Introduction
Lymphoma is the name given to a group of blood cell
tumors that begin in lymphocytes (a type of white blood cell), the
incidence rate of which has increased slightly among men in recent
years (1–3). In recent years, mortality rates for
lymphoma have been decreasing; however, certain types of lymphoma
are more aggressive and these patients have a lower survival rate
(2,3).
Therefore, investigating effective therapeutic approaches is
necessary to prolong survival of patients with lymphoma.
Tumors are caused by abnormal cell proliferation,
differentiation and apoptosis, which are due to the activation of
certain proto-oncogenes, inactivation of tumor suppressors and
alterations to apoptosis-associated genes (4,5).
Apoptosis, a programmed cell death, exerts a notable role by
allowing cells to respond appropriately to environmental stimuli
(6). Tumorigenesis and progression of
a number of types of human cancer are associated with the
dysregulation of the apoptotic process (7,8).
A number of chemotherapeutic agents have been
demonstrated to serve antitumor roles by inducing cell apoptosis,
resulting in the death of cancer cells (9–11). The
application of arsenic trioxide (ATO), an inorganic compound with
the formula As2O3, is controversial owing to
the high toxicity of arsenic compounds (12). Despite the toxicity of arsenic,
arsenic trioxide has been used in a number of traditional Chinese
medicines, and has been used to treat cancer (13). Previous studies have identified that
ATO induces apoptosis in acute promyelocytic leukemia (APL) cells
(14,15), glioblastoma cells (16) and gastric cancer cells (17), and inhibits cell growth in breast
cancer (18). The use of ATO is also
being evaluated for the treatment of certain malignancies,
including lung cancer (19),
hepatocellular cancer (20) and basal
cell cancer (21). Although ATO
exhibits a significant antitumor function in multiple cancer types,
its exact effect on lymphoma and the underlying mechanism of action
remain under investigation.
Nuclear factor-κB (NF-κB) is a protein complex that
controls the transcription of DNA, cytokine production and cell
survival (22). The dysfunctional
regulation of NF-κB has been associated with cancer, inflammatory
and autoimmune diseases, septic shock, viral infection and improper
immune development (23–26). NF-κB has been recognized as one of the
dominant oncogenes associated with lymphoma (27). NF-κB has emerged as a central player
in the development and maintenance of lymphoma (28). It has been revealed that the
constitutive activation of the canonical and non-canonical NF-κB
signaling pathways has been observed in Hodgkin and Reed/Sternberg
cells in classical Hodgkin lymphoma (27). It has also been reported that the
inhibition of NF-κB activity is involved in the apoptosis of
lymphoma cells (27).
The present study investigated the effect of ATO on
the proliferation and apoptosis in human lymphoma cells was
investigated and the underlying molecular mechanism, with
particular focus on intracellular expression and localization of
NF-κB, and levels of apoptosis-associated proteins B-cell
lymphoma-2 (Bcl-2), Bcl-2-associated X protein (Bax) and
caspase-3.
Materials and methods
Cell cultures and treatments
The human B lymphoma Raji cell line and human T
lymphoma Jurkat cell line were purchased from Shanghai Institute of
Pharmaceutical Industry (Shanghai, China). Cells were cultured in
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal bovine serum (FBS; Hangzhou Sijiqing
Biological Engineering Materials Co., Ltd., Hangzhou, China) and
100 µg/ml penicillin and streptomycin (Sigma-Aldrich, St. Merck,
KGaA, Darmstadt, Germany). Cell lines were cultured at 37°C in a 5%
CO2 incubator.
Cell viability assay
Cell viability was evaluated using Cell Counting
kit-8 (CCK-8; Beyotime Institute of Biotechnology, Haimen, China).
A total of 2×105 cells per well were seeded in a 96-well
plate and cultured at 37°C with 5% CO2 to 80%
confluence, and then were treated with ATO (Sigma-Aldrich; Merck
KGaA) at 0.5, 1, 2, 4, 10 and 30 µM for 24, 48 and 72 h. Next,
cells were incubated with CCK-8 reagent according to the
manufacturer's protocol and the number of viable cells was measured
by recording the optical density by using a Rainbow microplate
reader (Tecan Group, Ltd., Mannedorf, Switzerland) at a wavelength
of 450 nm, as described previously (29). The cell growth inhibition rate was
calculated according to the formula: Growth inhibition rate
(%)=[1-A (experimental group)/A (control group)] ×100, where A is
the absorbance value. The half-maximal inhibitory concentration
(IC50) value was adopted as the concentration of drug
intervention in future experiments in vitro.
Cell morphology observation
A total of 1×105 Raji cells or Jurkat
cells per well were seeded in a 24-well plate and cultured at 37°C
with 5% CO2 to reach ~80% confluence, and then were
treated with or without 2 or 3.5 µM ATO for 0, 24, 48, and 72 h.
Cells were harvested and centrifuged in 1,000 × g for 15 min at
4°C. Cells were washed with PBS three times, and then were placed
onto a glass slide and the slides were stained with Wright's stain
(Magnil Dye Chem, Maharashtra, India) according to manufacturer's
protocol. The glass slides were rinsed briefly in running deionized
water (pH 7.2) and dried in air thoroughly before capturing images
by a microscope (Olympus BX51, Olympus Corporation, Tokyo, Japan)
at ×1,000 magnification.
Detection of early cell apoptosis and
necrosis
Raji cells or Jurkat cells were grown to 80%
confluence and treated with 2 or 3.5 µM ATO, respectively, for 0,
24, 48 and 72 h at 37°C. ATO for different time periods. Apoptosis
was quantified by an Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) assay (BD Biosciences, Franklin Lakes,
NJ, USA) following the manufacturer's protocol. The Annexin
V-FITC/PI assay detects the quantity of phosphatidylserine on the
outer surface of the plasma membrane (a biochemical alteration
unique to membranes of apoptotic cells) and the quantity of PI, a
dye that readily enters dead cells or cells undergoing late-stage
apoptosis and binds to DNA but does not bind to the plasma membrane
of viable cells. Cytoclasis rate (Necrosis rate) was detected by PI
single staining. Fluorescence was detected using a FACSCalibur flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and data were
analyzed using CellQuest version 3.2 software (BD Biosciences).
Cells with phosphatidylserine on their surface were considered to
be apoptotic.
Western blot analysis
Cells were collected and lysed in lysis buffer (50
mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% NP40, 150 mM NaCl, 10 mM NaF
and 1 mM Na3VO4) containing a protease
inhibitor cocktail (Roche Applied Science, Penzberg, Germany).
