images were analyzed per specimen.
The results are presented in Table
IV. A comet assay was also performed. Cytochalasin B (6 µg/ml,
at 37°C for 28 h), used to induce cell DNA damage, and ethidium
bromide were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). A Nikon 90i fluorescence microscope was purchased from
Nikon Corporation (Tokyo, Japan). The chromosome image analysis
system GK-1303 was purchased from Leica Microsystems GmbH (Wetzlar,
Germany) and the Sanyo MCO-20AIC CO2 incubator was purchased from
SANYO Semiconductor Manufacturing Co., Ltd. (Sakata, Japan). The
normal-melting-point agarose was obtained from Biowest USA
(Riverside, MO, USA) and the low-melting-point agarose was from
Promega Corporation (Madison, WI, USA). Tris-HCl, dimethyl
sulfoxide (DMSO) and Triton X-100 were purchased from
Sigma-Aldrich, Merck KGaA. Lymphocyte-separated medium (lymphoprep)
was purchased from Axis Shield Diagnostics, Ltd. (Dundee, UK). The
horizontal-strip electrophoresis apparatus was from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA). The comet slides were from
Bio-comet (Institute for Surgical Research and Hospital Management,
Basel, Switzerland). The digital imaging system DMS300 was
purchased from Leica Microsystems GmbH.
 | Table I.Mutation of FANCD2 in the
patients. |
Table I.
Mutation of FANCD2 in the
patients.
|
| Mutation
information |
|
|---|
|
|
|
|
|---|
| Gene | Chromosome
location | Base change | AA change | MAF | Prediction | Disease |
|---|
| FANCD2 | Chr3:10106532 |
c.2141C>T(E23) | p.714, P>L | 0.085 | Harmful | Fanconi anemia |
|
| Chr3:10130159 | p.3493>T(E35) | p.1165, R>W | 0 | Harmful |
|
| Table III.Hereditary information of a
FANCD2 allele (allele 2) of the twins. |
Table III.
Hereditary information of a
FANCD2 allele (allele 2) of the twins.
| Family member | Gene | Mutation type | NA changes | AA changes | Prediction |
|---|
| Twins | FCND2 | Heterozygous |
c.3493C>T(E35) | p.R1165W | Harmful |
| Father | FCND2 | Heterozygous |
c.3493C>T(E35) | p.R1165W | Harmful |
| Mother | FCND2 | Normal | C.3493C
(wild-type) |
| Normal |
| Table IV.Chromosome breakage test. |
Table IV.
Chromosome breakage test.
| A, Number of abberant
cells per 100 cells. |
|---|
|
|---|
|
| MMC (ng/ml) |
|---|
|
|
|
|---|
| Sample | 0 | 50 | 100 |
|---|
| Twin 1 | 0.0 | 72.0±3.6 | 90.0±1.7 |
| Twin 2 | 0.0 | 70.5±1.8 | 87.0±3.6 |
| Control | 3.0±1.0 | 18.6±2.1 | 65.9±6.9 |
|
| B, Number of
breakage chromosomes per 100 cells. |
|
|
| MMC
(ng/ml) |
|---|
|
|
|
| Sample | 0 | 50 | 100 |
|
| Twin 1 | 0.0 | 172.0±6.0 | 990.0±30.8 |
| Twin 2 | 0.0 | 188.0±3.6 | 964.0±15.1 |
| Control | 3.0±0.6 | 19.0±1.7 | 114.8±5.3 |
The comet assay was performed under neutral
conditions, as described by Banath et al (15), but with a slight modification;
specifically, special comet slides were used rather than general
slides. There are gaps in comet slides to contain the agarose.
Furthermore, less agarose was used in the procedure than originally
described by Banath et al yielding a thinner gel agarose to
enable clearer viewing under a fluorescence microscope. First, the
comet slides was coated with 100 µl normal-melting-point agarose
(0.075%); then, once the first agarose layer was coagulated, a
mixture of 75 µl low-melting-point agarose (0.075%) and 25 µl
lymphocyte suspension was applied as the second layer. The comet
slides were immersed in cold fresh lysis solution (2.5 M NaCl, 1%
N-sodium lauryl sarcosinate, 30 mM Na2EDTA, 10 mM Tris, 1% Triton
X-100 and 10% DMSO) for 1.5 h at 4°C. Following lysis, slides were
placed in a horizontal electrophoresis tank pre-filled with cold
fresh Tris-borate-EDTA (TBE, Medicago, Inc., Durham, NC, USA)
buffer for 20 min at 4°C to loosen the tight double-helical
structure of DNA for electrophoresis. Electrophoresis was then
performed at 200 mA for 20 min in TBE buffer at room temperature.
