Introduction
Lièvre et al (1) first disclosed colorectal cancer (CRC)
with KRAS mutation as a predictor of poor response to
anti-epidermal growth factor receptor (anti-EGFR) at 2006. The OPUS
trial (2) in 2008 and the CRYSTAL
trial (3) in 2009 reported KRAS
mutations occured at a frequency of 42 and 36.5% among metastatic
CRCs. Those patients were insensitive to anti-EGFR therapy.
Furthermore, BRAF mutations in CRCs were reported at a rate of 8.7%
(4) and a 10% (5). The evidence identities that ~50% of CRCs
exhibit no response to anti-EGFR therapy, including cetuximab or
panitumumab (1–11). KRAS mutations occur at an increased
frequency, compared with BRAF mutations, and commonly occur at
codon 12 (G12D) and 13 (G13D) of exon 2 in the KRAS gene (12–16);
whereas, 90% of BRAF mutations occur in exon 15 (V600E) (17–19).
According to previous studies, reduced expression of microRNA-378
(miR-378) may serve a crucial role in CRC, which is considered as
an independent prognostic factor, and inhibits cell growth as well
as invasion in tumor cells (20–23). It is
known that miR-378 acts as an inhibitor in the mitogen-activated
protein kinase (MAPK) pathway, which affect extracellular
signal-regulated kinase (ERK) genes, such as ERK1/2; therefore, it
is involved in cellular proliferation, differentiation, and
transcription regulation and development (24). In our previous study, a reduced
expression level of miR-378 was commonly observed in KRAS- or
BRAF-mutant cells, compared with the wild-type CRC or normal
control cells; however, following transfection of miR-378 into
mutant CRC cells, to increase the expression level, it was observed
that drug sensitivity to cetuximab was significantly restored and
cell death was induced (25). The
present data coincided with information from the databases
TargetScanHuman (www.targetscan.org) and miRbase (www.mirbase.org), and further confirmed that miR-378
targets the 3′-untranslated region (UTR) of the ERK1/2 coding gene.
Feng et al (26) also
demonstrated that miR-378 suppressed the antigrowth protein
transducer of Erb-B2 receptor tyrosine kinase, which serves as a
transcriptional repressor of cyclin D1, a downstream effector of
the human epidermal growth factor receptor 2-Ras-ERK pathway. The
precursors of miR-378/378* are derived from the first intron of
host gene peroxisome proliferator-activated receptor γ coactivator
1 β (PGC-1β) (27). Fatty acids can
directly stimulate the gene PGC-1β expression, and as a result
increase the co-expression of miR-378, which was demonstrated by
our previous study (25).
Furthermore, a previous study indicated that PGC-1β
serves a function in lipid metabolism, in which the genes coding
mitochondrial fatty acid oxidation and oxidative phosphorylation
were diminished in liver specific-PGC-1β knock out mice (28). A number of miR-378/378* target genes
are associated with lipometabolism, including carnitine
O-acetyltransferase, mediator complex subunit 13 and glucose
transporter type 4 genes, and may also affect the development of
lipogenesis in fatty cells (27,29).
Additionally, a number of studies demonstrated that fatty acids
could significantly upregulate the expression of the PGC-1β gene,
in order to affect the metabolism of mitochondrial biogenesis
(27,28,30).
EPA is one of omega-3 fatty acids commonly found in
fish, including cod liver oil, salmons, herrings, sardines and
various edible seaweeds. Based on a report by the European Food
Safety Authority, a suggested dosage of intake for adults of
EPA/docosahexaenoic acid (DHA) is 200 to 600 mg per day, and 40 to
250 mg/day for infants >6 months old, children and adolescents
(31). Additionally, there are
0.2–1.2 mM free fatty acids in the human body (32,33) which
provided an estimate of the EPA concentration selection in current
study. A number of studies demonstrated that EPA and DHA can
trigger the majority of the activities of the caspase family
members, including caspase-8, which are associated with proteases
and cell apoptosis, which has been indicated in CRC and pancreatic
cancer cells (34,35). Notably, it has been observed that
neoplastic oral keratinocyte cells are significantly suppressed by
EPA through the inhibition of the expression of total protein
ERK1/2, which increased the ERK1/2 phosphorylation (36). Based on our previous study, following
restoring the expression level of miR-378, the CRC cells have a
significant response to EGFR inhibitor cetuximab (25). In the present study, the aim was to
firstly uncover the mechanism underlying the association between
miR-378 and the MAPK pathway. Secondly, whether the gene expression
of PGC-1β was also induced by EPA was investigated, with this gene
indirectly increasing miR-378 co-expression and restoring cetuximab
sensitivity. The results may directly benefit the treatment of
patients with CRC containing KRAS or BRAF mutations.
Materials and methods
Cell lines and cell culture
The CRC cell lines included in the present study
were SW480, HCT116, HT29, and Caco-2. SW480 and HCT116 contain KRAS
mutations (G5571T and G5574A, respectively), HT29 contains a BRAF
mutation (T171429A) and Caco-2 is a wild-type (without gene KRAS or
BRAF mutations) CRC cell line, which was used as the internal
control in the experiments, when required. All cell lines were
cultured according to our previous study (25). In brief, the cells were cultured in
high glucose Dulbecco's modified Eagle's medium (DMEM) containing 4
mM glutamine, penicillin (12.5 U/ml), streptomycin (6 µg/ml), 1 mM
sodium pyruvate and 10% fetal bovine serum (all from Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), then incubated at 37°C
in an atmosphere containing 5% CO2. SW480, HCT116, HT29
and Caco-2 were selected in the present study due to them
exhibiting aggressive growth, compared with other cell lines.
