Introduction
Malignant glioma is the most common type of primary
central nervous system tumor, accounting for 80% all brain and
central nervous system tumors (1,2). Although
major advancements have been made in surgical treatment,
postoperative chemotherapy, and radiation, the prognosis for
patients with malignant gliomas remains poor, with an average
survival of roughly 1 year after diagnosis (3). According to Chinese glioma genome atlas
(CGGA) statistics, the overall survival (OS) time for glioblastoma
[GBM, World Health Organization (WHO) grade IV] patients is only
14.4 months, while the 6-month, 1-, 3- and 5-year OS rates were 87,
61, 15 and 9%, respectively (4). The
aggressiveness of glioma arises from the proliferation and invasion
capabilities of malignant glioma cells (5). Thus, controlling these characteristics
is a potential therapeutic strategy for glioma treatment.
Temozolomide (TMZ) is a prodrug that forms
O6-methylguanine (O6-MeG) adducts, causing cytotoxicity via
mismatch with deoxythymidine residues, inducing apoptosis following
processing by the mismatch repair system (6). TMZ is converted to its active compound
3-methyl-(triazen-1-yl) imidazole-4-carboxamide at a physiological
pH (7). The activated form of TMZ
readily crosses the blood-brain barrier, and is, therefore,
commonly used for the treatment of primary and secondary
intracranial neoplasms (7). Despite
the clinical success of TMZ, a large number of patients respond
poorly to this agent, at least in part due to intrinsic resistance
of tumor cells to damage-induced cell death (8,9).
Therefore, the identification of a novel combined strategy for
glioma treatment is required.
Gliomas are highly vascular and rich in vascular
endothelial growth factor (VEGF), which promotes angiogenesis.
Therefore, the anti-VEGF monoclonal antibody, bevacizumab, and
anti-VEGF receptor (VEGFR) tyrosine kinase inhibitors (TKIs),
including sunitinib and sorafenib, have been used for treatment of
patients with glioma (10). Apatinib
is an orally administered second-generation inhibitor of the
phosphorylation of the tyrosine residues within the intracellular
domain of VEGF receptor 2 (VEGFR2) (11). Apatinib prevents VEGF-induced
phosphorylation of VEGFR2 and the subsequent downstream signaling
responsible for the biological effects of VEGF (12,13).
Preclinical studies have demonstrated that apatinib exhibits
antitumor activity in patients with different types of malignant
tumors (14,15). In the present study, it was
investigated whether apatinib has therapeutic potential in glioma.
The inhibitory activity of apatinib on cell proliferation and
invasion was investigated in vitro using the cell counting
kit-8 assay, colony formation assay, Matrigel-based invasion assay
and wound-healing assay. The potential of the combination of
apatinib with TMZ for glioma therapy was also studied.
Materials and methods
Cell culture and treatment
The U251MG and U-87MG ATCC cell lines were purchased
from the American Type Culture Collection (ATCC) and cultured in
Dulbecco's modified Eagle's medium (DMEM) with nutrient mixture:
F12 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), without antibiotics. U-87MG ATCC cells were of
CNS origin and are likely to be a bonafide human glioblastoma cell
line considering their mRNA expression profile. Thus, U-87MG ATCC
cells can be used in glioma research and are distinct from U87
Uppsala cells established in 1968 at the University of Uppsala
(16). Cells were maintained at 37°C
in a humidified atmosphere containing 5% CO2. Apatinib
(cat. no. S2221) and TMZ (cat. no. S1237) were purchased from
Selleck Chemicals (Houston, Texas, USA) and used to treat U251MG
and U-87MG ATCC cells. A stock solution was prepared in dimethyl
sulfoxide at 10 mM and was stored at −20°C.
Western blot analysis
Cells were washed twice with ice-cold PBS and lysed
in ice-cold RIPA lysis buffer (Beyotime Institute of Biotechnology,
Beijing, China) containing a protease/phosphatase inhibitor
cocktail (Beyotime Institute of Biotechnology). The protein
concentration was determined by BCA assay (Thermo Fisher
Scientific, Inc.). Cell lysates were separated by 6 or 10% SDS-PAGE
[6% for VEGFR2, phospho- (p-)VEGFR2 and 10% for Akt, p-Akt,
extracellular signal-regulated kinase (Erk), p-Erk and GAPDH] and
transferred onto polyvinylidene fluoride membranes (Merck KGaA,
Darmstadt, Germany). After blocking with 5% non-fat milk in
Tris-buffered saline with Tween (TBST), the blots were incubated
with the following primary antibodies: P-VEGFR2 (dilution, 1:600;
cat. no. 2478), VEGFR2 (dilution, 1:1,000; cat. no. 9658), Akt
(dilution, 1:1,000; cat. no. 4691), p-Akt (dilution, 1:1,000; cat.
no. 4060), ERK (dilution, 1:1,000; cat. no. 4695), p-ERK (dilution,
1:1,000; cat. no. 4370) and GAPDH (dilution, 1:5,000, cat. no.
2118; all Cell Signaling Technology, Inc., Danvers MA, USA) at 4°C
for overnight. Next, the membraned were incubated with a
horseradish peroxidase-conjugated goat anti-mouse or rabbit
secondary IgG antibody (cat. nos. ZDR5305 and ZDR5307; 1:10,000;
OriGene Technologies, Inc., Beijing, China) at 37°C for 1 h.
Immunoreactive bands were visualized with a Immobilon ECL
chemiluminescent reagent (EMD Millipore, Billerica, MA, USA). The
band densities were quantified using ImagePro Plus (version 6.0;
Media Cybernetics, Inc., Rockville, MD, USA).
Cell viability assay
U251MG and U-87MG ATCC cells were plated in 96-well
plates at 1×103 cells/well), following treatment with 0, 5, 10 or
20 µM apatinib, and 20 µM TMZ as a previous study indicated
(17) treatment for 48 h, the
viability was determined using Cell Counting Kit-8 (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan). The absorbance was
measured at 450 nm and relative cell viability was calculated.