Following centrifugation at 12,000 × g for 10 min at 4°C, the
supernatant was collected and quantified using a Bicinchoninic acid
(BCA) quantification kit (Beyotime Institute of Biotechnology,
Haimen, China). Protein samples (50 µg) were separated by 10%
SDS-PAGE (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
transferred to polyvinylidene fluoride membranes (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% non-fat
dried milk in Tris-buffered saline containing 0.05% Tween 20 for 1
h at room temperature, and incubated with the following specific
primary antibodies overnight at 4°C: Rabbit polyclonal IgG to NF-κB
(1:1,000; cat no. ab16502; Abcam, Cambridge, UK); mouse monoclonal
IgG to Bcl-2 (1:500; vat no. sc7382; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA); rabbit polyclonal IgG to Bax (1:500; cat
no. sc493; Santa Cruz Biotechnology, Inc.); mouse monoclonal IgG to
β-actin (1:1,000; cat no. sc47778; Santa Cruz Biotechnology, Inc.);
rabbit polyclonal IgG to cleaved-caspase-3 (1:1,000; cat no. 9661S;
Cell Signaling Technology, Inc., Danvers, MA, USA), followed by
horseradish peroxidase-conjugated secondary antibodies goat
anti-mouse (1:2,000; cat no. sc-2005; Santa Cruz Biotechnology,
Inc.) and goat anti-rabbit IgG (1:2,000; cat no. sc-2004; Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature. An Enhanced
Chemiluminescence detection reagent (GE Healthcare, Chicago, IL,
USA) was used for development. Additionally, IRDye® 800
CW-labeled secondary antibodies goat anti-mouse (1:10,000; cat no.
ab216772; Abcam, Cambridge, MA) and anti-rabbit immunoglobulin G
(1:10,000; cat no. ab216773; Abcam) were used to incubation for 1 h
at room temperature. Membranes were visualized and imaged using an
Odyssey infrared imaging system (LI-COR, Lincoln, NE, USA). The
gray value of the targeted bands was quantified with QuantityOne
software version 4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) following incubation, with β-actin used as the internal
reference.
Immunofluorescence and confocal
microscopy
The procedure was performed as previously described
(30). Raji cells or Jurkat cells
were fixed with 3% formaldehyde in PBS for 20 min at room
temperature. After three washes with PBS containing 50 mM
NH4Cl, the cells were soaked in a blocking solution (PBS
containing 5% fetal calf serum, FCS; Hangzhou Sijiqing Biological
Engineering Materials Co., Ltd., Hangzhou, China) for 15 min at
room temperature and then permeabilized with 0.1% Triton X-100 in
PBS for 5 min. Cells were incubated with anti-NF-κB antibody
(1:1,000; cat no. 8242; Cell Signaling Technology, Inc., Danvers,
MA, USA) overnight at 4°C, and stained with a
rhodamine-fluorescence labeled goat anti-rabbit secondary antibody
(1:1,000; cat no. 15076; Active motif, Inc., Carlsbad, CA, USA) in
1% blocking solution (PBS containing 1% FCS) and incubated for 1 h
at room temperature. Confocal imaging was performed with an LSM
META510 confocal scanning laser microscope (Carl Zeiss AG,
Oberkochen, Germany) at ×1,000 magnification. The images were
subsequently analyzed using the freely available image processing
software ImageJ Version 1.46 (National Institute of Health,
Bethesda, Maryland, USA). The observations were made in
triplicates, and representative images are presented here.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from Raji cells or Jurkat
cells with TRIzol reagent (cat no. 15596-026; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. Total RNA isolated by TRIzol reagent was
free of protein and DNA contamination. For RT-qPCR, the isolated
RNA was treated with amplification grade DNase I (cat no.
18068-015; Invitrogen, Thermo Fisher Scientific, Inc.). cDNA was
obtained by RT using the RevertAid™ First Strand cDNA synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc.). The relative mRNA
expression levels of target genes were detected with a specific
TaqMan® Gene Expression assay (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with fluorogenic FAM-labeled
probes. Primers for NF-κB and internal control RNA polymerase II
(RPII) were designed (31,32), and were synthesized by Shanghai
Institute of Pharmaceutical Industry (Shanghai, China). The primers
sequences were as follows: NF-κB, forward 3′-GGATTTCGTTTCCGTTATG-5′
and reverse 3′-GGTTTGCGAAGCCGACCA-5′; RPII (internal control),
forward 3′-GCACCACGTCCAATGACAT-5′ and reverse
3′-GTGCGGCTGCTTCCATAA-5′.
The two genes were amplified by a first step of 120
sec at 95°C, followed by 45 cycles of 30 sec at 95°C, 30 sec at
60°C and 30 sec at 72°C. The real-time fluorescence detection was
performed using the ABI PRISM 7700 Sequence detector (PerkinElmer,
Inc., Waltham, MA, USA). NF-κB expression was normalized to the
expression of the reference RPII and was calculated using the
2ΔΔCq method (33).
Statistical analysis
At least three replicates were performed within the
same experiment. The statistical package SPSS version 16.0 (SPSS,
Inc., Chicago, IL, USA) was used to assay the experimental data.
Differences were analyzed using one-way analysis of variance, and
P<0.05 was considered to indicate a statistically significant
difference.
Results
ATO treatment inhibits the growth of
lymphoma Raji and Jurkat cell lines
Human lymphoma Raji and Jurkat cell lines were
treated with ATO at 0.5, 1, 2, 4, 10 and 30 µM for 24, 48 and 72 h.
