Introduction
Chemotherapy has contributed significantly to the
development of cancer treatment. However, it has some unpleasant
features, such as side-effects at therapeutic dosages and multidrug
resistance (MDR). As a frontline chemotherapeutic agent, the use of
Adriamycin (ADM) can never avoid these problems. To date,
researchers have found that a variety of mechanisms are responsible
for MDR, including increased drug efflux, reduced drug intake and
the induction of anti-apoptotic mechanisms. Inefficient drug
absorption is partly due to the overexpression of adenine
triphosphate (ATP)-binding cassette (ABC) transporters, which can
pump therapeutic agents from cancer cells.
Ginseng has been used in Asian countries for
thousands of years as an herbal medicine to prevent and treat
diseases (1). Its active ingredients,
ginsenosides, have appealed to researchers for their promising
anticancer activity and low toxicity (2,3).
20(S)-protopanaxadiol (PPD) is a major gastrointestinal metabolic
product and possesses many pharmacological activities, including
anti-inflammatory (4), antidepressant
(5) and anticancer properties
(6–8).
However, although PPD acts as an active ingredient and major
metabolic product, the amount of PPD in natural herbal medicine is
far from meeting clinical needs, which has limited its drug
efficiency and clinical application. We have tried to better
evaluate its effectivity and improve its industrialized production.
On the basis of the literature, the oxidation of the 24,25-double
bond is the predominant metabolic pathway of PPD (9,10).
Previously, we modified the 24,25-double bond and designed and
synthesized a series of PPD derivatives (11). Furthermore, we found that PPD12 could
sensitize MDR cancer cells to chemotherapeutic agents in
vitro via ABC subfamily B member 1 (ABCB1), which is an
important member of the ABC family (12). The administration of 100 mg/kg PPD12
significantly enhanced the inhibitory effect of ADM against a
multidrug-resistant tumor in the xenograft model.
However, further details regarding PPD12 remain
unclear. The reverse ability of PPD12 on MDR requires further
investigation. Many chemosensitizing agents have had preliminary
success but are precluded from clinical use for their toxicity at
the doses required to have a therapeutic effect. For PPD12, we
briefly explored its reversibility on MDR, but its toxicity has not
been established. Previous studies suggested that in clinical
settings, the co-administration of a ginsenoside with other
therapeutic agents might lead to ginseng drug interactions
(13,14). Thus, the toxicity of a single
treatment of PPD12 and the role that PPD12 plays in the ADM
toxicity must be investigated. In addition to in vitro and
in vivo toxicity studies, we investigated PPD12's
pharmacokinetics.
Materials and methods
Cells and regents
The human oral carcinoma cell line KB were obtained
from the American Type Culture Collection. The KB cells was
originally thought to originate from an oral epidermal carcinoma,
but has now been shown to be a HeLa derivative (15,16) and
vincristine-selected cell line KB/VCR was subscribed by professor
Lihong Hu from Shanghai Institute of Materia Medica of Chinese
Academy of Sciences. All cell lines have been under cell line
authentication service and its result showed cell lines could be
used for research. Cells were maintained in DMEM supplemented with
10% fetal bovine serum, 100 units/ml penicillin and 100 µg/ml
streptomycin at 37°C and 5% CO2. PPD12 was synthesized
by our laboratory as previously reported (11). ADM, midazolam, testosterone,
phenacetin, bupropion, amodiaquine, diclofenac, s-mephenytoin,
dextromethorphan, Ketoconazol, α-Naphthoflavone, Ticlopidine,
Quercetin, Sulfaphenazole, Ticlopidine, Quinidine,
13C2,15N-acetaminophen and
d9−1′-hydroxybufuralol were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Acetonitrile and methanol
[high-performance liquid chromatography (HPLC) grade] were obtained
from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). All other
chemicals were of the highest quality available. A TUNEL staining
assay kit was purchased from Wuhan Boster Biological Technology,
Ltd. (Wuhan, China).
Cell growth and survival assays
Cell viability was assessed using the MTT assay as
previously described (17). Cells
were seeded in 96-well microculture plates overnight to allow cell
attachment and then incubated with various concentrations of PPD12
and different concentrations of chemotherapeutic agents. Then, MTT
reagent was added to each well. After 4 h of incubation, we
photometrically quantified the colored formazan products at 490 nm
using a multiwell plate reader (Bio-Rad Laboratories, Hercules, CA,
USA).
Animal toxicity studies
All animal studies were performed in accordance with
the Guide for Care and Use of Laboratory Animals (The Ministry of
Science and Technology of China, 2006) and the related regulations
of the Ninth People's Hospital (Shanghai, China). Forty SPF Balb/c
mice (age 4–5 weeks old) were supplied by the Shanghai Laboratory
Animal Center (Shanghai, China) and housed under pathogen-free
conditions in the animal care facilities of the Ninth People's
Hospital affiliated with Shanghai Jiao Tong University. Half of the
mice were female, and the other half were male. The mice were
divided into four groups, with 5 females and 5 males in each group.
Weight-matched groups of mice were used. The control group was
treated with a related vehicle. The PPD12 group was treated with
PPD12 at 100 mg/kg via gavage twice weekly for 2 weeks. The ADM
group was given ADM at 20 mg/kg through intraperitoneal injection
on the 6th day from the start of the experiment. After pretreatment
with PPD12 at 100 mg/kg twice a week for one week, the combined
group received ADM at 20 mg/kg via intraperitoneal injection on the
6th day and continued receiving PPD12 at 100 mg/kg twice a week for
another week. This study continued for 3 weeks. Animals were
weighed every day. At the end of this experiment, mice were
euthanized for subsequent necropsy.
H&E and TUNEL assay
Animals' tissues were fixed in 4% formaldehyde,
dehydrated with gradient ethanol and embedded in paraffin. The
tissue sections (4 µm) were dewaxed and rehydrated per a standard
protocol. For histological analysis, the sections were stained with
hematoxylin and eosin. Once cell got damaged, cells became
edematous and deformed and finally got necrosis with its structure
disorganized and nuclear fragmentation. And at least 3 tissue
samples and 5 fields of view were analyzed to determine the cell
damages in each experimental group. H&E assay could partly
indicate cell damage but the damaged cells need a specific
staining. So we performed TUNEL assay which can specifically
distinguish apoptotic cells. For the TUNEL assay, an in situ
apoptosis detection kit (Wuhan Boster Biological Technology, Ltd.)
was used to detect apoptotic cells in heart tissues. The positive
cells were identified, counted (three random fields per slide) and
analyzed through ECLIPSE 80i light microscopy (Nikon, Tokyo,
Japan).