Supplements and reagents
Ultra-pure free fatty acid of EPA (>99%) was
purchased from Nu-Chek-Prep, Inc. (Elysian, MN, USA). The stock
solution was diluted in 99% ethanol to a concentration of 1 mM and
aliquoted in dark-colored glass vials stored at −20°C until used.
EPA was added to DMEM to produce different testing concentrations
from 0–40 µM (0, 2, 5, 10, 20, 30 and 40 µM), and was then used to
treat the all cell lines for 24 h. at 37°C. The anti-EGFR cetuximab
(Erbitux®) was purchased from Merck KGaA (Darmstadt,
Germany) and was added into the DMEM to produce final testing
concentrations from 0–0.2 µM (0, 0.01, 0.05, 0.1 and 0.2 µM), and
then incubated with all cell lines at 37°C for 48 h. The optimal
EPA concentration was determined according to the IC50
calculation, for which growth inhibition was observed in half of
the tested cells, and it was determined as 40 µM.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
The primer sequences were as follows: MiR-378,
5′-CTCAACTGGTGTCGTGGAGT-3′ and 5′-GGGACTGGACTTGGAGTC-3′ (37); RNU44, 5′-CCTGGATGATGATAGCAAATGC-3′ and
5′-GAGCTAATTAAGACCTTCATGTT-3′ (38).
The miR-378 expression levels in cells were detected prior to
treatment, and then after 24 h treatment with 40 µM EPA/DMEM. RNA
extraction with MirVana™ miRNA Isolation kit (Ambion;
Thermo Fisher Scientific, Inc.), An optical density of 260/280 nm
absorbance ratio between 1.9–2.0 confirmed the quality of RNA
(BioPhotometer plus; Eppendorf, Hamburg, Germany).
TaqMan® MicroRNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) was used to transcript
RNA to cDNA-according to the manufacturer's protocols. In brief,
the thermocycling conditions were: 5 min at 85°C; 5 min at 60°C;
and lowered to 4°C which was immediately transferred to qPCR; or
long term storage at −20°C. RT-qPCR of miR-378 was performed with
the TaqMan Universal Master Mix II (no UNG) (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and TaqMan® MicroRNA
Assays kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) was
used for qPCR. The thermocycling conditions were: 10 min at 95°C
for the first stage; 15 sec at 95°C for the second stage; and then
1 min at 60°C; and total reaction for 40 cycles at 25°C for 30 sec.
The relative expression levels of targeted genes in cells were
calculated by normalizing with RNU-44 expression levels through the
comparative Cq method (39). Finally,
collected data were analyzed using the BioRad iCycle iQ system
software, version 3.1 (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Comparing the cell viability
efficiency using an MTS assay
All cell lines were grouped into four sets based on
different treatments: Original cells treated with 1X PBS; cells
treated with EPA only; cells treated with cetuximab only; and cells
treated with EPA and cetuximab. CRC cells were firstly treated in
gradient concentrations of EPA (0, 2, 5, 10, 20, 30, 40 µM)/DMEM
mixtures, and incubated separately using a 96-microplate (BD
Biosciences, Franklin Lakes, NJ, USA) at 37°C for 24 h, followed by
treating with grading concentrations of cetuximab (0, 0.01, 0.05,
0.1, 0.20 µM) at 37°C for 48 h. The cell viability was assessed
using the CellTiter 96® AQueous One Solution Cell
Proliferation Assay kit according to the manufacturer's protocols
(Promega Corporation, Madison, WI, USA), and the absorbance was
measured at 490 nm.
ERK1/2 expression level and
phosphorylation status detection by an ELISA assay
To observe the MAPK pathway, the expression level of
key protein ERK1/2 was determined. Following all cell lines being
treated with 40 µM EPA at 37°C for 24 h, ERK1/2 (Total/Phospho)
InstantOne™ ELISA kit (cat. no. ABIN1981832;
eBioscience; Thermo Fisher Scientific, Inc.), was used to detect
total ERK1/2 protein expression and phosphorylation status
(p-ERK1/2). In brief, the manufacturer's protocols of the ERK1/2
(Total/Phospho) InstantOne ELISA kit were followed with, and the
absorbance was measured at 450 nm (Fig.
2).
Statistical analysis
SPSS 15.0 statistics software (SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis in the present study.
Data are expressed as the mean ± standard error of the mean and
represent the fold differences among cells treated with EPA only,
cells treated with cetuximab only and cells treated with EPA and
cetuximab. Firstly, all three groups of cells were compared to
their original cells, which were normalized at a base line of 100%.