Cell viability was evaluated using a CCK-8 assay and the cell
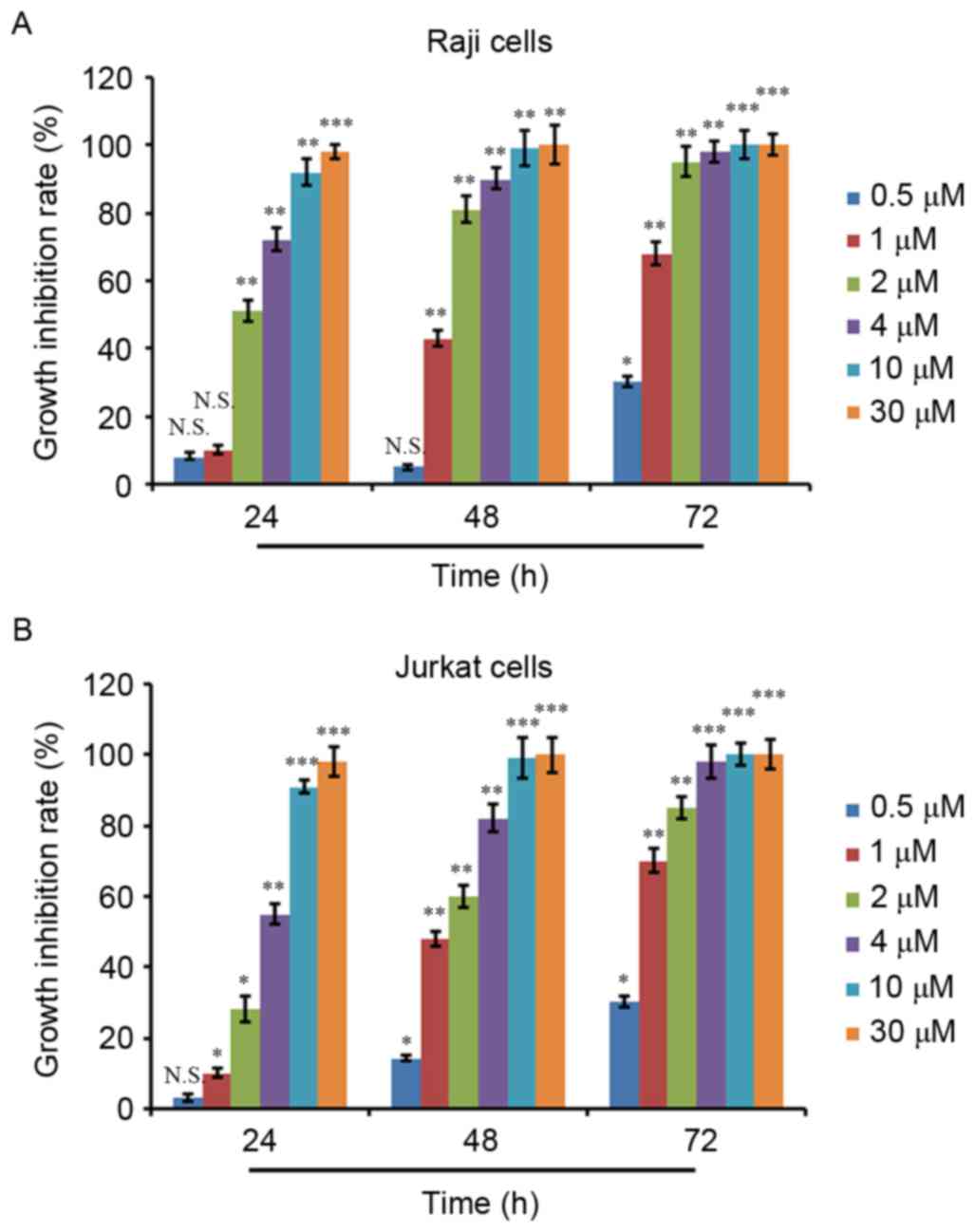
growth inhibition rate was calculated. As depicted in Fig. 1A and B, ATO treatment inhibited the
proliferation of Raji and Jurkat cells in a dose- and
time-dependent manner. ATO treatment at 0.5 µM for 24 and 48 h did
not result in significant proliferation inhibition in Raji cells,
whereas ATO treatment at other concentrations for 24 and 48 h and
ATO treatment at all concentrations for 72 h exhibited significant
proliferation inhibition activity (Fig.
1A). Additionally, 0.5 µM ATO treatment for 24 h did not show a
significant proliferation inhibition in Jurkat cells, whereas ATO
treatment at the other concentrations for 24 h and ATO treatment at
all concentrations for 48 and 72 h showed significant proliferation
inhibition activity (Fig. 1B). The
IC50 values of the two cell lines were then calculated,
which were 2.06 µM for Raji cells and 3.75 µM for Jurkat cells at
24 h.
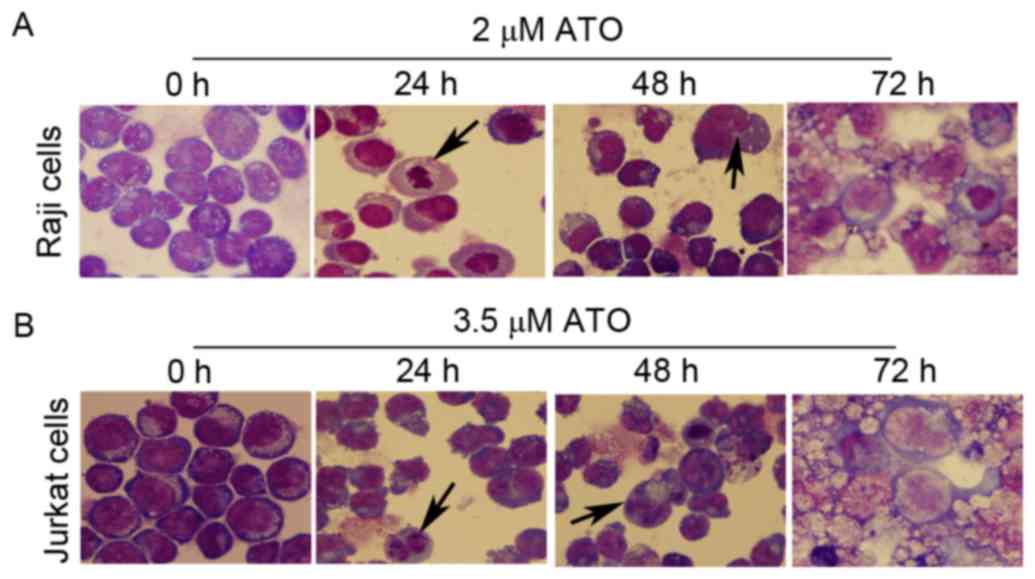
ATO treatment induces morphological
changes in Raji and Jurkat cells
The survival capabilities of Raji cells treated with
2 µM ATO and Jurkat cells treated with 3.5 µM ATO for 0, 24, 48 and
72 h, were compared by observing their morphology using Wright's
staining. The two cell lines exhibited significant apoptotic
features following ATO treatment for 24 and 48 h, with enlarged
cells, enlarged vacuole, reduced nucleoplasm, rough nuclear
chromatin, reduced nucleolus and pyknosis, and nuclear apoptotic
bodies, compared with untreated control cells (Fig. 2A and B). The two cell lines treated
with ATO for 72 h exhibited significant morphological alterations
with cellular debris for dead cells (Fig.
2A and B).