Pharmacokinetic study
The protocol utilized is based on a previous report
(18). PPD12 was administered as a
single dose of 10 mg/kg by tail vein injection or oral gavage into
adult male SD rats. At predose and at 0.083, 0.25, 0.5, 1, 2, 4, 8,
and 24 h post-dose, blood was collected from three male rats and
immediately processed for plasma by centrifugation for 10 min at
3,000 × g. The resulting plasma was frozen on dry ice, and the
samples were stored at −80°C until analysis. Proper measures were
taken to minimize any pain and discomfort the rats experienced. All
experimental procedures were performed in accordance with the
National Institutes of Health Guide for Care and Use of Laboratory
Animals (revised 2006). The experiments were performed in
compliance with ethical regulations, and the institute's committee
approved the protocols. For mouse plasma sample analysis of PPD12,
a 0.1-ml aliquot of plasma sample was treated with 0.3 ml of
methanol containing 250 nM internal standard (IS) for direct
deproteinization. After vortex mixing for 1 min and centrifuging
for 5 min at 10,000 × g, 0.2 ml of supernatant was transferred to a
sample vial, and 5 µl of the sample was injected onto the LC/MS/MS.
The LC was performed on an Agilent 1200 HPLC system (Agilent
Technologies, Inc., Santa Clara, CA, USA), and separation was
performed at 30°C using an Xterra column (2.1×50 mm, 3.5 µm; Waters
Corporation, Milford, MA, USA).
Inhibitory effects of PPD12 on seven
major CYP activities in human liver microsomes
The inhibitory potencies (IC50 values) of
PPD12 on CYP3A4, CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2B6 and CYP2D6
activities were evaluated in pooled human liver microsomes using a
cocktail of seven CYP substrates and liquid chromatography-tandem
mass spectrometry (LC-MS/MS). The incubation mixtures consisted of
pooled human liver microsomes (0.1 mg/ml), 1.3 mM NADPH, 3.3 mM
MgCl2, various concentrations of PPD12 or selective inhibitors in
dimethyl sulfoxide (DMSO; final concentrations of PPD12 of 0.1–33
µM; DMSO <1% v/v) and a cocktail of eight CYP
probe substrates, as described previously (19). The CYP substrates were used at
concentrations approximating their respective Km values:
5 µM Midazolam, 120 µM testosterone, 50 µM Phenacetin, 50 µM
Bupropion, 5 µM Amodiaquine, 7 µM Diclofenac, 40 µM S-Mephenytoin,
or 7 µM Dextromethorphan. After a 3-min preincubation at 37°C, the
reactions were initiated by the addition of NADPH; incubation
proceeded for 15 min at 37°C in a shaking water bath. The reaction
was stopped by placing the tubes on ice and adding 100 µl of
ice-cold methanol containing IS (13C2,
15N-acetaminophen for acetaminophen and
desethylamodiaquine, d9−1′-hydroxybufuralol for
4′-hydroxydiclofenac, 4′-hydroxy-mephenytoin and
1′-hydroxymidazolam). The incubation mixtures were then centrifuged
(13,000 rpm for 4 min at 4°C). All assays were performed in
triplicate, and the mean values were used in subsequent
calculations.
The metabolites formed from the seven substrates
were simultaneously quantified using our previously described
LC-MS/MS method. To this end, we employed a tandem mass
spectrometer (TSQ Quantum Access; Thermo Fisher Scientific, Inc.)
coupled to a Nanospace SI-2 LCsystem (Shiseido, Tokyo, Japan). The
column and auto sampler temperatures were 50°C and 6°C,
respectively. The mass spectrometer was equipped with an
electrospray ionization (ESI) source and operated in positive ion
mode. The ESI source settings for metabolite ionization were as
follows: Capillary voltage, 4200 V; vaporizer temperature, 350°C;
Capillary temperature, 330°C; sheath gas pressure, 35 psi; and
auxiliary gas pressure, 15 psi. Quantification was performed by
selected reaction monitoring (SRM) of the [M+H]+ion and the related
product ion for each metabolite: Acetaminophen, 152.1>110.3;
N-desethylamodiaquine, 328.1>283.0; 7-hydroxycoumarin,
163.0>107.2; 4′-hydroxydiclofenac, 312.0>231.1;
4′-hydroxy-mephenytoin, 235.1>150.1; 1′-hydroxybufuralol,
278.1>186.1, 1′-hydroxymidazolam, 341.9>324.0;
13C2-15N-acetaminophen 155.1>111.2; and d9-1′-hydroxybufuralol,
287.2>187.0. Analytical data were processed using the
Xcalibur® software (Thermo Fischer Scientific,
Inc.).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. The body weight data and percentage of apoptotic cells
were analyzed using one-way analysis of variance (ANOVA) followed
by Dunnett's post hoc test. The IC50 data were analyzed
using an unpaired t-test. Statistical analyses were performed with
GraphPad Prism, and P<0.05 was considered to indicate a
statistically significant difference. Plasma concentration data
were used to calculate the PK parameters by a noncompartmental
i.v.-bolus input model (WinNonlin 5.0; Pharsight Corporation,
Mountain View, CA, USA).
Results
Effect of PPD12 on reversing drug
resistance in KB/VCR cells with increasing time
PPD12 has been evaluated for its ability to reverse
MDR in several cell lines in a dose-dependent manner (20). To better investigate the relationship
between the anti-MDR ability and the duration of drug action,
KB/VCR and its parent cell line KB were incubated with ADM alone or
in combination with 2.5 µM PPD12 for different durations of time
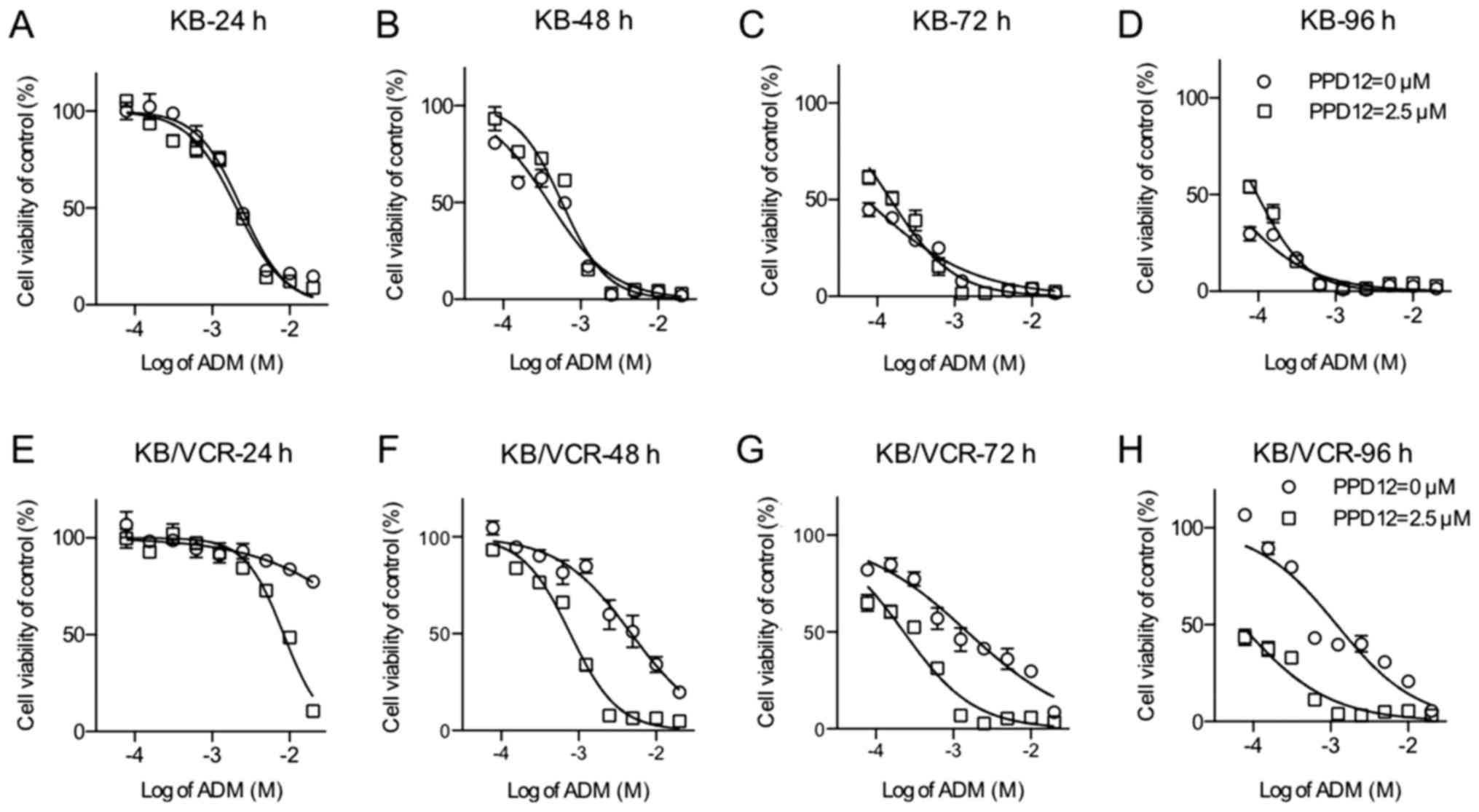
(24, 48, 72 and 96 h) (Fig. 1).