Secondly, to understand the efficiency of cetuximab with/without
EPA, EPA-treated cells were further normalized and used as a base
line for comparison. Comparisons within each cell group was
assessed by one-way analysis of variance. Individual comparisons
among subgroups were analyzed with Tukey's post-hoc test. The
miR-378, ERK1/2 protein and p-ERK1/2 expression levels prior to and
following treatment with EPA were analyzed with Student's t test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
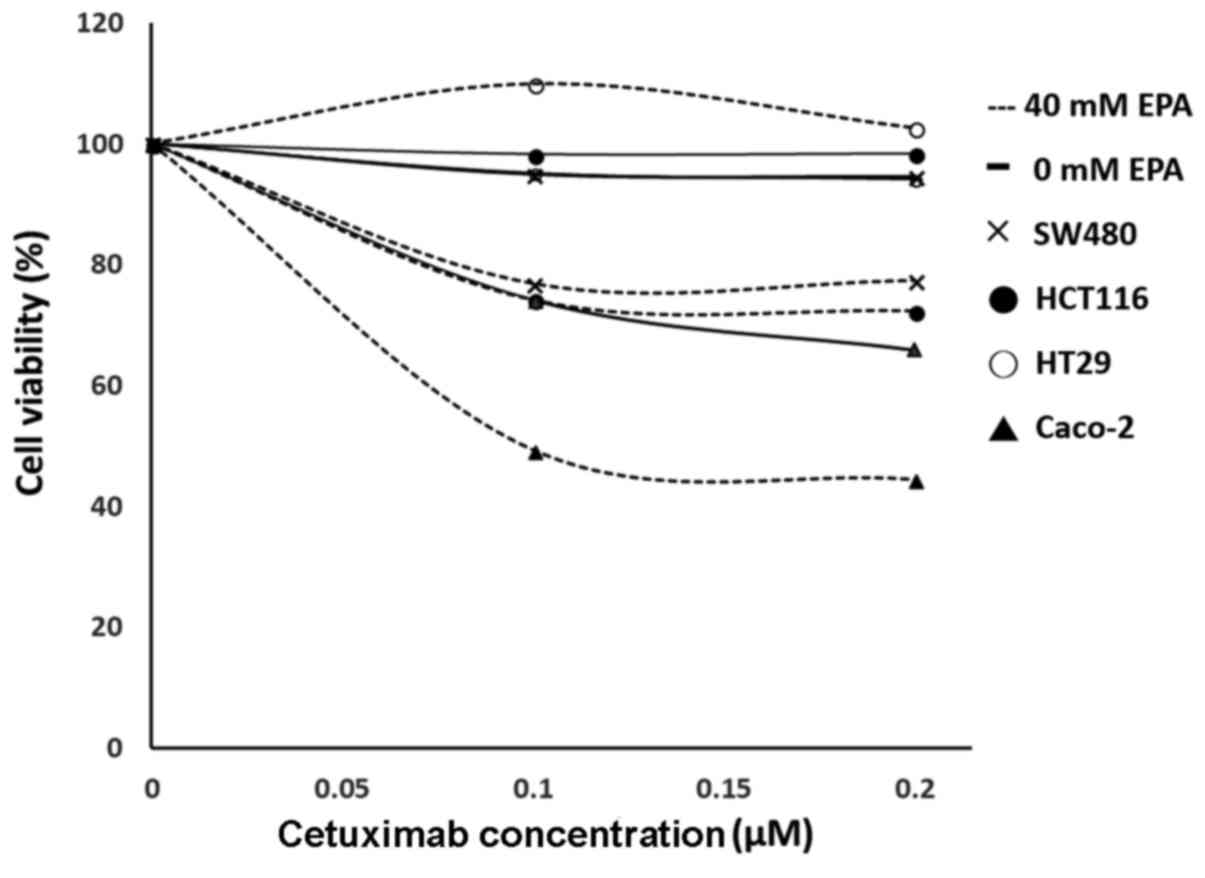
Efficiency of EPA on the viability of
CRC cells viability
Different concentrations of EPA (0, 2, 5, 10, 20, 30
and 40 µM) were produced by mixing with DMEM and used to treat all
cells lines. The results demonstrated that Caco-2 and SW480 cells
remained more stable when the EPA concentration increased, compared
with HCT116 and HT-29 cells. The cell viabilities indicated a range
between 95.9–111.5% and 98.0–112.0%, respectively. The cell lines
HCT116 and HT29 that were treated with 0–20 µM EPA had cell
viability ranges of 95.7–102.7%, and 93.4–102.0%, respectively;
however, when the concentration increased to 30–40 µM EPA, the cell
viabilities decreased to 71.9–53.3%, and 81.3–79.0%, respectively.
Notably, cells treated with 40 µM EPA had the greatest dosage
response to cetuximab.
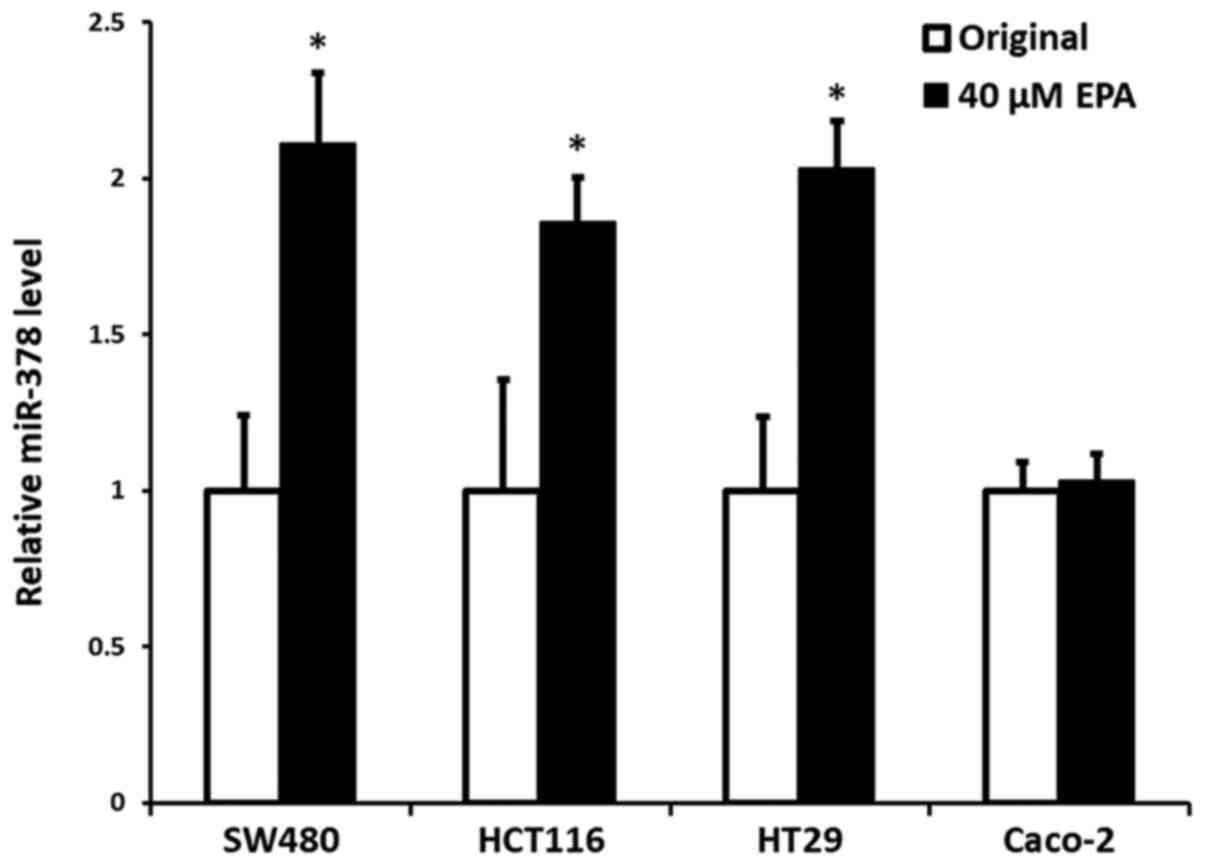
Efficiency of EPA on the expression of
miR-378