The slides were then rinsed twice with distilled water and were
stained with ethidium bromide (2 µg/ml) for 10 min at 25°C. All of
the above procedures were performed in the dark to avoid
supernumerary DNA damage. The comet slides were viewed using a
Nikon 90i fluorescence microscope (magnification, ×400) and images
of 100 comets were collected for each group using a digital imaging
system (DMS300; Leica Microsystems GmbH). Cells that overlapped
were not counted. All of the comet images were analyzed using Comet
Assay Software Pect (cat. no., CASP 1.2.3 beta 1; CASPLab,
University of Wroclaw, Institute of Theoretical Physics, Wroclaw,
Poland), and the percentage of DNA in the comet tail, the tail
length, the tail moment and the olive tail moment were recorded to
characterize the lymphocytic DNA damage. Statistical analyses were
performed using one-way analysis of variance and Dunnett's test
with SPSS13.0 (SPSS Inc., Chicago, IL, USA), and P<0.05 was
considered to indicate a statistically significant difference. The
results, presented in Table V,
revealed the Tail Moment of the comet experiment for twins were
higher than that of the control group, which was statistically
significant and therefore considered a positive result. Therefore,
on the basis of these data, the twins were definitively diagnosed
with FA.
| Table V.Comet assay. |
Table V.
Comet assay.
| Group | Head DNA, % | Tail DNA, % | Tail length
(pixel) | Comet length
(pixel) | Tail moment | Olive tail
moment |
|---|
| Twin 1 | 94.39 | 5.61 | 13.33 | 65.11 | 0.79 | 0.51 |
| Twin 2 | 94.03 | 5.97 | 14.02 | 65.95 | 0.97 | 0.66 |
| Control | 98.02 | 1.98 | 8.92 | 64.03 | 0.50 | 0.60 |
| P-value for Twin
1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | 0.135 |
| P-value for Twin
2 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | 0.112 |
Discussion
FA is a rare hereditary autosomal recessive disease
caused by mutations in FA genes that are primarily involved in DNA
damage response or repair (16). It
is characterized by congenital malformations, progressive bone
marrow failure, skin lesions and susceptibility to cancer (17). Early symptoms of FA include multiple
malformations, including skeletal and ear deformities, renal
malformation and abnormal skin pigmentation patterns, including
café au lait spots and chromatosis (18). These are followed by progressive bone
marrow failure, which begins to occur, on average, at 7 years of
age (19), with the majority of
patients eventually suffering from cancer (20). As these clinical manifestations lack
specificity, it is often difficult to diagnose FA, particularly in
patients who do not exhibit surface deformities or skin lesions.
For this reason, tests including chromosome breakage analysis and
comet assays are often performed to reach a definitive diagnosis.
FA diagnosis is also confirmed by the subtyping of FA (i.e.,
determination of the complementation group) (21,22). At
present, sequencing of the FA genes has also become an important
and accurate method of diagnosing the disease.
In the present case of the twins, the physical
examination, necessary laboratory and imaging tests and lack of
abnormal skin pigmentation patterns gave no indication of FA. Only
changes in the levels of certain blood components were observed,
without abnormal bone marrow cell morphology. Therefore, it was
difficult to diagnose the cause of the abnormal blood results and
even to select follow-up tests. Fortunately, their symptoms were
similar to those of IBMFS, thereby providing a clue for the
direction to follow. Next-generation gene sequencing was then
performed and revealed that the two patients exhibited double
heterozygous mutations in the FANCD2 gene, confirming that
each of the parents was an obligate carrier. With this information,
the twins were conclusively diagnosed with FA. In addition, the
chromosome breakage experiment and comet assays demonstrated
positive results, verifying the diagnosis.
The FANCD2 gene, located on chromosome 3 at
position 3p25.3 (including 44 exons), codes for 2 isoforms of
proteins (FANCD2-S and FANCD2-L) consisting of 1,451 amino acids,
with molecular masses of 155 and 162 kDa, respectively.