Indeed, KB/VCR cells had a higher tolerance of ADM than did KB
cells. For KB cells, the ADM dosage-effect curve was close to the
PPD co-treatment curves. In addition, in the first 24 h, the
absence of PPD12 could significantly sensitize KB to ADM. However,
for incubations of longer duration, this type of sensitivity did
not exist.
In contrast to the KB's response, KB/VCR cells were
much more vulnerable to co-treatment with PPD12 and ADM. A summary
of the IC50 values is shown in Table I. As the incubation time increased,
the IC50 values of KB cells to ADM treatment ranged from
2.383 to 0.0351 µM, whereas the IC50 values of KB cells
to the combination of ADM and PPD12 varied from 2.064 to 0.0968 µM.
In contrast to the response observed in KB cells, within 24 h of
incubation, the addition of PPD12 sharply decreased the
IC50 value of KB/VCR cells to ADM treatment from 150.9
to 8.431 µM, which is nearly an 18-fold reversal. After 48, 72, and
96 h of incubation, the IC50 values of KB/VCR to ADM
decreased from 4.782 to 1.193 µM, and those of KB/VCR to the
combination of ADM and PPD12 were all less than 1 µM; they were all
near the values of the KB cells to ADM alone or the combination of
ADM and PPD12. PPD12 helped ADM agents reach a satisfactory
anti-MDR activity level within a short period of time and keep its
effectiveness in the long term.
 | Table I.Effect of PPD12 on ADM-MDR reversal
in KB/VCR and KB cells. |
Table I.
Effect of PPD12 on ADM-MDR reversal
in KB/VCR and KB cells.
|
| IC50 ±
SD (µM)a (fold
reversalb) |
|---|
|
|
|
|---|
| Compound | Incubation
time | KB | KB/VCR |
|---|
| ADM | 24 h | 2.3830±0.0304
(1.00) | 150.900±0.2891
(1.00) |
|
| 48 h | 0.3898±0.0373
(1.00) | 4.782±0.0476
(1.00) |
|
| 72 h | 0.0713±0.0510
(1.00) | 1.521±0.0492
(1.00) |
|
| 96 h | 0.0351±0.0736
(1.00) | 1.193±0.0540
(1.00) |
| ADM+2.5 µM
PPD12 | 24 h | 2.0640±0.0242
(1.15)c | 8.4310±0.0265
(17.90)f |
|
| 48 h | 0.5732±0.0265
(0.68) | 0.7795±0.0213
(6.13)e |
|
| 72 h | 0.1493±0.0321
(0.48) | 0.2292±0.0328
(6.64)d |
|
| 96 h | 0.0968±0.0241
(0.36) | 0.0692±0.0550
(17.24)d |
Pharmacokinetic interactions of PPD12
with CYP450 in human liver microsomes
It is postulated that most metabolic drug
interactions can be attributed to inhibition or induction of the
drug-metabolizing cytochrome P450 (CYP or P450) enzymes (21). However, ginseng and its extract seem
to have different impacts on CYP, and the effect that PPD12 has on
CYP450 is unknown. To evaluate the influence of PPD12 on the CPY450
enzymes, their activities were examined with pooled human liver
microsomes, and selective inhibitors were used as a positive
control. For the selective inhibitors, the IC50 values
were all less than 0.01 µM. For PPD12, the values had a wide range.
PPD12 potently inhibited CYP2B6-catalyzed bupropion hydroxylation
and CYP3A4-catalyzed midazolam 1′-hydroxylation, with
IC50 values of 2.21 and 1.03 µM, respectively (Table II). PPD12 moderately inhibited
CYP3A4-mediated testosterone 6-β-hydroxylation and CYP2C8-mediated
amodiaquine N-deethylation with IC50 values of 8.43 and
9.57 µM, respectively. PPD12 weakly inhibited CYP2C19-catalyzed
s-mephenytoin4′-hydroxylation and CYP2D6-catalyzed dextromethor
phandextrorphan, with IC50 values of 15.04 and 11.85 µM,
respectively. At 33 µM, PPD12 produced a negligible inhibition of
CYP1A2-mediated phenacetin 0-deethylation and CYP2C9-mediated
diclofenac4′-hydroxylation.
| Table II.Inhibitory effect of PPD12 on major
CYP metabolic activities in human liver microsomes. |
Table II.