Our previous data (24) demonstrated that mutant cells, either
KRAS or BRAF, had significantly reduced expression of miR-378,
compared with wild-type CRC cells; however, the present data
indicated that the cells with an increased expression of miR-378
were notably associated with the addition of EPA, particularly at
the concentration of 40 µM, with the exception of Caco-2 wild-type
cells. The results indicated a significant increase in expression
of miR-378 in the cells of SW480, HCT116 and HT29, with an increase
by 0.98- (P=0.005), 0.88- (P=0.016) and 1.05-fold (P=0.004),
respectively, compared with their original control cells.
Contrarily, miR-378 expression in wild-type Caco-2 cells did not
demonstrate a significant difference to its original control cells
(P=0.317) (Fig. 1).
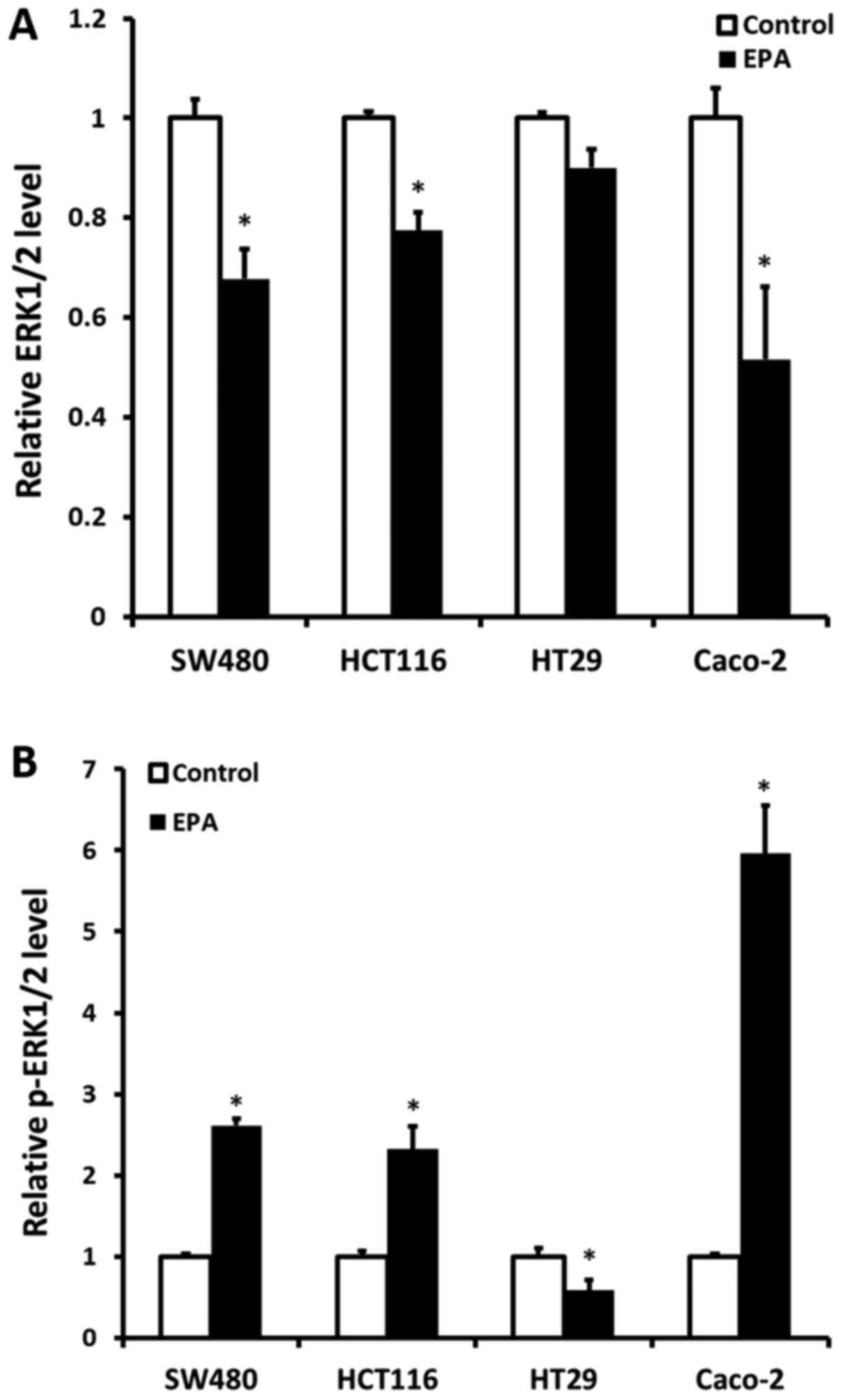
Cell ERK1/2 protein expression and
p-ERK1/2 status following treatment with 40 µM EPA
ERK1/2 protein expression and the p-ERK1/2 status
were detected following all CRC cell lines being treated with 40 µM
EPA mixture for 24 h. The results demonstrated that ERK1/2 protein
expression was significantly decreased, compared with their
original cells, except for BRAF-mutant HT29 cells. The KRAS-mutant
CRC cells SW480 and HCT116 had a decreased ERK1/2 protein
expression of 0.323- (P=0.035) and 0.226-fold (P=0.035),
respectively. Furthermore, Caco-2 cells were also determined to
have a decreased ERK1/2 protein expression of 0.484-fold (P=0.022).