Ubiquitination of the FANCD2-encoded protein is of crucial
importance in the pathogenesis of FA, as the ubiquitinated protein
normally binds with Fanconi Anemia Complementation Group I and
BRCA2, DNA Repair Associated, to repair damaged DNA. In the case of
the two patients, next-generation sequencing revealed a
c.2141C>T mutation in one of the alleles of FANCD2,
termed Allele 1, which changes the 714th amino acid of this protein
from a proline to a leucine. Protein structure prediction software
(23) identified this change to be
harmful. In addition, by testing this same FANCD2 site in
the genome of the parents of the twins, it was identified that this
mutation was passed on through the mother. In the other allele of
FANCD2, termed Allele 2, a c.3493C>T mutation was
identified, which changes the 1,165th amino acid of this protein
from an arginine to a tryptophan. Protein structure prediction
software also identified this change to be harmful. Testing of this
site on the genomes of the parents revealed that this mutation
originated from the father. On the basis of these results, it was
concluded that harmful mutations in the two alleles of
FANCD2 had been passed on to the twins, and they were
definitively diagnosed with FA.
In summary, the following were the characteristics
of the twins: Firstly, the two boys presented with the same
symptoms during the early stages and appeared to be suffering from
chronic illnesses for >2 years. Secondly, the twins indicated no
discomfort upon hospital presentation and no positive results from
the physical examination. Thirdly, specific abnormal blood test
results and a positive comet assay result were observed, and double
heterozygous mutations of FANCD2 were revealed. Fourthly,
there were no other diseases aside from FA that accurately matched
the symptoms and disease presentation of the twins. Therefore, on
the basis of these results, the twins were diagnosed with FA.
Analysis of the study may cause the following to be questioned:
Firstly, although the twins had abnormal routine blood test
results, physical and imaging examinations revealed no
malformations, and no abnormal skin pigmentation patterns were
identified. The results of bone marrow morphology analysis were
also identified to be normal, whereas the majority of cases of FA
demonstrate all of the above symptoms. A possible explanation for
this inconsistency is that the twins were in the early stage, or
suffered from a mild subtype, of FA. Secondly, next-generation gene
sequencing of FANCD2 does not appear to be sufficient to
support the diagnosis of FA, as in the case of Allele 1. Although
the protein structure prediction software predicted the change to
be harmful, the minor allele frequency (MAF) for this mutation is
5.9% according to the Exome Aggregation Consortium (24). However, as the MAF for this mutation
is as high as 5.9%, this means that the homozygous mutation may be
as high as 0.35%, but as is not consistent with the incidence of
FA, whether this type of mutation leads to FA is questionable. Our
interpretation is that the present MAF value of Allele 1 is not
representative of all existing data. An additional possible
explanation is that these types of homozygous mutations, including
that of Allele 1 in FA, result in mild cases FA or are not
pathogenic, but when they exist together with a second mutation,
for example a double heterozygous mutation, the symptoms become
more marked. In addition, as the mutation of Allele 2 has not been
identified previously for this gene, its MAF value is unknown and
it may be a novel mutation. Nevertheless, based on protein
structure prediction software, it was deduced that this mutation
was also harmful. Unfortunately, the present study was not able to
examine the protein structure and its mechanism.
It is well-known that hematopoietic stem cell
transplantation (HSCT) is presently the only way to improve the
hematopoietic environment and the long-term disease-free survival
rate of patients with FA (25).
Considering the fact that an early diagnosis of FA is difficult,
and alongside the known problems of transplantation failure,
graft-versus-host disease and numerous other
transplantation-associated issues, there is no unified consensus on
when to initiate HSCT for patients with FA (26). In a retrospective analysis of the
prognosis of patients with FA who had undergone allogeneic HSCT,
the European Group for Blood Marrow Transplantation identified that
a patient age of >10 years with a history of >20 blood
transfusions and hematopoietic system failure (absolute neutrophil
count <500×109/l, platelet count
<20×109/l, hemoglobin <80 g/l) would be not
conducive to the early reconstruction of the hematopoietic system
(27). In the present case of the
twin boys, due to their mild symptoms of tolerance according to the
guidelines of FA (28), no additional
tests or treatments were performed. The last out-patient follow-up
time was December 28th, 2017, the twins had no symptoms at that
time. The final routine blood tests revealed a WBC of
3.6×109/l, HGB of 116 g/l, PLT 310×1012/l,
and NE of 0.9×109/l for the elder brother, and a WBC of
3.0×1109/l, HGB of 112 g/l, PLT of 258×1012/l
and NE of 0.8×109/l for the younger brother. However,
follow-up will be continued, and HSCT will be a viable treatment
option for these patients in the future. Next-generation gene
sequencing was performed for the parents, and they were provided
with complete genetic information concerning FA, which may provide
support in pre-implantation genetic diagnosis for future
pregnancies if the couple wants to have another child.