Inhibitory effect of PPD12 on major
CYP metabolic activities in human liver microsomes.
|
|
|
| IC50
(µM) |
|---|
|
|
|
|
|
|---|
| CYP enzyme | Catalysis | Inhibitor | Inhibitor | PPD12 |
|---|
| 3A4 | Midazolam
1′-hydroxylation | Ketoconazole | 0.0040 | 1.03 |
| 3A4 | Testosterone
6-β-hydroxylation | Ketoconazole | 0.0154 | 8.43 |
| 1A2 | Phenacetin
0-deethylation |
α-Naphthoflavone | 0.0168 | >33 |
| 2B6 | Bupropion
hydroxylation | Ticlopidine | 0.0823 | 2.21 |
| 2C8 | Amodiaquine
N-deethylation | Quercetin | 0.7319 | 9.57 |
| 2C9 |
Diclofenac4′-hydroxylation | Sulfaphenazole | 0.0856 | >33 |
| 2C19 |
S-mephenytoin4′-hydroxylation | Ticlopidine | 0.5120 | 15.04 |
| 2D6 |
Dextromethorphandextrorphan | Quinidine | 0.0235 | 11.85 |
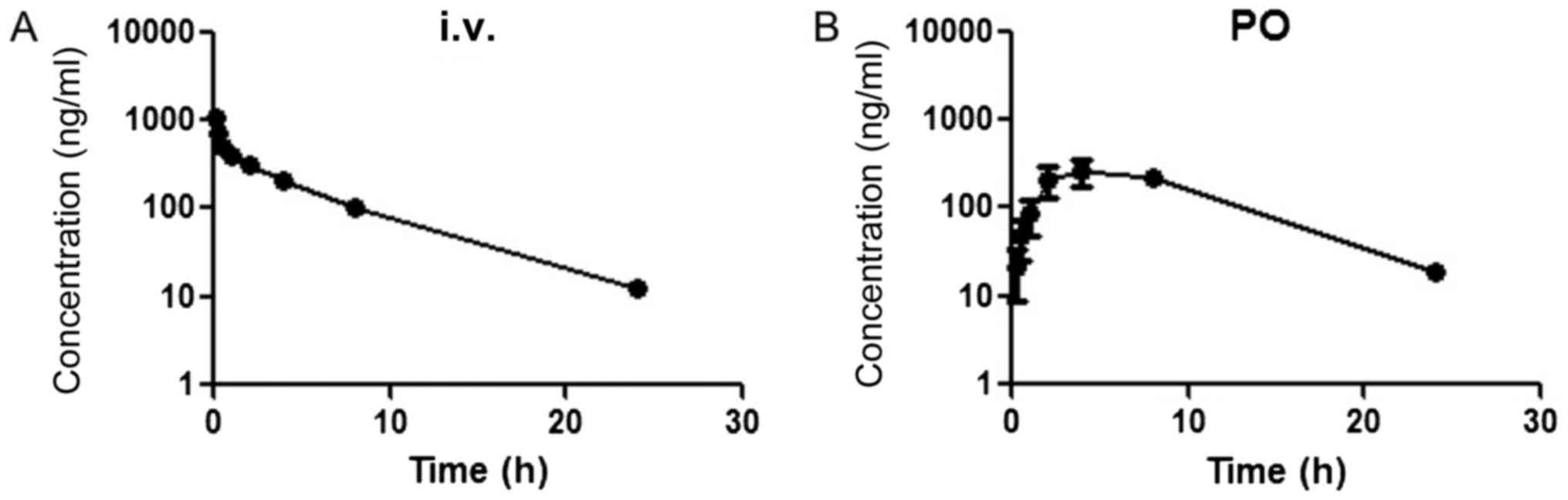
Plots of the plasma concentrations vs. time of PPD12
after oral and i.v. administration to male SD rats are shown in
Fig. 2. For intravenous (i.v.)
administration, the plasma concentration of PPD12 rapidly declined
within the first 2 h but then was maintained at a high level,
followed by a gradual linear decrease. The oral absolute
bioavailability was 114.41±22.24%.
Evaluation of the toxicity of PPD12 in
mice
To determine the maximum tolerant dosage of a single
PPD12 treatment in the mice, we administered a high dosage of 1
g/kg PPD12 to Balb/c mice. Interestingly, we did not observe any
lethal effects, even at such a high level, which suggests that a