Contrarily, a significant increase in p-ERK1/2 was observed in all
cell lines, except in HT29 cells (Fig.
2A). The expression of p-ERK1/2 in SW480, HCT116 and Caco-2
cells was increased by 1.61- (P=0.006), 1.32- (P=0.047) and
4.96-fold (P=0.033), respectively. Notably, a 0.42-fold decrease in
p-ERK1/2 was determined in HT29 cells (P=0.029) (Fig. 2B).
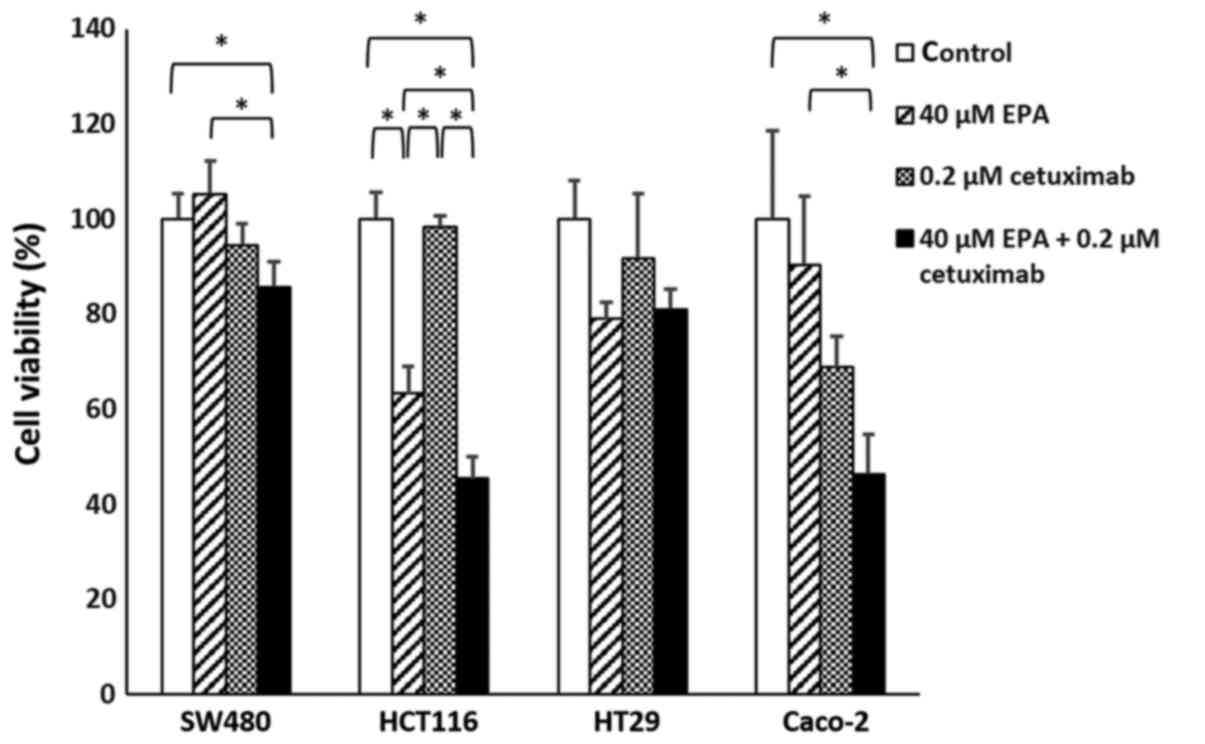
Cetuximab effect on EPA-treated
cells
Significant differences were observed in SW480
(P=0.0217), HCT116 (P<0.001) and Caco-2 (P=0.0064) cell groups.
With the exception of HT29 cells (P=0.1185), the cell viability of
CRC cells treated with EPA and cetuximab were significantly
decreased, compared with their controls. Furthermore, the combined
EPA with cetuximab-treated cells were compared with cells treated
with EPA only, in which all cell lines except HT29 cells were
observed to have significantly decreased cell viability (Fig. 3; P<0.05). With cetuximab in a
combination with EPA, the efficiency of cell growth inhibition was
greater than when treated with cetuximab alone for the wild-type
Caco-2 cells. Additionally, the response rate to cetuximab
following treatment with 0.1 or 0.2 µM cetuximab in the SW480,
HCT116 and Caco-2 cells treated with 40 µM EPA were significantly
improved, compared with their controls (P=0.0062, P=0.0176 and
P=0.0054, respectively); however, the BRAF-mutant cell line HT29
(P=0.2116) remained insensitive to cetuximab following 40 µM EPA
treatment (Fig. 4).