Finally, it is necessary to state the reason the
case of the present study was described, which was to share the
results obtained. Firstly, the case was very rare, as each parent
was a carrier for FA. Unfortunately, the illnesses of the twins
were caused as a result of their double heterozygosity; however, it
was notable that neither of them exhibited any typical symptoms of
FA. Secondly, one novel mutation in the FADCN2 gene that may
lead to FA was identified. Thirdly, the case demonstrated that
difficulties in clinical diagnosis may be overcome by including
genetic screening tests into the range of available diagnostic
tests, which may also reveal unexpected results.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Sciences Foundation of China (grant nos. 81570154, 81100359
and 81500231).
Availability of data and materials
The datasets generated and analyzed in the present
study are included in this published article.
Authors' contributions
WD and MZ performed the data analysis and wrote the
manuscript. LC contributed to the conception of the study. YL
contributed significantly to data analysis and manuscript
preparation. MY helped perform the data analysis and conducted
constructive discussions. All authors approved the final
manuscript.
Ethics and consent to participate
The present study was approved by the Ethical
Committee of Xiangya Hospital and written informed consent was
obtained from the patient, or from the parent or guardians, as
appropriate.
Consent for publication
Written informed consent was obtained for the
publication of data and materials.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fang Ye, Li'nan Zhang, Jianli Sun, et al:
Analysis of clinical features of leukopenia patients in a three
level general hospital in Beijing. China Medical Herald. 14:60–63.
2017.
|
|
2
|
Crawford D and Dearmun A: Fanconi anaemia.
Nurs Child Young People. 29:172017. View Article : Google Scholar
|
|
3
|
Rosenberg PS, Tamary H and Alter BP: How
high are carrier frequencies of rare recessive
syndromes/contemporary estimates for Fanconi Anemia in the united
states and Israe. Am J Med Genet A. 155A:1–1883. 2011.PubMed/NCBI
|
|
4
|
Pivovar A, Furquim CP, Bonfim C and
Torres-Pereira CC: Mouth examination performance by children's
parents and by adolescents in Fanconi Anemia. Pediatr Blood Cancer.
64: View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mohanty D: Understanding complexity of
Fanconi anaemia. Indian J Med Res. 143:132–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheung RS and Taniguchi T: Recent insights
into the molecular basis of Fanconi Anemia: Genes, modifiers, and
drivers. Int J Hematol. 106:335–344. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fagerholm R, Sprott K, Heikkinen T,
Bartkova J, Heikkilä P, Aittomäki K, Bartek J, Weaver D, Blomqvist
C and Nevanlinna H: Overabundant FANCD2, alone and combined with
NQO1, is a sensitive marker of adverse prognosis in breast cancer.
Ann Oncol. 24:2780–2785. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Winter JP, van der Weel L, de Groot J,
Stone S, Waisfisz Q, Arwert F, Scheper RJ, Kruyt FA, Hoatlin ME and
Joenje H: The Fanconi Anemia protein FANCF forms a nuclear complex
with FANCA, FANCC and FANCG. Hum Mol Genet. 9:2665–2674. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hess J, Unger K, Orth M, Schötz U,
Schüttrumpf L, Zangen V, Gimenez-Aznar I, Michna A, Schneider L,
Stamp R, et al: Genomic amplification of Fanconi Anemia
complementation group A (FancA) in head and neck squamous cell
carcinoma (HNSCC): Cellular mechanisms of radioresistance and
clinical relevance. Cancer Lett. 386:87–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Svojgr K, Sumerauer D, Puchmajerova A,
Vicha A, Hrusak O, Michalova K, Malis J, Smisek P, Kyncl M, Novotna
D, et al: Fanconi Anemia with biallelic FANCD1/BRCA2 mutations -
Case report of a family with three affected children. Eur J Med
Genet. 59:152–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Loizidou MA, Hadjisavvas A, Tanteles GA,
Spanou-Aristidou E, Kyriacou K and Christophidou-Anastasiadou V:
Fanconi Anemia-D1 due to homozygosity for the BRCA2 gene Cypriot
founder mutation: A case report. Oncol Lett. 11:471–473. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen X, Bosques L, Sung P and Kupfer GM: A
novel role for non-ubiquitinated FANCD2 in response to
hydroxyurea-induced DNA damage. Oncogene. 35:22–34. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coordinating Study Group of Nine Cities on
Physical Growth and Development of Children, ; Capital Institute of
Pediatrics, . A national survey on growth of children under 7 years
of age in nine cites of China, 2005. Zhonghua Er Ke Za Zhi.