severe and high-dosage treatment of PPD12 is well tolerated by and
non-toxic to animals. After a chronic, low-dosage and long-term
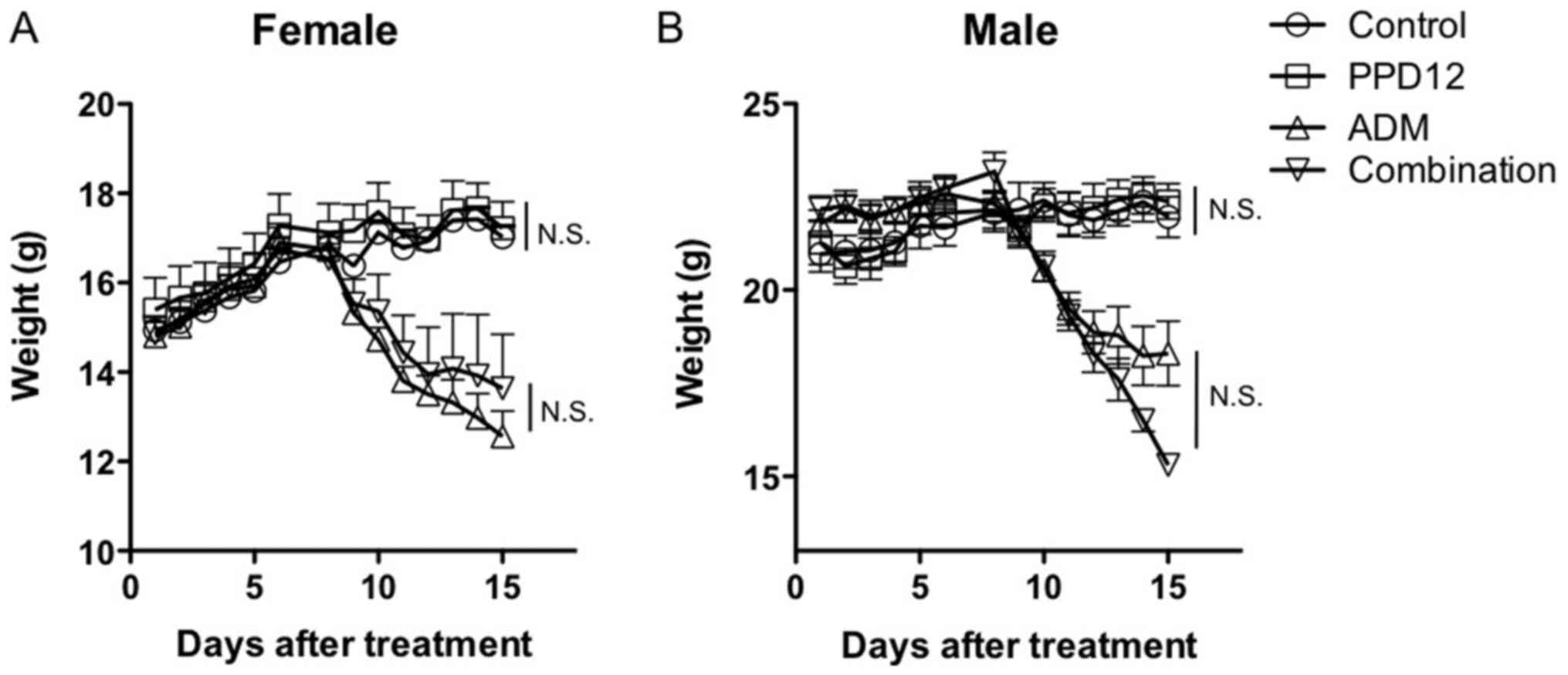
administration of PPD12 at 100 mg/kg for 3 weeks, we found that
PPD12 did not markedly reduce the body weight in either female or
male mice compared with the control group, as shown in Fig. 3. Similarly, compared with the control
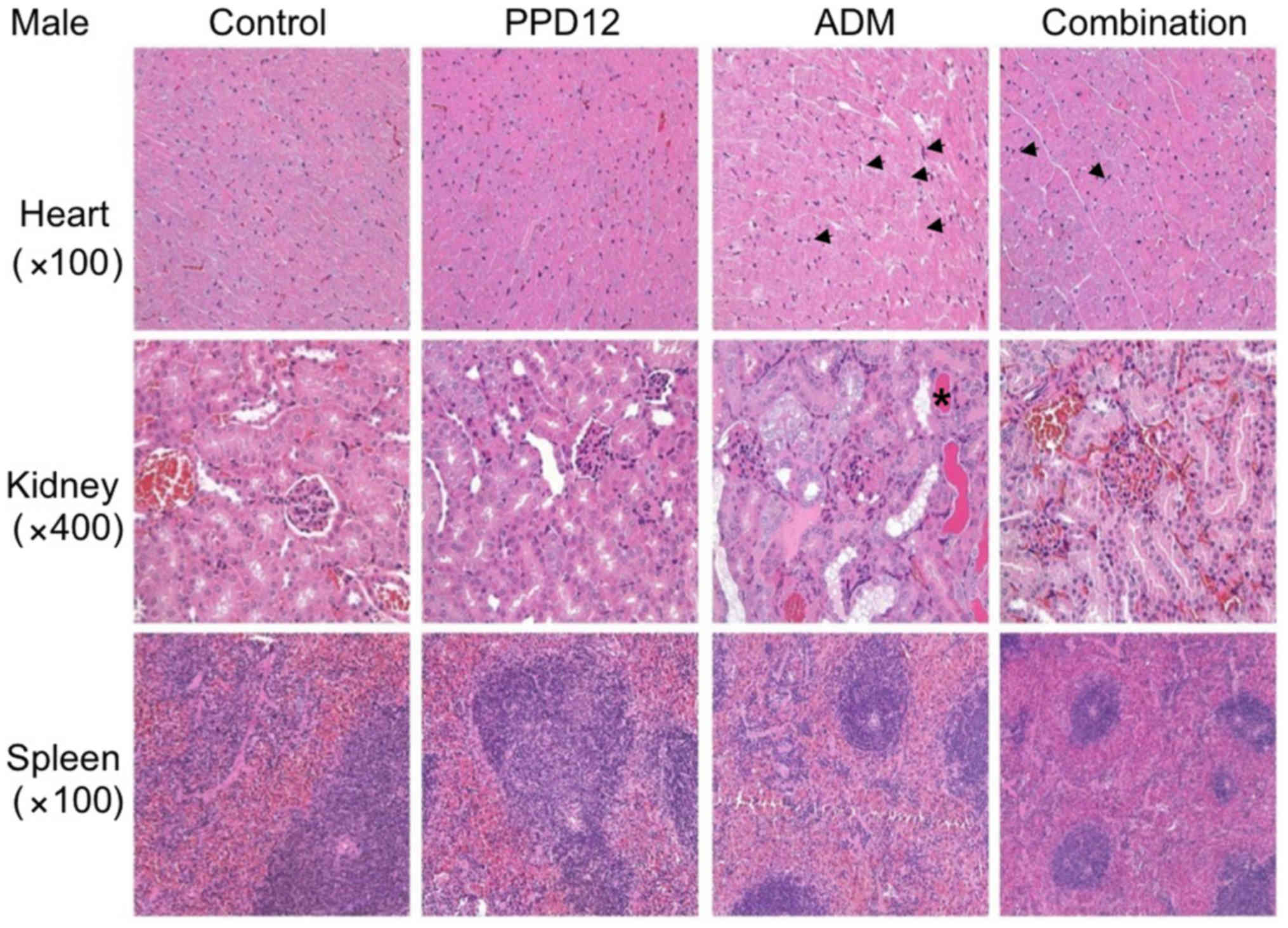
group, PPD12 incurred either no damage or, at most, an acceptable
amount of damage to organs such as the heart, kidney, spleen and
others that are not shown in this paper (Figs. 4 and 5).
These results all suggested that PPD12 exhibits low
toxicity with either short- or long-term administration.
Evaluation of the combination
treatment of PPD12 and ADM in mice
To further evaluate the role that PPD12 plays in ADM
toxicity, we used the same method used for the single PPD12
treatment. Mice received ADM or a combination of ADM and PPD12, and
the body weight, morphology of different tissues and apoptosis were
examined. As shown in Fig. 3A, for
female mice, a significant reduction in body weight was observed in
both the ADM group and the ADM plus PPD12 group compared to the
control group. However, neither the female nor the male mice
exhibited significant evaluated body weight loss when the
combination group was compared to the ADM alone group.