Colony formation assay
A 0.2-ml layer of 0.5% low-melt agarose was diluted
in complete growth medium (DMEM medium with 10% FBS, Gibco; Thermo
Fisher Scientific, Inc.) and plated in 6-well plates. A total of
1×103 U251MG or U-87MG ATCC cells were per well, with
was DMEM medium containing 10% FBS. Following 10 µM apatinib and 20
µM TMZ treatment for 12 days, the plate was fixed with 4%
paraformaldehyde and stained with 10% crystal violet (Beyotime
Institute of Biotechnology) at room temperature (from 22 to 25°C)
for 15 mins. Visible plots were defined as a colony and the number
of colonies in 3 separate wells were counted subsequent to scanned
by light stereo microscope (magnification, ×4).
Apoptosis detection assay
U251MG and U-87MG ATCC cells were plated in a 6-well
plate at 4×105 cells/well. Following 10 µM apatinib and 20 µM TMZ
treatment for 48 h, the cells were harvested and stained using a
Annexin V-FITC/PI Apoptosis Detection kit (Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China), according to the manufacturer's
protocol. A flow cytometer (Acea Biosciences Inc., Zhejiang, China)
was used to identify apoptotic cells and the apoptotic cells were
analyzed using De Novo Software (version 1.2.4; Acea Biosciences
Inc.).
Cell invasion assay
In vitro analysis of invasion was assessed using
Matrigel (BD Biosciences, Frankling Lakes, NJ, USA) and 8-µm
Transwell inserts (BD Biosciences). The cell invasion assay was
performed as previously described (18). Briefly, 50 µl Matrigel (diluted 1:5
with serum-free DMEM) was plated onto the Transwell insert. Then a
cell suspension of 5×103 U251MG or U-87MG ATCC cells was
added. Treatments of 10 µM apatinib and 20 µM TMZ were made for 24
h at 37°C. The invasive cells were fixed with 4% paraformaldehyde
(Beyotime Institute of Biotechnology) and stained with 10% crystal
violet (Beyotime Institute of Biotechnology) at room temperature
(from 22 to 25°C) for 15 mins. The invasive cells were photographed
by light microscopy (DTX500; Nikon Corporation, Tokyo, Japan) with
a magnification of ×40 and the cells in three random fields of view
were counted.
Wound-healing assay
U251MG and U-87MG ATCC cells were grown to 90%
confluency in 6-well plates. A wound was made by dragging a plastic
pipette tip across the cell surface. Phase contrast images of the
wounds were taken at 0 and 48 h of treatment with 10 µM apatinib
and 20 µM TMZ. Image J software (National Institutes of Health,
Bethesda, MD, USA) was used to evaluate the migration rate of
U251MG and U-87MG ATCC cells.
Statistical analysis
All data are presented as the mean ± standard
deviations. All experiments were performed ≥3 times independently.
Statistical analyses were performed using SPSS (version 19.0; IBM
Corp., Armonk, NY, USA). Student t-tests were performed to compare
differences between 2 groups. One-way analysis of variance followed
by Tukey's test was used for multiple comparisons analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
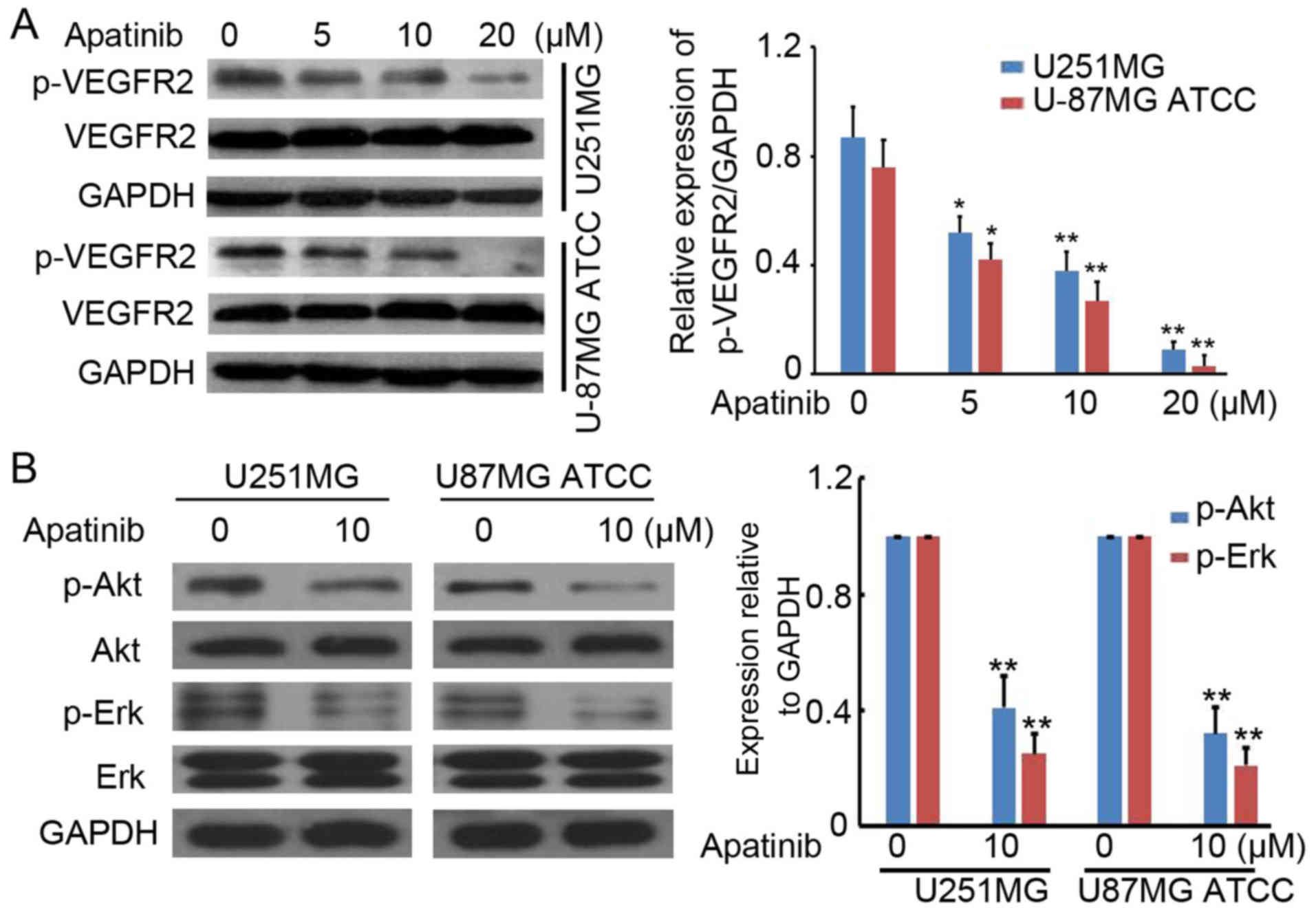
Apatinib suppresses activation of
VEGFR2 in glioma cells
To determine the potential role of apatinib in
glioma-cell proliferation, p53- and EGFR-mutated (U251MG) and
wild-type (U-87MG ATCC) cells were treated with 0, 5, 10 and 20 µM
apatinib. Western blotting was performed to detect p-VEGFR2 and
VEGFR2 protein expression. Apatinib significantly inhibited
p-VEGFR2 protein expression in a concentration-dependent manner in
U251MG and U-87MG ATCC cells (Fig.