45:609–614. 2007.(In Chinese). PubMed/NCBI
|
|
14
|
International Atomic Energy Agency, .
Dicentric analysis[M]/Cytogenetic analysis for radiation dose
assessment: A manual. Vienna: International Atomic Energy Agency;
2001, pp. 27–59
|
|
15
|
Banath JP, Fushiki M and Olive PL:
Rejoining of DNA single-and double-strand breaks in human white
blood cells exposed to ionizing radiation. Int J Radiat Biol.
73:649–660. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burgos-Moron E, Calderon-Montano JM, Orta
ML, Guillen-Mancina E, Mateos S and Lopez-Lazaro M: Cells Deficient
in the Fanconi Anemia protein FANCD2 are hypersensitive to the
cytotoxicity and DNA damage induced by coffee and caffeic acid.
Toxins (Basel). 8: View Article : Google Scholar : PubMed/NCBI
|
|
17
|
D'Andrea AD: Susceptibility pathways in
Fanconi's Anemia and breast cancer. N Engl J Med. 362:1909–1919.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Webb ML, Rosen H, Taghinia A, McCarty ER,
Cerrato F, Upton J and Labow BI: Incidence of Fanconi Anemia in
children with congenital thumb anomalies referred for diepoxybutane
testing. J Hand Surg Am. 36:1052–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hanenberg H, Roellecke K and Wiek C: Stem
cell genetic therapy for Fanconi Anemia - a New Hope. Curr Gene
Ther. 16:309–320. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rosenberg PS, Greene MH and Alter BP:
Cancer incidence in persons with Fanconi Anemia. Blood.
101:822–826. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aslan D, Ameziane N and De Winter JP:
Molecular diagnosis of Fanconi Anemia with next-generation
sequencing in a case with subtle signs and a negative chromosomal
breakage test. Turk J Pediatr. 57:282–285. 2015.PubMed/NCBI
|
|
22
|
Solomon PJ, Margaret P, Rajendran R,
Ramalingam R, Menezes GA, Shirley AS, Lee SJ, Seong MW, Park SS,
Seol D and Seo SH: A case report and literature review of Fanconi
Anemia (FA) diagnosed by genetic testing. Ital J Pediatr.
41:382015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nepal M, Che R, Ma C, Zhang J and Fei P:
FANCD2 and DNA Damage. Int J Mol Sci. 18:E18042017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
MacMillan ML and Wagner JE: Haematopoeitic
cell transplantation for Fonconi anaemia-when and how? Br I
Haematol. 149:14–21. 2010. View Article : Google Scholar
|
|
26
|
Ayas M: Hematopoietic cell transplantation
in Fanconi Anemia and dyskeratosis congenita: A minireview. Hematol
Oncol Stem Cell Ther. 10:285–289. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guardiola P, Pasquini R, Dokal I, Ortega
JJ, van Weel-Sipman M, Marsh JC, Ball SE, Locatelli F, Vermylen C,
Skinner R, et al: Outcome of 69 allogeneic stem cell
transplantations for Fanconi Anemia using HLA-matched unrelated
donors: A study on behalf of the European Group for Blood and
Marrow Transplantation. Blood. 95:422–429. 2000.PubMed/NCBI
|
|
28
|
Blanche P; Diagnosis evaluation of FA[M],
; Mary EE, Dave F, Lynn F, et al: Fanconi Anemia: Guidelines for
diagnosis and management. 3rd. Eugene: Fanconi Anemia Res Funf Inc;
2008, pp. 34–53
|
|
29
|
Końca K, Lankoff A, Banasik A, Lisowska H,
Kuszewski T, Góźdź S, Koza Z and Wojcik A: A cross-platform public
domain PC image-ayalysis program for the comet assay. Mutat Res.
534:15–20. 2003. View Article : Google Scholar : PubMed/NCBI
|