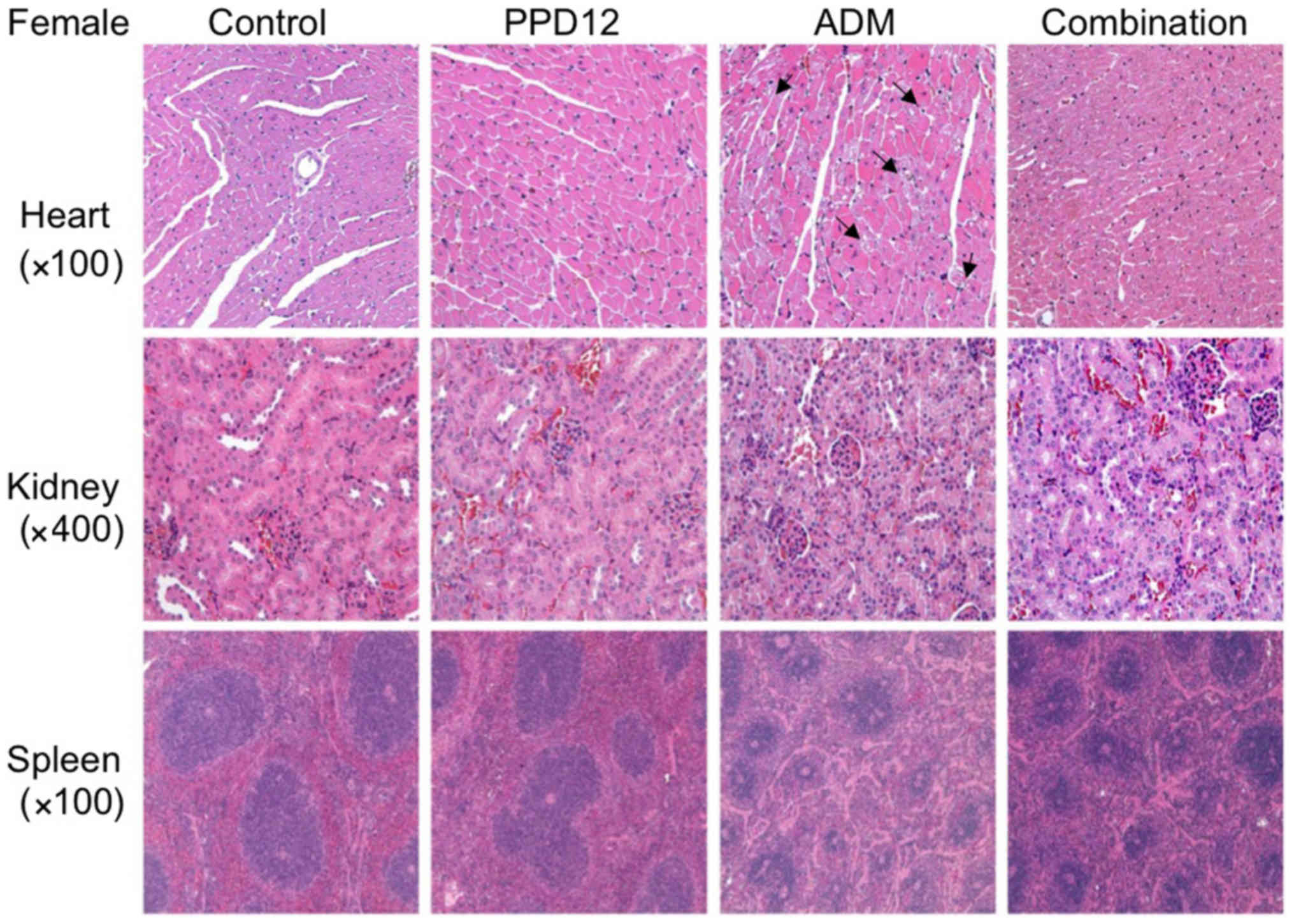
For tissue observations, we first
observed the morphological changes in the hearts
We found that in the ADM group, the interstitial
tissue was widened, and some myocardial cells became edematous,
with pale red H&E staining. Moreover, with ADM alone, much more
obvious necrosis was observed in the treated female mice than in
the male mice. However, these damages were slightly reduced by the
combination of PPD12 and ADM. Furthermore, it seemed that PPD12
could reverse the cardiac toxicity of the female group to a level
similar to that of the male group.
We next focused on the kidney. In the ADM group,
kidney epithelial cells were enlarged, and the cytoplasm was loose
and contained vacuoles, and some of it was even necrotic. In
contrast to the hearts of female and male mice, ADM did more damage
to the kidneys of the male group, and a hyaline cast was much more
common in male mice than in female mice. Nevertheless, co-treatment
with PPD12 and ADM visibly reversed ADM's toxicity on the kidney
PPD12, especially in the male group.
The spleen was also examined in this study. Whereas
ADM alone treatment severely shrank the splenic nodule, narrowed
the medullary cord and dilated and congested the splenic sinus,
many types of lesser damage were observed following co-treatment
with PPD12 and ADM, such as a larger spleen nodule.
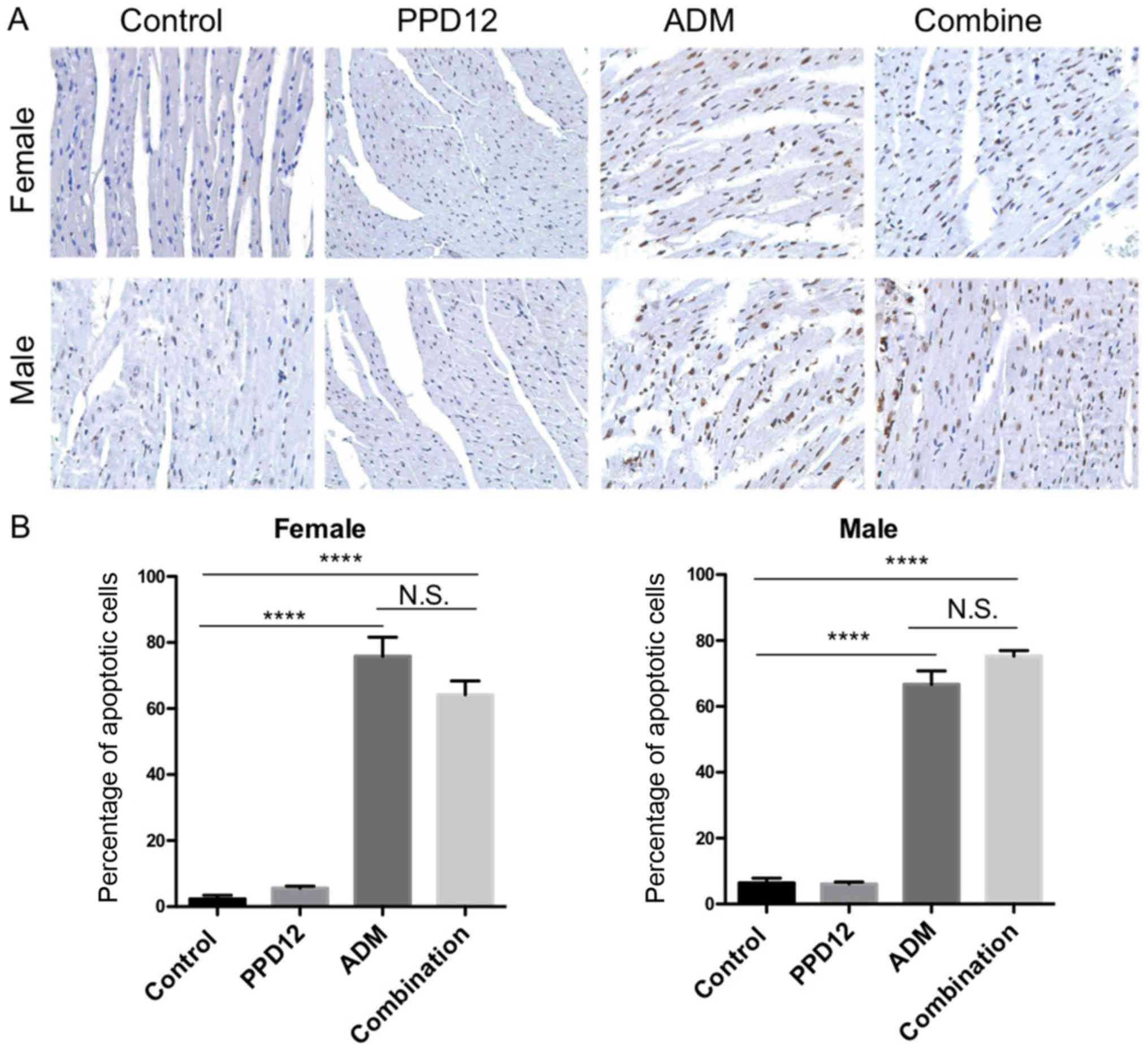
To better distinguish damaged cells, we also
performed specific staining to evaluate apoptosis. Cardio toxicity
was the most common and serve adverse effect of ADM so the
apoptosis in heart was analyzed. In the TUNEL assay of heart
tissues (Fig. 6), there was no
obvious difference in apoptotic cells between the control and PPD12
group but a significant increase in apoptosis in the single ADM
group. The toxicity of PPD12 was well tolerated compared to ADM.