ATO treatment induces apoptosis of
lymphoma Raji and Jurkat cell lines
Raji and Jurkat cell lines were treated with 2 µM or
3.5 µM ATO for 24, 48 and 72 h. Cell apoptosis was evaluated by
flow cytometric analysis. The results revealed that the apoptosis
of Raji and Jurkat cells was significantly increased by ATO
treatment for 24 and 48 h compared with ATO-untreated control
cells. However, ATO-induced cell apoptosis at 72 h was lower than
that at 48 h (Fig. 3A). Additionally,
the cytoclasis rate was increased by ATO treatment for 48 and 72 h
compared with ATO-untreated control cells (Fig. 3A). The expression levels of
apoptosis-associated proteins were detected. Western blot analysis
demonstrated that ATO treatment for 48 h reduced the expression of
the anti-apoptotic protein Bcl-2 and elevated that of the
pro-apoptotic protein Bax and the levels of cleaved caspase-3
(Fig. 3B). These data indicated that
ATO inhibited the growth of lymphoma Raji and Jurkat cells,
possibly by inducing cell apoptosis, with the abnormal expression
of apoptosis-associated proteins.
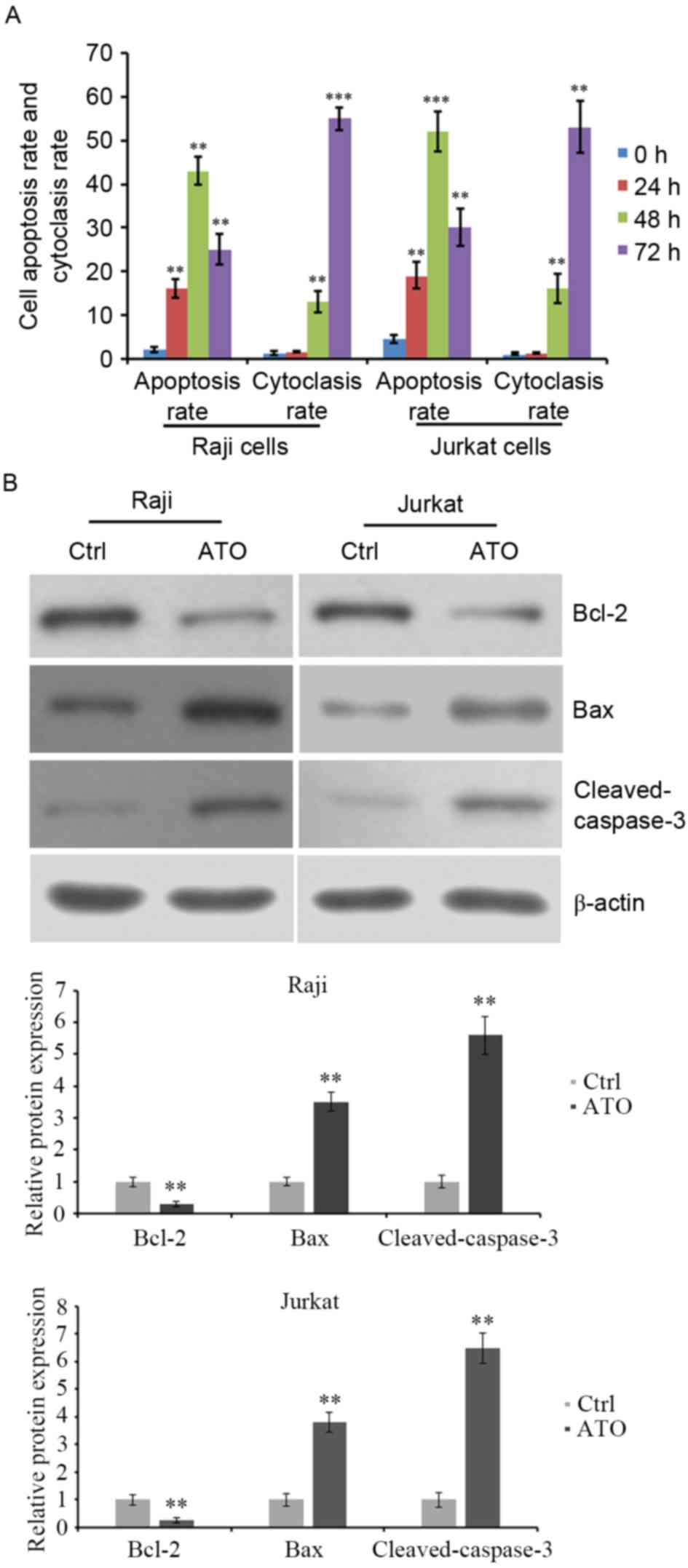 | Figure 3.Apoptosis and cytoclasis rate of Raji
and Jurkat cells following ATO treatment. (A) Raji and Jurkat cells
were treated with 2 and 3.5 µM ATO, respectively, for 0, 24, 48 and
72 h. The rate of cell apoptosis and cytoclasis were evaluated by
flow cytometry, (B) Cells were treated with ATO treatment for 48 h
and the levels of anti-apoptotic protein Bcl-2, pro-apoptotic
protein Bax and cleaved caspase-3 were detected by western blot
analysis. β-actin was detected as internal reference. **P<0.01
and ***P<0.001 vs. ATO-untreated cells. ATO, arsenic trioxide;
Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X; Ctrl,
control. |
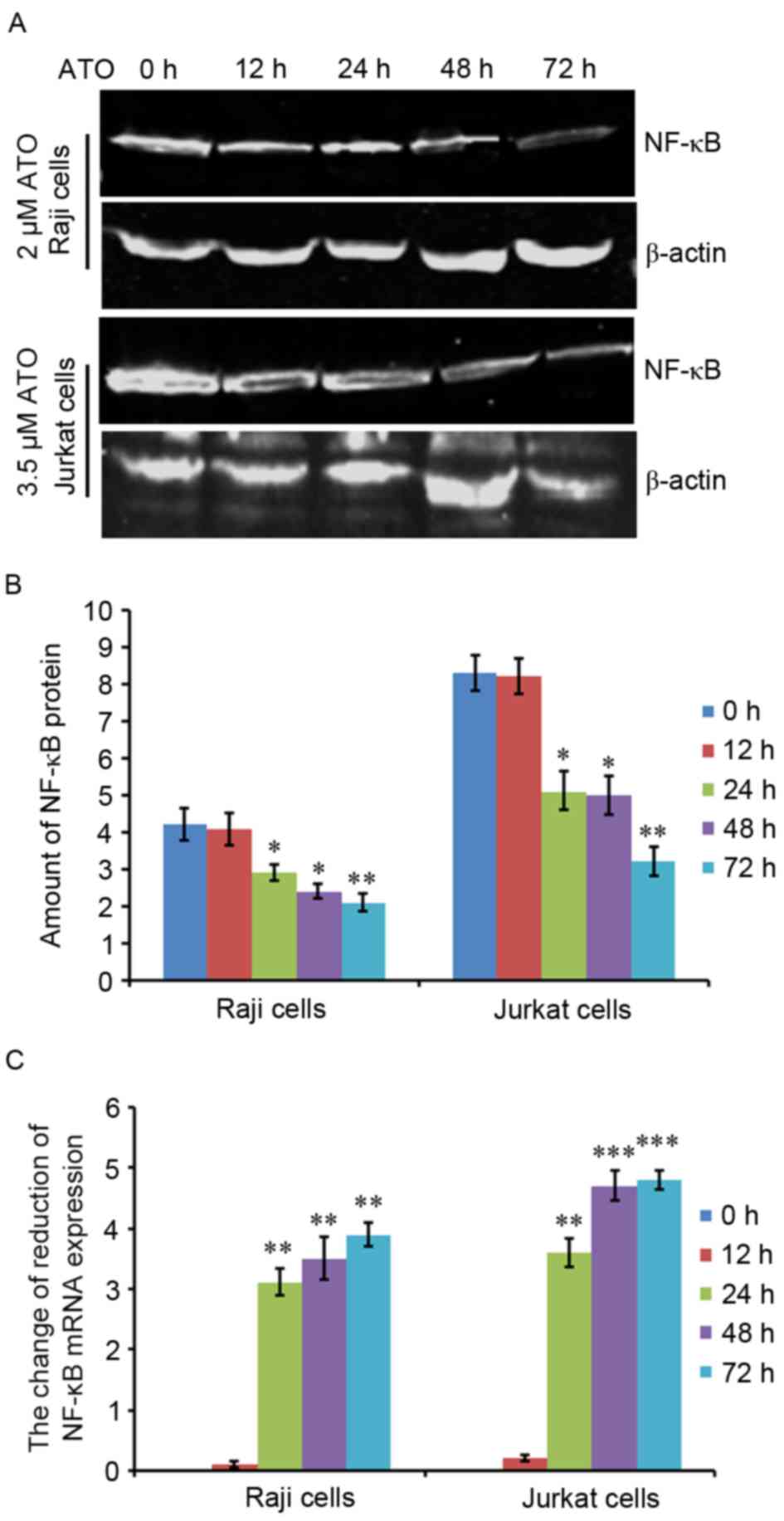
ATO inhibits protein and mRNA
expression levels of NF-κB
To investigate the underlying mechanism behind the
ATO-dependent induction of apoptosis in Raji and Jurkat cells, the