Discussion
The crucial data of the present study indicated that
40 µM EPA combined with 0.2 µM cetuximab can significantly reduce
the cell viabilities of KRAS-mutant CRC cell lines, with the
exception of HT29 cells. Secondly, following treating cells with 40
µM EPA, the cells exhibited significantly reduced the expression of
protein ERK1/2 but increased the expression of phosphorylation
ERK1/2 in KRAS-mutant CRC cells and also the wild-type CRC cells.
Notably, contradicting data was observed in BRAF-mutant HT29 cells.
Additionally, following treatment with EPA, the expression levels
of miR-378 were determined to be significantly increased in all
cell lines, with the exception of wild-type Caco-2 cells. These
data were consistent with our previous study, in which the cells
were treated with Lauric acid (25).
Based on the present results, the expression of
miR-378 directly mediates the expression of ERK1/2, which is
consistent to the TargetScanHuman database, where it is predicted
that the 3′UTR of mRNA ERK1/2 is one of the miR-378 target binding
sites; therefore, this results in the inhibition of protein ERK1/2
expression (24,26). However, the opposite results were
determined for the ERK1/2 phosphorylation status, with the
expression being increased in KRAS- and BRAF-mutant cells.
Undetermined cellular mechanism may affect the MAPK signaling
pathway, which may therefore trigger cell proliferation;
nevertheless, the present results demonstrated a significant
decrease in cell viabilities. Similar data has been reported by
Nikolakopoulou et al (36), in
which they produced a model regarding the reason why total protein
ERK1/2 had the opposite expression level to the phosphorylation of
ERK1/2. They concluded that EPA may be an important element to the
EGFR ligand, and EPA may selectively inhibit the growth of
premalignant and malignant keratinocytes by inducing a sustained
activation of ERK1/2, but not have an effect on normal cells.
Additionally, a number of studies also indicate that increased
phosphorylation status of ERK1/2 is associated with cell death. For
example, Yang et al (40)
indicated that if the total protein expression level of ERK1/2
remained the same, the phosphorylation of ERK1/2 could still be
increased, and consequently resulted in cell death. Similar data
have been determined in CRC cells, lung cancer cells and cervical
carcinoma studies, and this data agree that ERK1/2 phosphorylation
could trigger the expression of caspase-3 and result in cell
apoptosis (41–43). Although the potential mechanism is
unknown between the ERK1/2 phosphorylation status and protein
expression, which may be a result of unknown kinase effects. The
MAPK/ERK pathway with a high phosphorylation ERK expression is
correlated with an increased rate of cell apoptosis, as
demonstrated by a number of studies (40–44). The
present study further clarified that EPA was associated with
miR-378. Regardless of suppressed expression of ERK1/2, the
activation of phosphorylation ERK1/2 still significantly inhibited
cell growth.
There are at least two major pathways,
phosphoinositide 3-kinase (PI3K) and MAPK pathways, that are
involved in anti-EGFR therapy (45,46).
According to the present results, it was speculated the KRAS-mutant
cells may be associated with the MAPK or PI3K/Akt pathways;
therefore, miR-378 can trigger phosphorylation of ERK1/2 and result
in cell apoptosis when the MAPK pathway is blocked or through an
unknown mechanism of the PI3K/Akt pathway. This may explain the
reason why the cells continued to survive following EPA treatment,
but eventually succumbed to anti-EGFR cetuximab treatment; however,
the BRAF-mutant cells exhibited a reduced expression level of
phosphorylated ERK1/2, which is consistent to reduced ERK1/2
protein expression following restoration of miR-378 expression.
Additionally, the BRAF-mutant HT29 cells exhibited no significant
response to anti-EGFR antibody following the EPA treatment. We
hypothesized that BRAF-mutant cells may harbor an unknown mechanism
underlying cell signaling pathways, which differ from the
KRAS-mutant cells that will require further research in the
future.
To conclude, the present results indicated that EPA
could significantly induce the expression level of miR-378 in CRC
mutant cells, but not in the wide-type cells. Restoration of the
anti-EGFR antibody sensitivity of the KRAS-mutant cells was
achieved following treating the cells with up to 40 µM of EPA,
which was not observed in the BRAF-mutant cells in the present
study. Notably, the status of phosphorylation of ERK1/2 may serve
an important role in mediating the cell response to cetuximab
therapy; however, the bio-mechanism behind this will require
further study. This could also further reveal the discrepancy of
clinical behaviors between KRAS- and BRAF-mutated colon cancer
types.
Acknowledgements
The authors would like to thank Mr. Chi-Chia Pang
from the Department of Chemical Engineering and Biotechnology,
Graduate Institute of Biochemical and Biomedical Engineering,
National Taipei University of Technology (Taipei, Taiwan) for
editing.
Funding
The present study was supported by the National
Taipei University of Technology, Mackay Memorial Hospital Joint
Research Program (NTUT-MMH-107-03).
Availability of data and materials
The datasets used and/or analyzed in this study are
available from the corresponding author on reasonable request.