1A). It was also suggested that the activation of downstream
targets of VEGFR2 signaling pathway (Akt and ERK) was also
inhibited by apatinib in glioma cells (Fig. 1B). These results confirm an inhibition
role of apatinib in VEGFR2-activation in glioma cells.
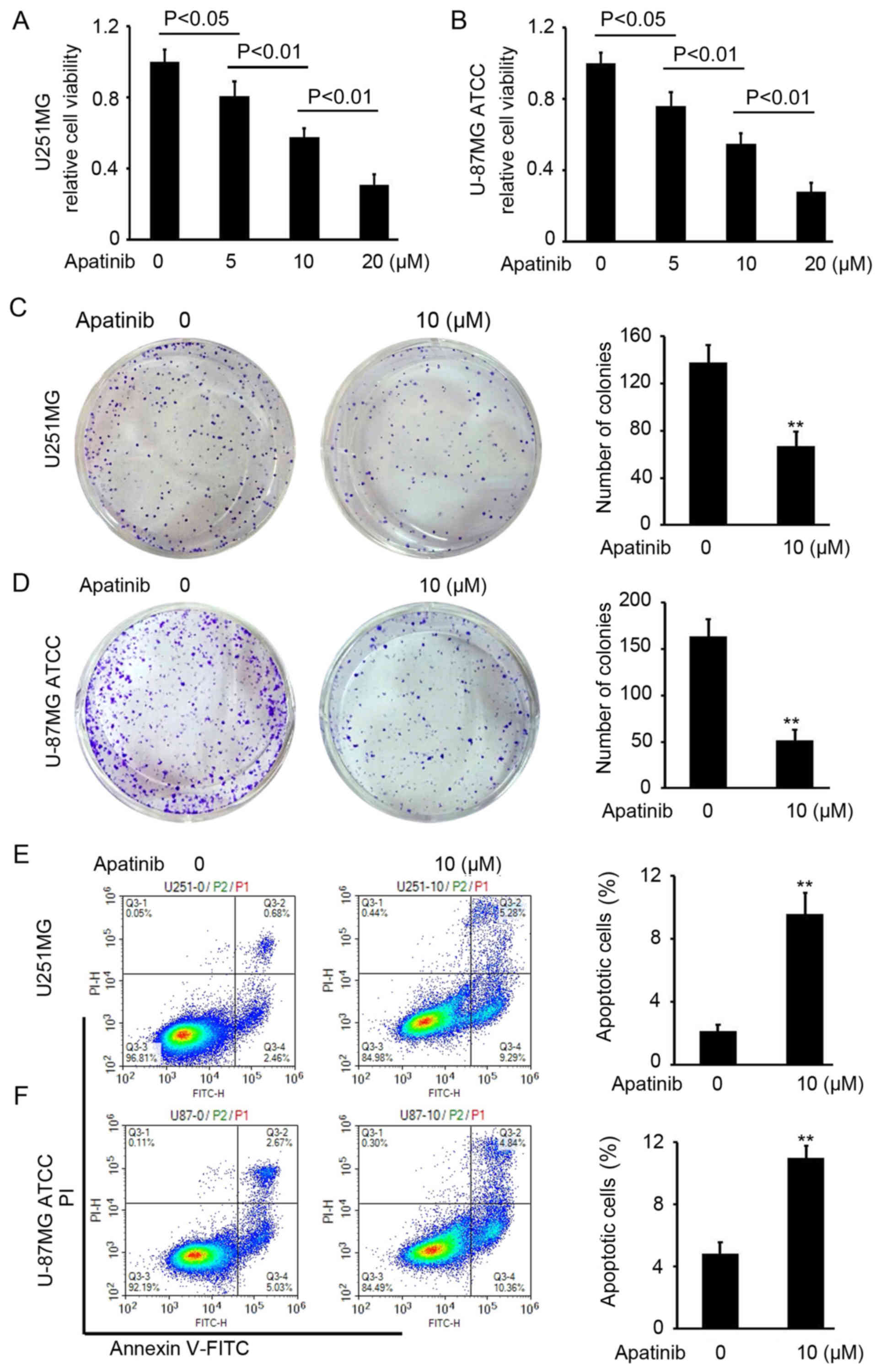
Apatinib suppresses cell proliferation
in glioma
To analyze the antitumor activity of apatinib in
glioma, a CCK-8 assay was performed to determine the cell viability
of U251MG and U-87MG ATCC cells following apatinib treatment at 0,
5, 10 and 20 µM for 48 h. Apatinib efficiently impaired U251MG and
U-87MG ATCC cell proliferation at concentrations of 10 and 20 µM,
an effect which was concentration-dependent (Fig. 1A and B). The IC50 of
apatinib was ~13 µM in U-87MG ATCC and U251MG cells. Therefore, a
concentration of 10 µM which was lower than IC50 of
apatinib was used for colony formation and apoptosis detection
assays. Inhibition of colony formation of U251MG and U-87MG ATCC
cells was also identified following apatinib treatment (P<0.01;
Fig. 2C and D). To determine the
mechanism underlying apatinib-mediated inhibition of glioma-cell
proliferation, flow cytometry was performed to investigate U251MG
and U-87MG ATCC-cell apoptosis with or without apatinib. The
results revealed an increased rate of apoptosis both in U251MG and
U-87MG ATCC cells treated with apatinib compared with untreated
cells (P<0.01; Fig. 2E and F).