two cells were treated with 2 or 3.5 µM ATO, respectively, for 0,
12, 24, 48 and 72 h. Next, NF-κB protein and mRNA expression levels
were evaluated by western blot analysis and RT-qPCR. The results
revealed that NF-κB protein expression in Raji and Jurkat cells was
decreased by ATO treatment in a time-dependent manner (Fig. 4A), and quantification analysis
supported these results (Fig. 4B).
NF-κB mRNA expression levels were also significantly reduced by ATO
treatment for 24, 48 and 72 h in the two cell lines compared with
ATO-untreated control cells. However, there was no significant
difference in the mRNA expression levels of NF-κB at 24, 48 and 72
h (Fig. 4C). These results indicated
that NF-κB protein and mRNA expression levels decreased during
ATO-induced cell apoptosis in lymphoma Raji and Jurkat cells.
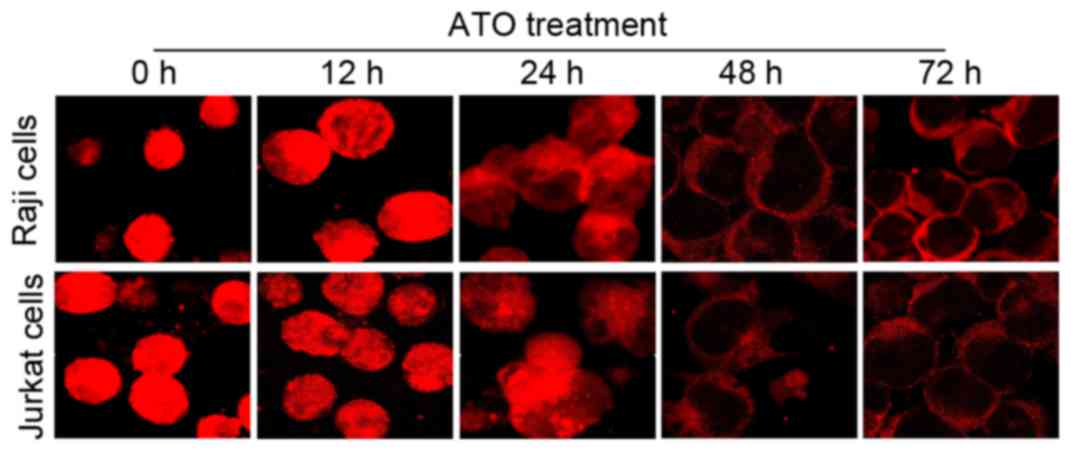
ATO treatment affects the sub-cellular
localization of NF-κB
To investigate the status of NF-κB during
ATO-induced cell apoptosis in Raji and Jurkat cells, the two cell
lines were treated with ATO (at the aforementioned concentrations)
for 0, 12, 24, 48 and 72 h, and then the sub-cellular localization
of NF-κB was detected by immunofluorescence analysis. The results
revealed that fluorescently labeled NF-κB was expressed in the
nucleus and cytoplasm of the two cell lines prior to ATO treatment,
whereas NF-κB expression diminished gradually following ATO
treatment for 12 and 24 h, and was only observed in the cytoplasm
after 48 and 72 h (Fig. 5). Nuclei
were stained using DAPI (data not shown). Fluorescence of NF-κB
protein expression was clearly observed all over the visible cells
under weak light prior to ATO treatment, while fluorescence was
only observed around the cell membranes following ATO treatment for
48 and 72 h. These data indicated that NF-κB may be associated with
ATO-induced cell apoptosis in lymphoma Raji and Jurkat cells, with
alterations of NF-κB protein expression levels and sub-cellular
localization from the nucleus to the cytoplasm.