Authors' contributions
WHW contributed to conception and design,
acquisition of data, analysis and interpretation of data; and
revising the article critically for important intellectual content;
and giving final approval of the version to be published. WHL
contributed to collecting samples, acquiring data and writing the
article. YJP performed the experiments, and contributed to the
acquisition of data and English editing. LWK performed the
experiments and contributed to the acquisition of data. HHH
contributed to the conception and design, and gave final approval
of the version to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lièvre A, Bachet JP, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bokemeyer C, Bondarenko I, Makhson A,
Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G,
Stroh C, et al: Fluorouracil, leucovorin, and oxaliplatin with and
without cetuximab in the first-line treatment of metastatic
colorectal cancer. J Clin Oncol. 27:663–671. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chien Chang CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. NEJM. 360:1408–1417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tol J, Nagtegaal ID and Punt CJ: BRAF
mutation in metastatic colorectal cancer. NEJM. 361:98–99. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Nicolantonio F, Martini M, Molinari F,
Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L,
Frattini M, Siena S and Bardelli A: Wild-type BRAF is required for
response to panitumumab or cetuximab in metastatic colorectal
cancer. J Clin Oncol. 26:5705–5712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baldus SE, Schaefer KL, Engers R, Hartleb
D, Stoecklein NH and Gabbert HE: Prevalence and heterogeneity of
KRAS, BRAF, and PIK3CA mutations in primary colorectal
adenocarcinomas and their corresponding metastases. Clin Cancer
Res. 16:790–799. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Di Fiore F, Blanchard F, Charbonnier F, Le
Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech
JJ, et al: Clinical relevance of KRAS mutation detection in
metastatic colorectal cancer treated by Cetuximab plus
chemotherapy. Br J Cancer. 96:1166–1169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frattini M, Saletti P, Romagnani E, Martin
V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F and
Mazzucchelli L: PTEN loss of expression predicts cetuximab efficacy
in metastatic colorectal cancer patients. Br J Cancer.
97:1139–1145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Roock W, Piessevaux H, De Schutter J,
Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL,
Peeters M, Humblet Y, et al: KRAS wild-type state predicts survival
and is associated to early radiological response in metastatic
colorectal cancer treated with cetuximab. Ann Oncol. 19:508–515.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watzinger F, Mayr B, Haring E and Lion T:
High sequence similarity within ras exons 1 and 2 in different
mammalian species and phylogenetic divergence of the ras gene
family. Mamm Genome. 9:214–219. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andreyev HJ, Ross PJ, Cunningham D and
Clarke PA: Antisense treatment directed against mutated Ki-ras in
human colorectal adenocarcinoma. Gut. 48:230–237. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bazan V, Migliavacca M, Zanna I, Tubiolo
C, Grassi N, Latteri MA, La Farina M, Albanese I, Dardanoni G,
Salerno S, et al: Specific codon 13 K-ras mutations are predictive
of clinical outcome in colorectal cancer patients, whereas codon 12
K-ras mutations are associated with mucinous histotype. Ann Oncol.
13:1438–1446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benvenuti S, Sartore-Bianchi A, Di
Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S and Bardelli
A: Oncogenic activation of the RAS/RAF signaling pathway impairs
the response of metastatic colorectal cancers to anti-epidermal
growth factor receptor antibody therapies. Cancer Res.
67:2643–2648. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esteller M, Gonzalez S, Risques RA,
Marcuello E, Mangues R, Germà JR, Herman JG, Capellà G and Peinado
MA: K-ras and p16 aberrations confer poor prognosis in human
colorectal cancer. J Clin Oncol. 19:299–304. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roskoski R Jr: RAF
protein-serine/threonine kinases: Structure and regulation. Biochem
Biophys Res Commun. 399:313–317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pakneshan S, Salajegheh A, Smith RA and
Lam AK: Clinicopathological relevance of BRAF mutations in human
cancer. Pathology. 45:346–356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng G, Bell I, Crawley S, Gum J, Terdiman
JP, Allen BA, Truta B, Sleisenger MH and Kim YS: BRAF mutation is
frequently present in sporadic colorectal cancer with methylated
hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin
Cancer Res. 10:191–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Callari M, Dugo M, Musella V, Marchesi E,
Chiorino G, Grand MM, Pierotti MA, Daidone MG, Canevari S and De
Cecco L: Comparison of microarray platforms for measuring
differential microRNA expression in paired normal/cancer colon
tissues. PLoS One. 7:e451052012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faltejskova P, Svoboda M, Srutova K,
Mlcochova J, Besse A, Nekvindova J, Radova L, Fabian P, Slaba K,
Kiss I, et al: Identification and functional screening of microRNAs
highly deregulated in colorectal cancer. J Cell Mol Med.
16:2655–2666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mosakhani N, Sarhadi VK, Borze I,
Karjalainen-Lindsberg ML, Sundström J, Ristamäki R, Osterlund P and
Knuutila S: MicroRNA profiling differentiates colorectal cancer
according to KRAS status. Genes Chromosomes Cancer. 51:1–9. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang YX, Zhang XY, Zhang BF, Yang CQ, Chen
XM and Gao HJ: Initial study of microRNA expression profiles of
colonic cancer without lymph node metastasis. J Dig Dis. 11:50–54.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ganesan J, Ramanujam D, Sassi Y, Ahles A,
Jentzsch C, Werfel S, Leierseder S, Loyer X, Giacca M, Zentilin L,
et al: MiR-378 controls cardiac hypertrophy by combined repression
of mitogen-activated protein kinase pathway factors. Circulation.