These results suggest that apatinib inhibits glioma-cell
proliferation and colony formation through induction of
apoptosis.
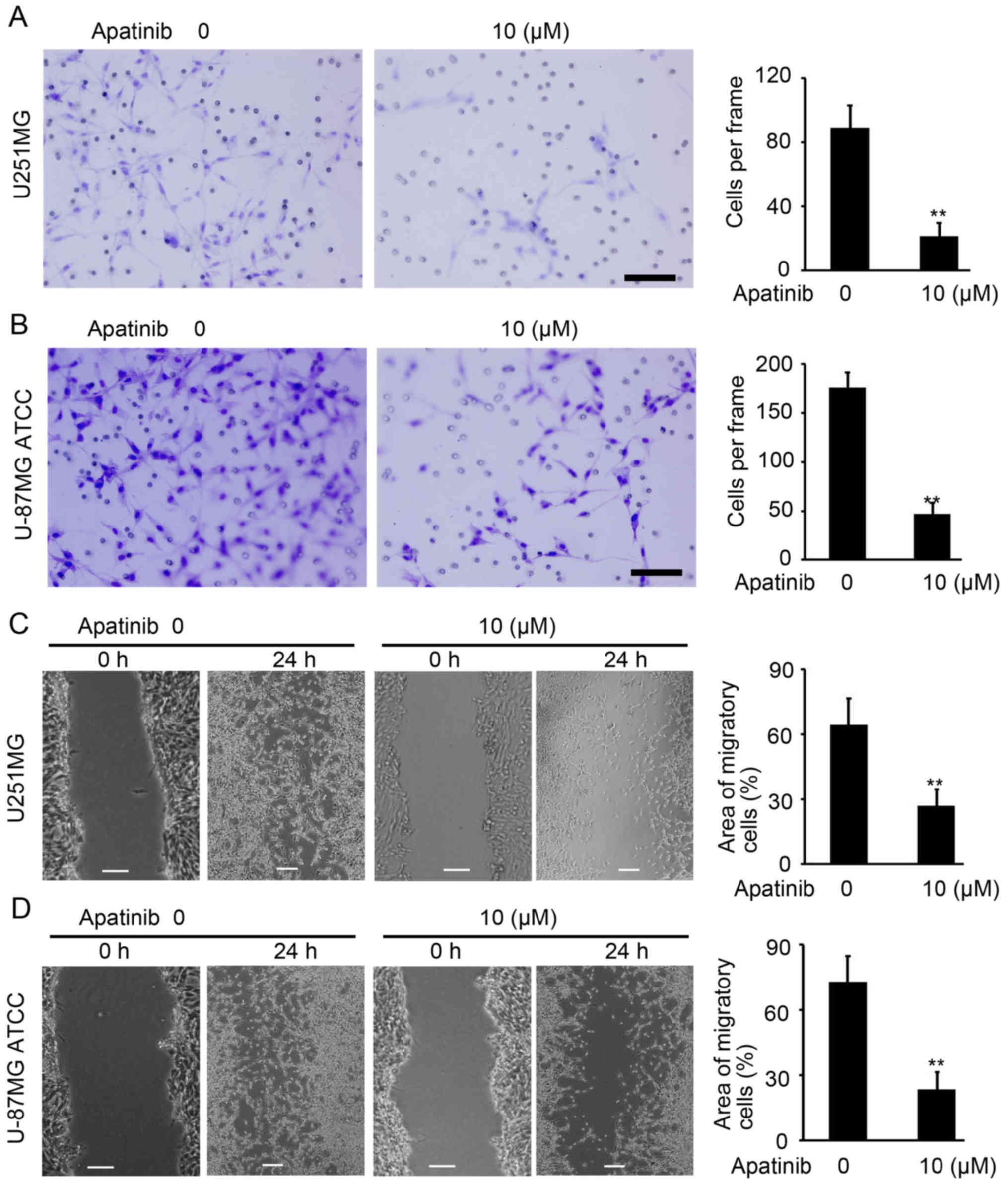
Apatinib suppresses cell invasion in
glioma
Invasion and wound-healing assays were performed to
investigate the invasion and migration abilities of U251MG and
U-87MG ATCC cells upon apatinib treatment. As demonstrated in
Fig. 3A and B, apatinib-treated
U251MG and U-87MG ATCC cells were less invasive than untreated
cells (P<0.01; Fig. 3A and B).
Furthermore, apatinib significantly inhibited the migratory ability
of U251MG and U-87MG ATCC cells compared with control (P<0.01;
Fig. 3C and D). Collectively, these
results demonstrate the inhibitory role of apatinib on cell
invasion in glioma.