Similar to the results of body weight loss, there was a significant
increase in apoptotic cells in the combined and single ADM group
compared to the control group. In addition, there was no obvious
difference between the combination group and ADM group.
Discussion
ADM is an ordinary chemotherapy agent, but some
treatments that use it result in failure. The MDR is one of the
main causes responsible for such failures (22). Although targeting special molecules
responsible for MDR, many clinical trials do not reach the third
stage because the agents cannot reach a satisfactory effect at
clinically tolerant doses.
Traditional herbal medicine, such as ginsenosides,
have recently been gaining more attention for their natural
constituents, biological activities and low toxicity (23). Among these ginsenosides, 20(S)-PPD is
the main ingredient responsible for anti-neoplastic effects
(24) and is the final active
metabolite (25). To better improve
its effectiveness, based on a structure analysis, we designed and
synthesized PPD derivates and found that PPD12 is a prominent
candidate. In a previous study, we demonstrated that PPD12 could
sensitize multidrug-resistant cancer cells to chemotherapeutic
agents in vitro and in vivo and that it worked to
reverse MDR partly by inhibiting ABCB1 activity.
In the present study, we further found that PPD12
did not obviously alter the effect of ADM in KB cells. However, for
the MDR cell line KB/VCR, PPD12 apparently strengthened the
anticancer activity of ADM, reversed MDR at early time points and
could maintain or even enhance this effect with increasing time.
After 24 h of incubation, the addition of PPD12 to ADM achieved
nearly an 18-fold reversal of MDR and attained a noteworthy
decrease in cell viability. These findings all suggest the perfect
anticancer and anti-MDR qualities of PPD12.
It is postulated that most metabolic drug-drug
interactions can induce an inhibition or drug-metabolization of
CYP450 enzymes (21) and that the
inhibition of P450 s by ginsenosides can be attributed to
ginseng-drug interactions (26).
Different ginsenosides or ginsenoside extracts have different
impacts on CYP450. In assays on a series of CYP450 activities, the
IC50 values for PPD12 were significantly higher than
those of the positive selective inhibitors that were used as
positive controls; furthermore, PPD12 showed slight inhibition of
the activities of CYP450 enzymes. PPD12 was also determined to be
well absorbed in the mouse model, and the bioavailability of PPD12
was nearly 100% by oral administration.
We next evaluated the maximum tolerant dosage in the
PPD12 alone treatment group. The results demonstrated that PPD12
was well tolerated in the mice tested, even in the 1 g/kg treatment
group. A chronic low-dose treatment of PPD12 also did not show any
obvious influence on body weight, tissue morphology and
apoptosis.
Furthermore, an additional treatment with PPD12 did
not increase the toxicity of the chemotherapy drug ADM but did
partly decrease the damage incurred by ADM. Interestingly, the
PPD12-reduced kidney toxicity of ADM was more obvious in male mice
than in female mice, whereas the PPD12-reduced cardio-toxicity of
ADM, as shown by the contrast phenomenon, was more significant in
female mice, in agreement with previous studies. Lipshultz et
al stated that ADM cardiac toxicity was more severe in female
patients than in male patients (27).
Further, ADM cardiac toxicity occurred more readily in females than
in males (28–30). In our studies, the cardiac toxicity of
ADM was more serious in female mice. Sakemi et al reported
that ADM treatment induced more massive nephropathy in male rats
compared with female rats and that castration significantly reduced
the nephropathy to an extent equal to the levels observed in female
rats (31). In addition, H&E
staining showed that the damage in the kidney was more serious in
male mice. Although the damage of ADM differed in the heart and
kidney of the different sexes, the morphological modifications in
the heart and kidney of the combination group remained at a similar
level in both female and male mice. The toxicity of ADM alone had a
wide range in female and male mice, which may have been due to sex
hormones. This range could be reversed by the additional
administration of PPD12. Based on the literature, ginsenosides
share a backbone similar to estrogen hormones, and some have even
been shown to bind to hormone receptors (32–35).
Studies confirmed that melatonin could kill breast cancer cells,
reduce cellular damage and mitochondrial degeneration of epirubicin
and protect the integrity of the subcellular structure in
doxorubicin-treated hearts via an estrogen signaling pathway
(27). Therefore, PPD12 possibly
helped to reduce the adverse effect of ADM because of the special
structure shared by PPD12 and estrogen.
In conclusion, PPD12 was able to achieve and
maintain satisfactory ADM-MDR reversal. PPD12 also exhibited low
toxicity and high bioactivity and did not further increase the
toxicity of ADM treatment. PPD12 is a promising candidate to help
overcome MDR in cancer chemotherapy and is suitable for further
clinical trials in tumor patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project of
the Shanghai Science and Technology Committee (grant no.
14431905800).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP and XW designed the study. SW performed the
experiments. XW, WC and LH analyzed the data and organized the
study. XW and SW wrote the manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Ninth People's Hospital [approval no. HKDL (2017) 126].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Attele AS, Wu JA and Yuan CS: Ginseng
pharmacology: Multiple constituents and multiple actions. Biochem
Pharmacol. 58:1685–1693. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiang YZ, Shang HC, Gao XM and Zhang BL: A
comparison of the ancient use of ginseng in traditional Chinese
medicine with modern pharmacological experiments and clinical
trials. Phytother Res. 22:851–858. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang W, Rayburn ER, Hao M, Zhao Y, Hill
DL, Zhang R and Wang H: Experimental therapy of prostate cancer
with novel natural product anti-cancer ginsenosides. Prostate.
68:809–819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee WM, Kim SD, Kim KS, Song YB, Kwak YS,
Cho JY, Park HJ, Oh JW and Rhee MH: Protopanaxadiol modulates
LPS-induced inflammatory activity in murine macrophage RAW264.7
cells. J Ginseng Res. 30:181–187. 2006. View Article : Google Scholar
|
|
5
|
Hui YZ, Yang ZR, Yang ZQ and Ge Q:
Inventors; CN-KnowHow intellectual property agent limited,
assignee. Antidepressant compositions containing
20(S)-protopanaxadiol extracted from ginsenoside. China Patent CN
200610027507.1. Jan 17–2007.
|
|
6
|
Qi LW, Wang CZ and Yuan CS: American
ginseng: Potential structure-function relationship in cancer
chemoprevention. Biochem Pharmacol. 80:947–954. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li G and Wang Z, Sun Y, Liu K and Wang Z:
Ginsenoside 20(S)-protopanaxadiol inhibits the proliferation and
invasion of human fibrosarcoma HT1080 cells. Basic Clin Pharmacol
Toxicol. 98:588–592. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu GY, Bu X, Yan H and Jia WW:
20S-protopanaxadiol-induced programmed cell death in glioma cells
through caspase-dependent and -independent pathways. J Nat Prod.