127:2097–2106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weng WH, Leung WH, Pang YJ and Hsu HH:
Lauric acid can improve the sensitization of cetuximab in KRA/BRAF
mutated colorectal cancer cells by retrivable microRNA-378
expression. Oncol Rep. 35:107–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feng M, Li Z, Aau M, Wong CH, Yang X and
Yu Q: Myc/miR-378/TOB2/cyclin D1 functional module regulates
oncogenic transformation. Oncogene. 30:2242–2251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carrer M, Liu N, Grueter CE, Williams AH,
Frisard MI, Hulver MW, Bassel-Duby R and Olson EN: Control of
mitochondrial metabolism and systemic energy homeostasis by
microRNAs 378 and 378*. Proc Natl Acad Sci USA. 109:15330–15335.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chambers KT, Chen Z, Crawford PA, Fu X,
Burgess SC, Lai L, Leone TC, Kelly DP and Finck BN: Liver-specific
PGC-1beta deficiency leads to impaired mitochondrial function and
lipogenic response to fasting-refeeding. PLoS One. 7:e526452012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gerin I, Bommer GT, McCoin CS, Sousa KM,
Krishnan V and MacDougald OA: Roles for miRNA-378/378* in adipocyte
gene expression and lipogenesis. Am J Physiol Endocrinol Metab.
299:E198–E206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crunkhorn S, Dearie F, Mantzoros C, Gami
H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC and
Patti ME: Peroxisome proliferator activator receptor gamma
coactivator-1 expression is reduced in obesity: Potential
pathogenic role of saturated fatty acids and p38 mitogen-activated
protein kinase activation. J Biol Chem. 282:15439–15450. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
EFSA: Scientific option on the tolerable
upper intake level of eicosapentaenoic acid (EPA), docosahexaenoic
acid (DHA) and docosapentaenoic acid (EPA). European Food Safety
Authority Journal. 10:28152012.
|
|
32
|
Ijzerman RG, Stehouwer CD, Serné EH,
Voordouw JJ, Smulders YM, Delemarre-van de Waal HA and van
Weissenbruch MM: Incorporation of the fasting free fatty acid
concentration into quantitative insulin sensitivity check index
improves its association with insulin sensitivity in adults, but
not in children. Eur J Endocrinol. 160:59–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mensink RP, Zock PL, Kester AD and Katan
MB: Effects of dietary fatty acids and carbohydrates on the ratio
of serum total to HDL cholesterol and on serum lipids and
apolipoproteins: A meta-analysis of 60 controlled trials. Am J Clin
Nutr. 77:1146–1155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Giros A, Grzybowski M, Sohn VR, Pons E,
Fernandez-Morales J, Xicola RM, Sethi P, Grzybowski J, Goel A,
Boland CR, Gassull MA, Llor X, et al: Regulation of colorectal
cancer cell apoptosis by the n-3 polyunsaturated fatty acids
Docosahexaenoic and Eicosapentaenoic. Cancer Prev Res (Phila).
2:732–742. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fukui M, Kang KS, Okada K and Zhu BT: EPA,
an omega-3 fatty acid, induces apoptosis in human pancreatic cancer
cells: Role of ROS accumulation, caspase-8 activation, and
autophagy induction. J Cell Biochem. 114:192–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nikolakopoulou Z, Nteliopoulos G,
Michael-Titus AT and Parkinson EK: Omega-3 polyunsaturated fatty
acids selectively inhibit growth in neoplastic oral keratinocytes
by differentially activating ERK1/2. Carcinogenesis. 34:2716–2725.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ding N, Sun X, Wang T, Huang L, Wen J and
Zhou Y: miR-378a-3p exerts tumor suppressive function on the
tumorigenesis of esophageal squamous cell carcinoma by targeting
Rab10. Int J Mol Med. 42:381–391. 2018.PubMed/NCBI
|
|
38
|
Bai B, Liu H and Laiho M: Small RNA
expression and deep sequencing analyses of the nucleolus reveal the
presence of nucleolus-associated microRNAs. FEBS Open Bio.
4:441–449. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD.: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang L, Su T, Lv D, Xie F, Liu W, Cao J,
Sheikh IA, Qin X, Li L and Chen L: ERK1/2 mediates lung
adenocarcinoma cell proliferation and autophagy induced by
apelin-13. Acta Biochmm Biophys Sin (Shanghai). 46:100–111. 2014.
View Article : Google Scholar
|
|
41
|
Randhawa H, Kibble K, Zeng H, Moyer MP and
Reindl KM: Activation of ERK signaling and induction of colon
cancer cell death by piperlongumine. Toxicol In Vitro.
27:1626–1633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu Y, Yang Y, Ye YC, Shi QF, Chai K,
Tashiro S, Onodera S and Ikejima T: Activation of ERK-p53 and
ERK-mediated phosphorylation of Bcl-2 are involved in autophagic
cell death induced by the c-Met inhibitor SU11274 in human lung
cancer A549 cells. J Pharmacol Sci. 118:423–432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Singh S, Upadhyay AK, Ajay AK and Bhat MK:
p53 regulates ERK activation in carboplatin induced apoptosis in
cervical carcinoma: A novel target of p53 in apoptosis. FEBS Lett.
581:289–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schweyer S, Soruri A, Meschter O, Heintze
A, Zschunke F, Miosge N, Thelen P, Schlott T, Radzun HJ and Fayyazi
A: Cisplatin-induced apoptosis in human malignant testicular germ
cell lines depends on MEK/ERK activation. Br J Cancer. 91:589–598.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu W, Ren H, Ren J, Yin T, Hu B, Xie S,
Dai Y, Wu W, Xiao Z, Yang X and Xie D: The role of
EGFR/PI3K/Akt/cyclinD1 signaling pathway in acquired middle ear
cholesteatoma. Mediators Inflamm. 2013:6512072013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lo HW and Hung MC: Nuclear EGFR signalling
network in cancers: Linking EGFR pathway to cell cycle progression,
nitric oxide pathway and patient survival. Br J Cancer. 94:184–188.
2006. View Article : Google Scholar : PubMed/NCBI
|