Discussion
Studies concerning acute leukemia have verified that
ATO serves a critical role in the inhibition of leukemia cell
proliferation, promotion of cell differentiation and stimulation of
cell apoptosis (14,15). ATO may also inhibit the proliferation
of various types of lymphoma cells (34–36),
including Burkitt lymphoma, by reducing mitochondrial membrane
potential (37,38), consuming ATP and prolonging the cell
cycle, downregulating the proto-oncogene Bcl-2, and upregulating
tumor protein p53 (39). Previous
studies have also revealed that ATO increases the proportion of
cells in the G2/M phase of the cell cycle in lymphoma
cells, which is sufficient to induce cell apoptosis (40).
The present study revealed that ATO might inhibit
the proliferation of B lymphoma Raji cells and T lymphoma Jurkat
cells, which was time- and drug-concentration-dependent. ATO at a
concentration of 0.5 µM (dosage for acute promyelocytic leukemia
(APL) apoptosis (41) showed little
inhibition on growth in the two cell lines, indicating that the
dosage for clinical ATO treatment of lymphoma may possibly exceed
the dosage for APL treatment. Cell morphology observation and flow
cytometric analysis indicated that the two cell lines exhibited
features of apoptosis following ATO treatment for 24 and 48 h,
whereas the cytosomes of the two cell lines underwent clear
necrosis that was shown by cytoclasis rate following ATO treatment
for 72 h. These data demonstrated that ATO might inhibit the
proliferation of lymphoma Raji and Jurkat cells by inducing
apoptosis. Additionally, it was found that the dosage of ATO that
was able to inhibit the growth of T lymphoma Jurkat cells by
inducing cell apoptosis was higher than that required for growth
inhibition in B lymphoma Raji cells.
NF-κB was initially identified in B lymphocytes and
served a role as a regulator of the κ-immunoglobulin gene (42). NF-κB is located in the cytoplasm in an
inactivated state. The activated NF-κB is translocated into the
nucleus where it binds to response elements (RE) specific sequences
of DNA, and then recruits co-activators and RNA polymerase to
regulate gene transcription and thus affect translation, resulting
in an alteration in cell function (22,43,44).
Previous studies have indicated that NF-κB activation is closely
associated with tumorigenesis and drug resistance in tumors
(45,46). Beuso-Ramos et al (47) demonstrated that expression of NF-κB in
acute medullocell leukemia cells was frequently upregulated, and
therefore the cells may have escaped from apoptosis via the
regulation of certain apoptosis-resistant genes through expression
of NF-κB. Hinz et al (48)
revealed that ATO inhibited NF-κB activation followed by its
degradation, and consequently induced the apoptosis of leukemia
cells. ATO was also reported to induce apoptosis and incapacitate
proliferation and invasive properties through possible
NF-κB-mediated inhibition of survivin and telomerase activity, and
NF-κB-dependent suppression of cathepsin B, matric
metalloproteinase (MMP)-2 and MMP-9 in U87-MG glioblastoma cells
(49). A further previous study
demonstrated that ATO may prevent NF-κB from nuclear translocation,
which thereby led to NF-κB inactivation, either by upregulation and
stabilization of expression of NF-κB inhibitory factor IκB, or by
suppression of IκB kinase, which blocked the degradation of IκB
(50). Immunofluorescence analysis
performed in the present study revealed that NF-κB existed in the
nucleus and cytoplasm of lymphoma cells prior to ATO treatment,
whereas nuclear NF-κB was gradually decreased following ATO
treatment, and was possibly inactivated or degraded. Endonuclear
NF-κB was almost completely degraded following ATO treatment for 48
h, whereas the expression of cytoplasmic NF-κB was observed, which
provided evidence of synchronism in T lymphoma Jurkat and B
lymphoma Raji cells. Thus, it was concluded that ATO inhibited the
proliferation of lymphoma cells by influencing the intracellular
localization of NF-κB.
The present study revealed that the NF-κB gene and
protein were highly expressed in lymphoma cells prior to ATO
treatment, with greater expression levels observed in in T lymphoma
cells than in B lymphoma cells, which was in accordance with
clinical therapeutic effectiveness and prognosis of T cell lymphoma
compared with B cell lymphoma identified in a preliminary study
(data not shown). Following ATO treatment for 24 h, NF-κB gene and
protein expression levels began to change and the rates of cell
apoptosis significantly increased in T and B lymphoma cells,
indicating that ATO inhibited the proliferation of lymphoma cells,
possibly by inducing apoptosis. In addition, it was found that in B
cell lymphoma cells, NF-κB gene and protein expression did not vary
greatly between the three ATO treatment time points (24, 48 and 72
h), whereas in T cell lymphoma cells, NF-κB gene expression did not
exhibit a change following ATO treatment for 48 h; NF-κB protein
expression was relatively stable between 24 and 48 h, but dropped
markedly after 72 h. These data demonstrated that NF-κB expression
in lymphoma altered following 24 h ATO treatment, and the
subsequent stable NF-κB expression between 24 and 48 h in the two
cell lines may imply that NF-κB is an upstream promoter of ATO
induced apoptosis pathway in lymphoma cells, and inhibition of
NF-κB activity would further regulate downstream signal
transduction molecules such as vascular endothelial growth factor
(VEGF). Previous studies have reported that NF-κB targets the VEGF
gene, which has a specific binding site for NF-κB on its promoter
(51,52). When NF-κB is activated by external
stimulation, it translocates into the nucleus; the activated κB
sequence then binds to the VEGF promoter to promote the
transcription to upregulate VEGF expression and thus induces tumor
neovascularization (51,52). Whether downregulated NF-κB influences
VEGF directly or through a series of signal transduction pathways
following ATO treatment remains unclear, with further investigation
required.
The present study revealed that NF-κB gene and
protein expression in T cell lymphoma was further downregulated,
after being stable for a certain time period following drug
treatment; however, the drug concentration required in T cell
lymphoma was greatly increased compared with B cell lymphoma,
indicating that the clinical treatment of T cell lymphoma with ATO
may require higher doses and a longer administration time to
achieve drug efficacy in T cell lymphoma, compared with B cell
lymphoma. However, further clinical research is required to verify
this hypothesis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ and FC were major contributors in the conception
and design of the research, and revised the manuscript for
important intellectual content. Acquisition of data was performed
by FX. LZ was the major contributor in the analysis and
interpretation of data and statistical analysis. Drafting the
manuscript was performed by FC.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Taylor Elizabeth J: Dorland's Illustrated
medical dictionary. (29th ed.). Philadelphia, Saunders: pp.