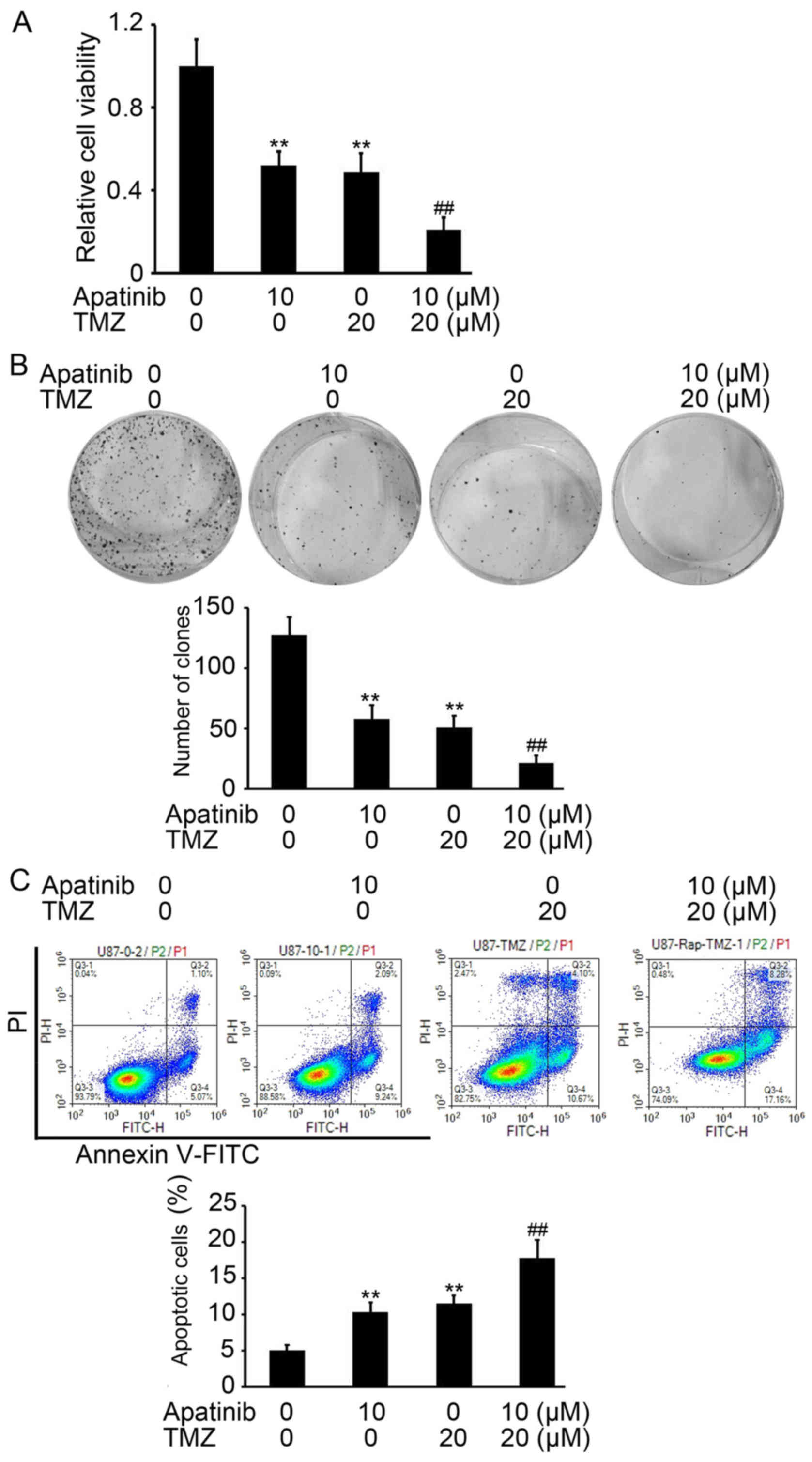
Apatinib promotes TMZ-mediated
inhibition of proliferation in glioma
Based on the above results, it was investigated
whether apatinib enhances TMZ-mediated inhibition of proliferation
of glioma cells. The cell viability of U-87MG ATCC cells after
treatment with apatinib with or without TMZ was determined by CCK-8
assay. TMZ was used at a concentration of 20 µM according to a
previous study by Nitta et al (17). Apatinib or TMZ treatments alone
efficiently inhibited proliferation of glioma cells, compared with
untreated cells (P<0.01; Fig. 4A).
However, in combination, apatinib significantly enhanced
TMZ-mediated inhibition of proliferation of U-87MG ATCC cells,
compared with cells treated with TMZ alone (P<0.01; Fig. 4A). Similar results were found in U251
cells (data not shown). A colony formation assay also demonstrated
the enhancing role of apatinib on TMZ-mediated antitumor activity
(P<0.01, compared with cells treated with TMZ alone; Fig. 4B). An increased proportion of
apoptotic U-87MG ATCC cells was also demonstrated in the group
treated with apatinib and TMZ compared with U-87MG ATCC cells
treated with TMZ alone (P<0.01; Fig.
4C). Collectively, these results indicate that apatinib
significantly improved the antitumor activity of TMZ in glioma.