70:259–264. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kasai R, Hara K, Dokan R, Suzuki N,
Mizutare T, Yoshihara S and Yamasaki K: Major metabolites of
ginseng sapogenins formed by rat liver microsomes. Chem Pharm Bull
(Tokyo). 48:1226–1227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li L, Chen X, Li D and Zhong D:
Identification of 20(S)-protopanaxadiol metabolites in human liver
microsomes and human hepatocytes. Drug Metab Dispos. 39:472–483.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu J, Wang X, Liu P, Deng R, Lei M, Chen
W and Hu L: 20(S)-Protopanaxadiol (PPD) analogues chemosensitize
multidrug-resistant cancer cells to clinical anticancer drugs.
Bioorg Med Chem. 21:4279–4287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen G, Liu J, Chen W, Xu Q, Xiao M, Hu L,
Mao L and Wang X: A 20(S)-protopanoxadiol derivative overcomes
multi-drug resistance by antagonizing ATP-binding cassette
subfamily B member 1 transporter function. Oncotarget. 7:9388–9403.
2016.PubMed/NCBI
|
|
13
|
Gurley BJ, Gardner SF, Hubbard MA,
Williams DK, Gentry WB, Cui Y and Ang CY: Cytochrome P450
phenotypic ratios for predicting herb-drug interactions in humans.
Clin Pharmacol Ther. 72:276–287. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Butterweck V and Derendorf H: Herb-drug
interactions. Planta Med. 78:13992012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nelson-Rees WA and Flandermeyer RR: HeLa
cultures defined. Science. 191:96–98. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Capes-Davis A, Theodosopoulos G, Atkin I,
Drexler HG, Kohara A, MacLeod RA, Masters JR, Nakamura Y, Reid YA,
Reddel RR and Freshney RI: Check your cultures! A list of
cross-contaminated or misidentified cell lines. Int J Cancer.
127:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiu JG, Zhang YJ, Li Y, Zhao JM, Zhang WJ,
Jiang QW, Mei XL, Xue YQ, Qin WM, Yang Y, et al: Trametinib
modulates cancer multidrug resistance by targeting ABCB1
transporter. Oncotarget. 6:15494–15509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai Q, Sun H, Peng Y, Lu J,
Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R,
et al: A potent and orally active antagonist (SM-406/AT-406) of
multiple inhibitor of apoptosis proteins (IAPs) in clinical
development for cancer treatment. J Med Chem. 54:2714–2726. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jeong HU, Kong TY, Kwon SS, Hong SW, Yeon
SH, Choi JH, Lee JY, Cho YY and Lee HS: Effect of honokiol on
cytochrome P450 and UDP-glucuronosyltransferase enzyme activities
in human liver microsomes. Molecules. 18:10681–10693. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma S, Li X, Dong L, Zhu J, Zhang H and Jia
Y: Protective effect of Sheng-Mai Yin, a traditional Chinese
preparation, against doxorubicin-induced cardiac toxicity in rats.
BMC Complement Altern Med. 16:612016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wienkers LC and Heath TG: Predicting in
vivo drug interactions from in vitro drug discovery data. Nat Rev
Drug Discov. 4:825–833. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vtorushin SV, Khristenko KY, Zavyalova MV,
Perelmuter VM, Litviakov NV, Denisov EV, Dulesova AY and
Cherdyntseva NV: The phenomenon of multi-drug resistance in the
treatment of malignant tumors. Exp Oncol. 36:144–156.
2014.PubMed/NCBI
|
|
23
|
Shi Z, Liang YJ, Chen ZS, Wang XW, Wang
XH, Ding Y, Chen LM, Yang XP and Fu LW: Reversal of
MDR1/P-glycoprotein-mediated multidrug resistance by vector-based
RNA interference in vitro and in vivo. Cancer Biol Ther. 5:39–47.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ren HC, Sun JG, Wang GJ, A JY, Xie HT, Zha
WB, Yan B, Sun FZ, Hao HP, Gu SH, et al: Sensitive determination of
20(S)-protopanaxadiol in rat plasma using HPLC-APCI-MS: Application
of pharmacokinetic study in rats. J Pharm Biomed Anal.
48:1476–1480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hasegawa H: Proof of the mysterious
efficacy of ginseng: Basic and clinical trials: metabolic
activation of ginsenosid: Deglycosylation by intestinal bacteria
and esterification with fatty acid. J Pharmacol Sci. 95:153–157.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hao M, Zhao Y, Chen P, Huang H, Liu H,
Jiang H, Zhang R and Wang H: Structure-activity relationship and
substrate-dependent phenomena in effects of ginsenosides on
activities of drug-metabolizing P450 enzymes. PLoS One.
3:e26972008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lipshultz SE, Lipsitz SR, Mone SM, Goorin
AM, Sallan SE, Sanders SP, Orav EJ, Gelber RD and Colan SD: Female
sex and higher drug dose as risk factors for late cardiotoxic
effects of doxorubicin therapy for childhood cancer. N Engl J Med.
332:1738–1743. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lanzarini L, Bossi G, Laudisa ML, Klersy C
and Aricò M: Lack of clinically significant cardiac dysfunction
during intermediate dobutamine doses in long-term childhood cancer
survivors exposed to anthracyclines. Am Heart J. 140:315–323. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Silber JH and Barber G: ‘Increased risk of
cardiac dysfunction after anthracyclines in girls’. Med Pediatr
Oncol. 25:130–131. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sorensen K, Levitt G, Sebag-Montefiore D,
Bull C and Sullivan I: Cardiac function in Wilms' tumor survivors.
J Clin Oncol. 13:1546–1556. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakemi T, Ohtsuka N, Tomiyoshi Y and
Morito F: Sex difference in progression of adriamycin-induced
nephropathy in rats. Am J Nephrol. 16:540–547. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gray SL, Lackey BR, Tate PL, Riley MB and
Camper ND: Mycotoxins in root extracts of American and Asian
ginseng bind estrogen receptors alpha and beta. Exp Biol Med
(Maywood). 229:560–568. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee YN, Lee HY, Chung HY, Kim SI, Lee SK,
Park BC and Kim KW: In vitro induction of differentiation by
ginsenoides in F9 teratocarcinoma cells. Eur J Cancer. 32A:1–1428.
1996.
|
|
34
|
Lee YN, Lee HY, Lee YM, Chung HY, Kim SI,
Lee SK, Park BC and Kim KW: Involvement of glucocorticoid receptor
in the induction of differentiation by ginsenosides in F9
teratocarcinoma cells. J Steroid Biochem Mol Biol. 67:105–111.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee Y, Jin Y, Lim W, Ji S, Choi S, Jang S
and Lee S: A ginsenoside-Rh1, a component of ginseng saponin,
activates estrogen receptor in human breast carcinoma MCF-7 cells.
J Steroid Biochem Mol Biol. 84:463–468. 2003. View Article : Google Scholar : PubMed/NCBI
|