10382000, ISBN 0721662544.
|
|
2
|
General Information About Adult Hodgkin
Lymphoma. National Cancer Institute 2014-04-23. Retrieved 20 June.
2014.
|
|
3
|
General Information About Adult
Non-Hodgkin Lymphoma. National Cancer Institute. 2014-04-25.
Retrieved 20 June. 2014.
|
|
4
|
Tsai SC, Lu CC, Lee CY, Lin YC, Chung JG,
Kuo SC, Amagaya S, Chen FN, Chen MY, Chan SF and Yang JS: AKT
serine/threonine protein kinase modulates bufalin-triggered
intrinsic pathway of apoptosis in CAL 27 human oral cancer cells.
Int J Oncol. 41:1683–1692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Umit UM, Berna T, Handan K, Ipek E, Berrak
Y, Can E and Bahadir GM: Role of melatonin and luzindole in rat
mammary cancer. J Invest Surg. 25:345–353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valk PJ, Verhaak RG, Beijen MA, Erpelinck
CA, van Waalwijk van Doorn-Khosrovani Barjesteh S, Boer JM,
Beverloo HB, Moorhouse MJ, van der Spek PJ, Löwenberg B and Delwel
R.: Prognostically useful gene-expression profiles in acute myeloid
leukemia. N Engl J Med. 350:1617–1628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cotter TG: Apoptosis and cancer: the
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reed JC: Dysregulation of apoptosis in
cancer. J Clin Oncol. 17:2941–2953. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Xing D, Chen Q and Chen WR:
Enhancement of chemotherapeutic agent-induced apoptosis by
inhibition of NF-kappaB using ursolic acid. Int J Cancer.
127:462–473. 2010.PubMed/NCBI
|
|
10
|
Shi M, Lu XJ, Zhang J, Diao H, Li G, Xu L,
Wang T, Wei J, Meng W, Ma JL, et al: Oridonin, a novel lysine
acetyltransferases inhibitor, inhibits proliferation and induces
apoptosis in gastric cancer cells through p53- and
caspase-3-mediated mechanisms. Oncotarget. 7:22623–22631.
2016.PubMed/NCBI
|
|
11
|
Yang P, Zhao J, Hou L, Yang L, Wu K and
Zhang L: Vitamin E succinate induces apoptosis via the PI3K/AKT
signaling pathways in EC109 esophageal cancer cells. Mol Med Rep.
14:1531–1537. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grund SC, Hanusch K and Wolf HU: Arsenic
and arsenic compounds, Ullmann's encyclopedia of industrial
chemistry. Weinheim: Wiley-VCH; 2005
|
|
13
|
Gielen M and Tiekink ER:
Metallotherapeutic drugs and metal-based diagnostic agents. Wiley;
pp. 2982005, ISBN 0-470-86403-6.
|
|
14
|
Zhou LY, Chen FY, Shen LJ, Wan HX and
Zhong JH: Arsenic trioxide induces apoptosis in the THP1 cell line
by downregulating EVI-1. Exp Ther Med. 8:85–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang S, Zhou M, Ouyang J, Geng Z and Wang
Z: Tetraarsenictetrasulfide and arsenic trioxide exert synergistic
effects on induction of apoptosis and differentiation in acute
promyelocytic leukemia cells. PLoS One. 10:e01303432015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghaffari SH, Yousefi M, Dizaji MZ, Momeny
M, Bashash D, Zekri A, Alimoghaddam K and Ghavamzadeh A: Arsenic
trioxide induces apoptosis and incapacitates proliferation and
invasive properties of U87MG Glioblastoma cells through a Possible
NF-κB-mediated mechanism. Asian Pac J Cancer Prev. 17:1553–1564.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun XP, Zhang X, He C, Qiao H, Jiang X,
Jiang H and Sun X: ABT-737 synergizes with arsenic trioxide to
induce apoptosis of gastric carcinoma cells in vitro and in vivo. J
Int Med Res. 40:1251–1264. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Wang L, Yin C, An B, Hao Y, Wei T,
Li L and Song G: Arsenic trioxide inhibits breast cancer cell
growth via microRNA-328/hERG pathway in MCF-7 cells. Mol Med Rep.
12:1233–1238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walker AM, Stevens JJ, Ndebele K and
Tchounwou PB: Evaluation of arsenic trioxide potential for lung
cancer treatment: Assessment of apoptotic mechanisms and oxidative
damage. J Cancer Sci Ther. 8:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu HT, Yao QJ, Meng YL, Li HL, Zhang H,
Luo JP, Guo CY and Geng X: Arsenic trioxide intravenous infusion
combined with transcatheter arterial chemoembolization for the
treatment of hepatocellular carcinoma with pulmonary metastasis:
Long-term outcome analysis. J Gastroenterol Hepatol. 32:295–300.
2016. View Article : Google Scholar
|
|
21
|
Ally MS, Ransohoff K, Sarin K, Atwood SX,
Rezaee M, Bailey-Healy I, Kim J, Beachy PA, Chang AL, Oro A, et al:
Effects of combined treatment with arsenic trioxide and
Itraconazole in patients with refractory metastatic basal cell
carcinoma. JAMA Dermatol. 152:452–456. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vlahopoulos SA, Cen O, Hengen N, Agan J,
Moschovi M, Critselis E, Adamaki M, Bacopoulou F, Copland JA,
Boldogh I, et al: Dynamic aberrant NF-κB spurs tumorigenesis: A new
model encompassing the microenvironment. Cytokine Growth Factor
Rev. 26:389–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Monaco C, Andreakos E, Kiriakidis S, Mauri
C, Bicknell C, Foxwell B, Cheshire N, Paleolog E and Feldmann M:
Canonical pathway of nuclear factor kappa B activation selectively
regulates proinflammatory and prothrombotic responses in human
atherosclerosis. Proc Natl Acad Sci USA. 101:5634–5639. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vidal PM, Lemmens E, Dooley D and Hendrix
S: The role of ‘anti-inflammatory’ cytokines in axon regeneration.