Apatinib improves TMZ-mediated
inhibition of cell invasion in glioma
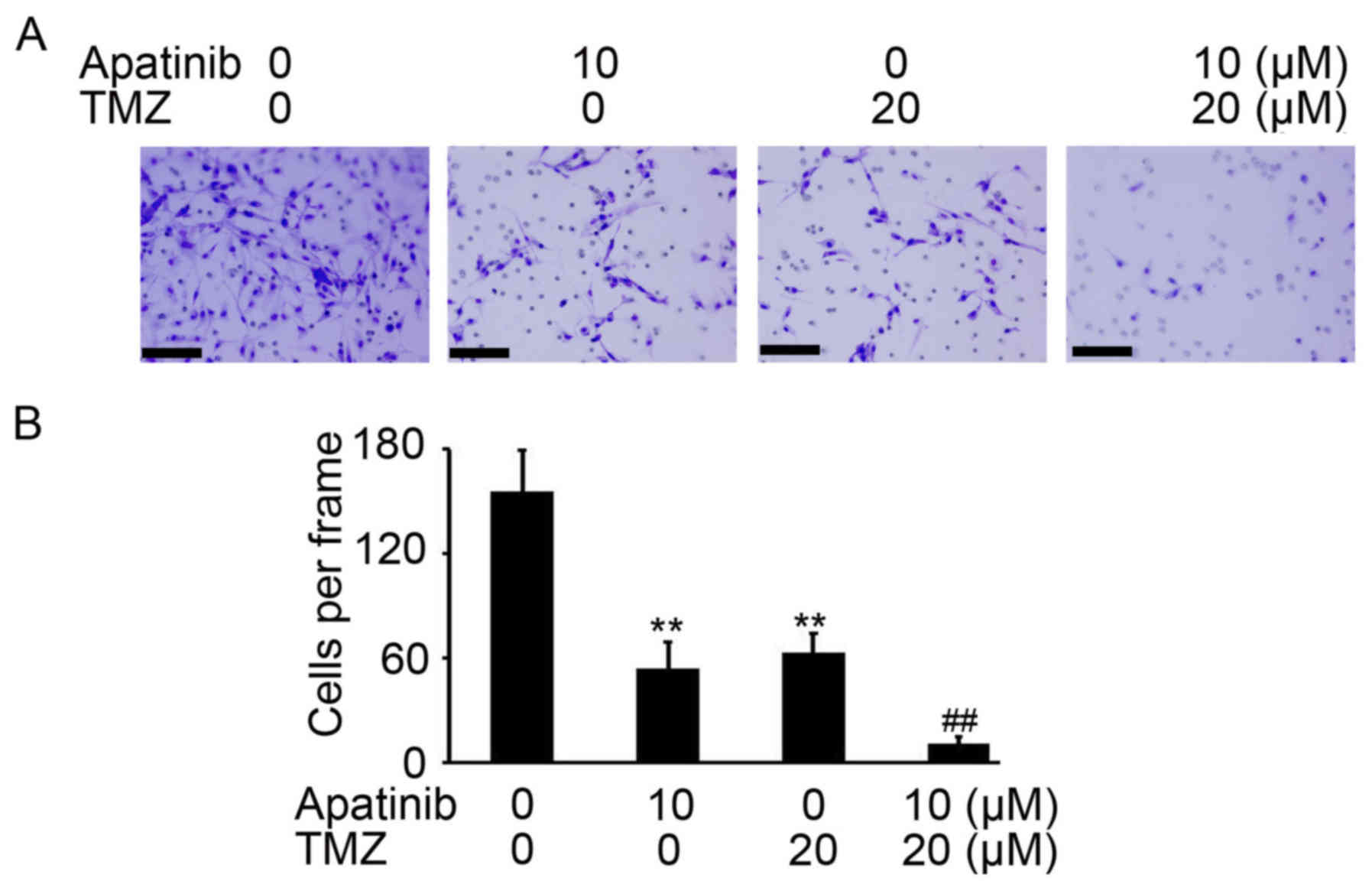
It was also investigated whether apatinib enhances
TMZ-mediated inhibition invasion of glioma cells. As demonstrated
Fig. 5A, a reduced number of invasive
U-87MG ATCC cells were counted in the groups treated with apatinib
and TMZ compared with control cells (P<0.01; Fig. 5A and B). Collectively, these results
indicate that apatinib significantly enhanced the antitumor
activity of TMZ in glioma cancer cells.
Discussion
Seeing as glioma is hypervascular in nature,
sunitinib and sorafenib have been used to control abnormal
vasculature in clinical trials (https://clinicaltrials.gov) (10). However, internal resistance restricts
the use of first-generation inhibitors of the VEGF-VEGFR pathway
(19). Previous clinical studies have
indicated the efficiency of apatinib treatment in recurrent glioma
and that apatinib can transcend the brain-blood barrier (11,20).
However, its mechanism of action remained unclear. In the present
study, antitumor activity was observed in apatinib-treated glioma
cells (significant inhibition of cell proliferation and invasion).
Apatinib was also demonstrated to improve the antitumor activity of
TMZ in glioma cells. All experiments were performed using two
glioma cell lines, which differ in mutation status. Collectively,
the present study suggests that apatinib may provide a novel
therapeutic strategy for glioma, particularly in combination with
TMZ.
VEGFR2 is expressed in endothelial cells and
upregulated in tumor vasculature (21). Phosphorylation of VEGFR2 by autocrine
or paracrine VEGF has been demonstrated to activate the ERK, Akt,
FAK, and MAPK pathways, which regulate cell proliferation and
migration (21). Seeing as VEGFR2 is
highly expressed in glioma cells (22), it is hypothesized that it serves a
critical role in invasion and proliferation. The VEGFR2 pathway
tyrosine kinase inhibitors (TKIs), sunitinib and sorafenib, have
been previously used to treat patients with glioma (11). In the present study, it was
demonstrated that VEGFR2 is highly expressed in glioma cells, and
apatinib was revealed to inhibit p-VEGFR2, p-Akt and p-ERK protein
expression in U251MG and U-87MG ATCC cells in a
concentration-dependent manner. Apatinib also significantly
inhibited glioma cell proliferation and colony formation, and
induced cell apoptosis in p53- and EGFR-mutated U251MG and
wild-type U-87MG ATCC cells. Furthermore, inhibition of glioma-cell
invasion and migration of glioma cells was observed in
apatinib-treated glioma cells. These results provide evidence of
the antitumor activity of apatinib in glioma cells. However,
further in vivo studies are required to confirm this
antitumor activity of apatinib.
Acquired resistance to temozolomide remains a
limitation in the treatment of glioblastoma; approximately 90%
recurrent glioblastoma cases are resistant to additional TMZ
therapy (23). Type-selective
endothelin receptor antagonists combined with chemotherapy have
been investigated as a therapeutic strategy to improve clinical
outcomes for patients with glioma (24). Macitentan, a dual endothelin receptor
antagonist, combined with temozolomide treatment is well-tolerated,
produces a durable response, and warrants clinical evaluation in
glioblastoma (25). Treatment of
glioblastoma cells with ER stress-inducing drugs has been
demonstrated to induce drug-sensitization of glioma cells to TMZ
through downregulation of O-6-methylguanine-DNA methyltransferase,
N-methylpurine DNA glycosylase and Rad51 recombinase (26). The present study suggests that the
addition of apatinib to TMZ may enhance the effect of chemotherapy.
Treatment of glioma cells with apatinib or TMZ alone resulted
increased apoptosis and decreased proliferation and invasion
compared with untreated cells. Notably, the combination of apatinib
and TMZ resulted in enhanced inhibition of cell proliferation and
invasion compared with TMZ treatment alone. These findings
demonstrated a synergistic antitumor effect of apatinib and TMZ in
glioma cells and may explain the clinical benefits observed
following combination treatment of recurrent glioma (24). Further in vivo studies are
required to demonstrate the synergistic antitumor effect of
apatinib and TMZ in glioma.
Previous studies have demonstrated enhanced
activation of Akt and ERK in TMZ-resistant glioma cells (27,28).
Overexpression of Akt suppressed the enhanced cytotoxic effect of
TMZ mediated by SRC-silencing (29).
Treatment with the estrogen receptor-β agonist (liquiritigenin) was
demonstrated to enhance TMZ-sensitivity of glioma cells by
inhibiting PI3K/Akt/mTOR pathway (30). TGF-β1-dependent activation of SMAD/ERK
signaling is involved in connective tissue growth factor-mediated
TMZ-resistance in glioma (31).
Targeting of the MEK-ERK-MDM2-p53 signaling pathway in combination
with TMZ has been suggested to be a novel and promising therapeutic
strategy in the treatment of glioblastoma (32). In the present study, inhibition of
Akt/ERK activation by apatinib was demonstrated in glioma cells,
which may contribute to the synergistic antitumor effect of
apatinib and TMZ in glioma.
In summary, apatinib demonstrated efficient
antitumor activity and enhanced the effect of TMZ in glioma cells,
which was associated with decreased cell proliferation, colony
formation, invasion and migration, and increased cell apoptosis.
This antitumor activity of apatinib was observed in p53- and
EGFR-mutated cells, as well as wild-type cells. These results
provide a basis for the clinical use of apatinib in glioma
treatment. Further in vivo studies are required to
characterize the clinical antitumor activity of apatinib in
glioma.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81602186) and the
China Postdoctoral Science Foundation (grant no. 2017M612216).
Availability of data and materials
All of the datasets generated/analyzed in the
present study are included in the published manuscript.
Authors' contributions
CW, MJ, XZ, HH, QL and ZY contributed to data
acquisition, the analysis and interpretation of data, were involved
in drafting and writing the manuscript, revising it critically for
important intellectual content and gave the final approval of the
version to be published. ZY and XZ were accountable for design of
this study and all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang Y, Rajappa P, Hu W, Hoffman C, Cisse
B, Kim JH, Gorge E, Yanowitch R, Cope W, Vartanian E, et al: A
proangiogenic signaling axis in myeloid cells promotes malignant
progression of glioma. J Clin Invest. 127:1826–1838. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wick W, Weller M, van den Bent M, Sanson
M, Weiler M, von Deimling A, Plass C, Hegi M, Platten M and
Reifenberger G: MGMT testing-the challenges for biomarker-based
glioma treatment. Nat Rev Neurol. 10:372–385. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watkins S and Sontheimer H: Unique biology
of gliomas: Challenges and opportunities. Trends Neurosci.
35:546–556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang T, Mao Y, Ma W, Mao Q, You Y, Yang
X, Jiang C, Kang C, Li X, Chen L, et al: CGCG clinical practice
guidelines for the management of adult diffuse gliomas. Cancer
Lett. 375:263–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Skalsky RL and Cullen BR: Reduced
expression of brain-enriched microRNAs in glioblastomas permits
targeted regulation of a cell death gene. PLoS One. 6:e242482011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ji Y, Vogel RI and Lou E: Temozolomide
treatment of pituitary carcinomas and atypical adenomas: Systematic
review of case reports. Neurooncol Pract. 3:188–195.
2016.PubMed/NCBI
|
|
7
|
Newlands ES, Stevens MF, Wedge SR,
Wheelhouse RT and Brock C: Temozolomide: A review of its discovery,
chemical properties, pre-clinical development and clinical trials.
Cancer Treat Rev. 23:35–61. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chan JA, Stuart K, Earle CC, Clark JW,
Bhargava P, Miksad R, Blaszkowsky L, Enzinger PC, Meyerhardt JA,
Zheng H, et al: Prospective study of bevacizumab plus temozolomide
in patients with advanced neuroendocrine tumors. J Clin Oncol.
30:2963–2968. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gilbert MR, Wang M, Aldape KD, Stupp R,
Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT,
et al: Dose-dense temozolomide for newly diagnosed glioblastoma: A
randomized phase III clinical trial. J Clin Oncol. 31:4085–4091.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khasraw M, Simeonovic M and Grommes C:
Bevacizumab for the treatment of high-grade glioma. Expert Opin
Biol Ther. 12:1101–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen JH, Yu GF, Jin SY, Zhang WH, Lei DX,
Zhou SL and Song XR: Activation of α2 adrenoceptor attenuates
lipopolysaccharide-induced hepatic injury. Int J Clin Exp Pathol.