Cytokine Growth Factor Rev. 24:1–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bonavita E, Galdiero MR, Jaillon S and
Mantovani A: Phagocytes as corrupted policemen in cancer-related
inflammation. Adv Cancer Res. 128:141–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weniger MA and Küppers R: NF-κB
deregulation in Hodgkin lymphoma. Semin Cancer Biol. 39:32–39.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pasqualucci L and Zhang B: Genetic drivers
of NF-κB deregulation in diffuse large B-cell lymphoma. Semin
Cancer Biol. 39:26–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Lv Y, Xie Y, Hong Q, Cai G, Zhang
S, Liu W and Chen X: Change of MAX interactor 1 expression in an
anti-Thy1 nephritis model and its effect on mesangial cell
proliferation. Cell Physial Biochem. 27:391–400. 2011. View Article : Google Scholar
|
|
30
|
Ohkubo T and Ozawa M: p120(ctn) binds to
the membrane-proximal region of the E-cadherin cytoplasmic domain
and is involved in modulation of adhesion activity. J Biol Chem.
274:21409–21415. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ,
Si GY, Jin XL, Tang W, Li XS, Xong SM, et al: In vitro studies on
cellular and molecular mechanisms of arsenic trioxide (As2O3) in
the treatment of acute promyelocytic leukemia: As2O3 induces NB4
cell apoptosis with downregulation of Bcl-2 expression and
modulation of PML-RAR alpha/PML proteins. Blood. 88:1052–1061.
1996.PubMed/NCBI
|
|
32
|
Bazarbachi A, El-Sabban ME, Nasr R,
Quignon F, Awaraji C, Kersual J, Dianoux L, Zermati Y, Haidar JH,
Hermine O and de Thé H: Arsenic trioxide and interferon-alpha
synergize to induce cell cycle arrest and apoptosis in human T-cell
lymphotropic virus type I-transformed cells. Blood. 93:278–283.
1999.PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu XH, Shen YL, Jing YK, Cai X, Jia PM,
Huang Y, Tang W, Shi GY, Sun YP, Dai J, et al: Apoptosis and growth
inhibition in malignant lymphocytes after treatment with arsenic
trioxide at clinically achievable concentrations. J Natl Cancer
Inst. 91:772–778. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou L, Jing Y, Styblo M, Chen Z and
Waxman S: Glutathione-s-trandferase pi inhibits As2O3-induced
apoptosis in lymphoma cells: Involvement of hydrogen peroxide
catabolism. Blood. 105:1198–1203. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Soignet SL, Maslak P, Wang ZG, Jhanwar S,
Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J,
Scheinberg DA, et al: Complete remission after treatment of acute
promyelocytic leukemia with arsenic trioxide. N Engl J Med.
339:1341–1348. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zamzami N, Hirsch T, Dallaporta B, Petit
PX and Kroemer G: Mitochondria implication in accidental and
programmed cell death: Apoptosis and necrosis. J Bioenerg Biomembr.
29:185–193. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Daj J, Weinberg RS, Waxman S and Jing Y:
Malignant cells can be sensitized to undergo growth inhibition and
apoptosis by arsenic trioxide through modulation of the glutathione
redox system. Blood. 93:268–277. 1999.PubMed/NCBI
|
|
39
|
Shen L, Chen TX, Wang YP, Lin Z, Zhao HJ,
Zu YZ, Wu G and Ying DM: As2O3 induces apoptosis of the human B
lymphoma cell line MBC-1. J Biol Regul Homeost Agents. 14:116–119.
2000.PubMed/NCBI
|
|
40
|
Korper S, Nolte F, Thiel E, Schrezenmeier
H and Rojewski MT: The role of mitochondrial targeting in arsenic
trioxide induced apoptosis in myeloid cell lines. Br J Haematol.
124:186–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ghaffari SH, Momeny M, Bashash D, Mirzaei
R, Ghavamzadeh A and Alimoghaddam K: Cytotoxic effect of arsenic
trioxide on acute promyelocytic leukemia cells through suppression
of NFkβ-dependent induction of hTERT due to down-regulation of Pin1
transcription. Hematology. 17:198–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaileh M and Sen R: NF-κB function in B
lymphocytes. Immunol Rev. 246:254–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brasier AR: The NF-kappaB regulatory
network. Cardiovasc Toxicol. 6:111–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun W, Guo L, Shao G, Liu X, Guan Y, Su L
and Zhao S: Suppression of LASP-1 attenuates the carcinogenesis of
prostatic cancer cell lines: Key role of the NF-κB pathway. Oncol
Rep. 37:341–347. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bentires-Alj M, Barbu V, Fillet M, Chariot
A, Relic B, Jacobs N, Gielen J, Merville MP and Bours V: NF-kappaB
transcription factor induces drug resistance through MDR1
expression incancer cells. Oncogene. 22:90–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bueso-Ramos CE, Rocha FC, Shishodia S,
Medeiros LJ, Kantarjian HM, Vadhan-Raj S, Estrov Z, Smith TL,
Nguyen MH and Aggarwal BB: Expression of constitutively active
nuclear-kappaB RelA transcription factor in blasts of acute myeloid
leukemia. Hum Pathol. 35:246–253. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hinz M, Lemke P, Anagnostopoulos I, Hacker
C, Krappmann D, Mathas S, Dörken B, Zenke M, Stein H and
Scheidereit C: Nuclear factor kappaB-dependent gene expression
profiling of Hodgkin's disease tumor cells, pathogenetic
significance, and link to constitutive signal transducer and
activator of transcription 5a activity. J Exp Med. 196:605–617.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ghaffari SH, Yousefi M, Dizaji MZ, Momeny
M, Bashash D, Zekri A, Alimoghaddam K and Ghavamzadeh A: Arsenic
trioxide induces apoptosis and incapacitates proliferation and
invasive properties of U87MG Glioblastoma cells through a possible
NF-κB-mediated mechanism. Asian Pac J Cancer Prev. 17:1553–1564.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee YJ, Hwang SM, Yoon JJ, Lee SM, Kyung
EH, Kim JS, Kang DG and Lee HS: Inhibitory effect of Thuja
orientalis on TNF-α-induced vascular inflammation. Phytother Res.
24:1489–1495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Godzich M, Hodnett M, Frank JA, Su G,
Pespeni M, Angel A, Howard MB, Matthay MA and Pittet JF: Activation
of the stress protein response prevents the development of
pulmonary edema by inhibiting VEGF cell signaling in a model of
lung ischemia-reperfusion injury in rats. Blood. 2:1519–1521.
2006.
|
|
52
|
Loennechen T, Mathisen B, Hansen J,
Lindstad RI, El-Gewely SA, Andersen K, Maelandsmo GM and Winberg
JO: Colchicine induces membrane-associated activation of matrix
metalloproteinase-2 in osteosarcoma cells in an s100A4-independent
manner. Biochem Pharmacol. 66:2341–2353. 2003. View Article : Google Scholar : PubMed/NCBI
|