8:10752–10759. 2015.PubMed/NCBI
|
|
12
|
Peng H, Zhang Q, Li J, Zhang N, Hua Y, Xu
L, Deng Y, Lai J, Peng Z, Peng B, et al: Apatinib inhibits VEGF
signaling and promotes apoptosis in intrahepatic
cholangiocarcinoma. Oncotarget. 7:17220–17229. 2016.PubMed/NCBI
|
|
13
|
Kim KL and Suh W: Apatinib, an inhibitor
of vascular endothelial growth factor receptor 2, suppresses
pathologic ocular neovascularization in mice. Invest Ophthalmol Vis
Sci. 58:3592–3599. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou N, Liu C, Hou H, Zhang C, Liu D, Wang
G, Liu K, Zhu J, Lv H, Li T and Zhang X: Response to apatinib in
chemotherapy-failed advanced spindle cell breast carcinoma.
Oncotarget. 7:72373–72379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang L, Wei Y, Shen S, Shi Q, Bai J, Li
J, Qin S, Yu H and Chen F: Therapeutic effect of apatinib on
overall survival is mediated by prolonged progression-free survival
in advanced gastric cancer patients. Oncotarget. 8:29346–29354.
2017.PubMed/NCBI
|
|
16
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re3532016. View Article : Google Scholar
|
|
17
|
Nitta Y, Shimizu S, Shishido-Hara Y,
Suzuki K, Shiokawa Y and Nagane M: Nimotuzumab enhances
temozolomide-induced growth suppression of glioma cells expressing
mutant EGFR in vivo. Cancer Med. 5:486–499. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dai L, Cui X, Zhang X, Cheng L, Liu Y,
Yang Y, Fan P, Wang Q, Lin Y, Zhang J, et al: SARI inhibits
angiogenesis and tumour growth of human colon cancer through
directly targeting ceruloplasmin. Nat Commun. 7:119962016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dietrich J, Norden AD and Wen PY: Emerging
antiangiogenic treatments for gliomas-efficacy and safety issues.
Curr Opin Neurol. 21:736–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu X, Zhang J, Xu B, Jiang Z, Ragaz J,
Tong Z, Zhang Q, Wang X, Feng J, Pang D, et al: Multicenter phase
II study of apatinib, a novel VEGFR inhibitor in heavily pretreated
patients with metastatic triple-negative breast cancer. Int J
Cancer. 135:1961–1969. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fontanella C, Ongaro E, Bolzonello S,
Guardascione M, Fasola G and Aprile G: Clinical advances in the
development of novel VEGFR2 inhibitors. Ann Transl Med.
2:1232014.PubMed/NCBI
|
|
22
|
Shankar A, Jain M, Lim MJ, Angara K, Zeng
P, Arbab SA, Iskander A, Ara R, Arbab AS and Achyut BR: Anti-VEGFR2
driven nuclear translocation of VEGFR2 and acquired malignant
hallmarks are mutation dependent in glioblastoma. J Cancer Sci
Ther. 8:172–178. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alshami J, Guiot MC, Owen S, Kavan P,
Gibson N, Solca F, Cseh A, Reardon DA and Muanza T: Afatinib, an
irreversible ErbB family blocker, with protracted temozolomide in
recurrent glioblastoma: A case report. Oncotarget. 6:34030–34037.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim SJ, Lee HJ, Kim MS, Choi HJ, He J, Wu
Q, Aldape K, Weinberg JS, Yung WK, Conrad CA, et al: Macitentan, a
dual endothelin receptor antagonist, in combination with
temozolomide leads to glioblastoma regression and long-term
survival in mice. Clin Cancer Res. 21:4630–4641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xipell E, Aragón T, Martinez-Velez N, Vera
B, Idoate MA, Martinez-Irujo JJ, Garzón AG, Gonzalez-Huarriz M,
Acanda AM, Jones C, et al: Endoplasmic reticulum stress-inducing
drugs sensitize glioma cells to temozolomide through downregulation
of MGMT, MPG, and Rad51. Neuro Oncol. 18:1109–1119. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han S, Li Z, Master LM, Master ZW and Wu
A: Exogenous IGFBP-2 promotes proliferation, invasion, and
chemoresistance to temozolomide in glioma cells via the integrin
β1-ERK pathway. Br J Cancer. 111:1400–1409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vo VA, Lee JW, Lee HJ, Chun W, Lim SY and
Kim SS: Inhibition of JNK potentiates temozolomide-induced
cytotoxicity in U87MG glioblastoma cells via suppression of Akt
phosphorylation. Anticancer Res. 34:5509–5515. 2014.PubMed/NCBI
|
|
29
|
Wang Z, Sun J, Li X, Yang S, Yue S, Zhang
J, Yang X, Zhu T, Jiang R and Yang W: Downregulation of Src
enhances the cytotoxic effect of temozolomide through AKT in
glioma. Oncol Rep. 29:1395–1398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu X, Wang L, Chen J, Ling Q, Wang H, Li
S, Li L, Yang S, Xia M and Jing L: Estrogen receptor β agonist
enhances temozolomide sensitivity of glioma cells by inhibiting
PI3K/AKT/mTOR pathway. Mol Med Rep. 11:1516–1522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zeng H, Yang Z, Xu N, Liu B, Fu Z, Lian C
and Guo H: Connective tissue growth factor promotes temozolomide
resistance in glioblastoma through TGF-β1-dependent activation of
Smad/ERK signaling. Cell Death Dis. 8:e28852017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato A, Sunayama J, Matsuda K, Seino S,
Suzuki K, Watanabe E, Tachibana K, Tomiyama A, Kayama T and
Kitanaka C: MEK-ERK signaling dictates DNA-repair gene MGMT
expression and temozolomide resistance of stem-like glioblastoma
cells via the MDM2-p53 axis. Stem Cells. 29:1942–1951. 2011.
View Article : Google Scholar : PubMed/NCBI
|